
Last Updated: March 27, 2026
Introduction to RNA and the Processes of Transcription
Transcription is the mechanism by which a template strand of DNA is utilized by specific RNA polymerases to generate one of the five distinct classifications of RNA. These five RNA classes are:
1. Messenger RNAs (mRNAs): This class of RNA is the genetic coding templates used by the translational machinery to determine the order of amino acids incorporated into an elongating polypeptide in the process of translation.
2. Transfer RNAs (tRNAs): This class of small RNA form covalent attachments to individual amino acids and recognize the encoded sequences of the mRNAs to allow correct insertion of amino acids into the elongating polypeptide chain.
3. Ribosomal RNAs (rRNAs): This class of RNA is assembled, together with numerous ribosomal proteins, to form the ribosomes. Ribosomes engage the mRNAs and form a catalytic domain into which the tRNAs enter with their attached amino acids. A unique function of the 28S rRNA of the large ribosomal subunit is catalytic. This rRNA catalyzes the formation of the peptide bond via the ribozyme (RNA-directed catalysis) activity.
4. Small Non-Coding RNAs (sncRNA): This class of RNA includes the small nuclear RNAs (snRNAs) involved in RNA splicing and the microRNAs (miRNAs) involved in the modulation of gene expression through the alteration of target mRNA activity.
5. Long Non-Coding RNAs (lncRNA): This class of RNA is defined by the distinction that these RNA molecules are greater than 200 nucleotides. Most of the lncRNAs are post-transcriptionally processed like mRNAs such that they are capped, polyadenylated, and spliced, yet unlike mRNAs the lncRNAs are not translated into protein.
All RNA polymerases are dependent upon a DNA template in order to synthesize RNA. The resultant RNA is, therefore, complimentary to the template strand of the DNA duplex and identical to the non-template strand. The non-template strand is called the coding strand because its sequences are identical to those of the mRNA. However, in RNA, U is substituted for T and the intronic DNA sequences are removed from the RNAs through the process of splicing.
Classes of RNA Polymerases
In prokaryotic cells, all three RNA classes are synthesized by a single polymerase. In eukaryotic cells there are three distinct classes of RNA polymerase, RNA polymerase (pol) I, II and III. Each polymerase is responsible for the synthesis of a different class of RNA. The capacity of the various polymerases to synthesize different RNAs was shown with the toxin α-amanitin. At low concentrations of α-amanitin synthesis of mRNAs are affected but not rRNAs nor tRNAs. At high concentrations, both mRNAs and tRNAs are affected. These observations have allowed the identification of which polymerase synthesizes which class of RNAs.
RNA pol I (RNAP I; also identified as RNA polymerase 7) is responsible for rRNA synthesis (excluding the 5S rRNA). The functional enzyme is a large (590 kDa) multi-subunit complex composed of 14 subunits. Twelve of the RNAP I subunits are identical to or related to subunits of the RNAP II complex. The genes that encode the subunits of the RNAP I complex are identified as POLR1 genes, with five distinct genes (POLR1A–POLR1E) expressed in humans. There are four major rRNAs in eukaryotic cells designated by their sedimentation size. The 28S, 5S, and 5.8S rRNAs are associated with the large ribosomal subunit and the 18S rRNA is associated with the small ribosomal subunit.
RNA pol II (RNAP II) in humans is a large 550kDa complex composed of 12 distinct subunits. Each of the 12 subunits of the RNAP II complex are identified as RBP1–RBP12 and the genes that encode these subunits are POLR2A–POLR2L. The RBP1 subunit is the largest subunit of the complex and is the actual RNA polymerizing activity of the complex. This subunit is encoded by the POLR2A gene. The function of RNAP II is to synthesize all of the mRNAs and some of the small nuclear RNAs (snRNAs) involved in RNA splicing, and several microRNAs. The POLR2A gene is located on chromosome 17p13.1 and is composed of 29 exons that encode a 1970 amino acid protein. The POLR2A encoded enzyme contains the C-terminal regulatory domain (CTD) that harbors the repeat sequences that play a critical role in the regulation of RNA pol II activity as discussed in the Processes of Transcription section below.
RNA pol III (RNAP III) is also a multi-subunit complex and is composed of at least 17 proteins. Ten of the RNAP III subunits are unique to this complex, two are common with subunits of RNAP I, and five are common to all three RNAP complexes. The genes encoding the RNAP III-specific proteins are identified as POLR3A–POLR3H. All of the RNAs transcribed by RNAP III are small stable untranslated RNAs. The products of RNAP III include all of the tRNAs, the 5S rRNA, several microRNAs, and the U6 small nuclear RNA (snRNA) of the splicing machinery.
Mechanisms of RNA Polymerases
Synthesis of RNA exhibits several features that are synonymous with DNA replication. RNA synthesis requires accurate and efficient initiation, elongation proceeds in the 5′ → 3′ direction (i.e. the polymerase moves along the template strand of DNA in the 3′ → 5′ direction), and RNA synthesis requires distinct and accurate termination. Transcription exhibits several features that are distinct from replication.
- Transcription initiates, both in prokaryotes and eukaryotes, from many more sites than replication
- There are many more molecules of RNA polymerase per cell than DNA polymerase
- RNA polymerase proceeds at a rate much slower than DNA polymerase (approximately 50–100 bases/sec for RNA versus near 1000 bases/sec for DNA)
- Finally, the fidelity of RNA polymerization is much lower than DNA. This is allowable since the aberrant RNA molecules can simply be turned over and new correct molecules made
Processes of mRNA Transcription
Signals are present within the DNA template that act in cis to stimulate the initiation of transcription. These sequence elements are termed promoters. Promoter sequences promote the ability of RNA polymerases to recognize the nucleotide at which initiation begins. Additional sequence elements are present within genes that act in cis to enhance polymerase activity even further. These sequence elements are termed enhancers. Transcriptional promoter and enhancer elements are important sequences used in the control of gene expression. The major defining differences between promoters and enhancers are that cis-acting promoter elements must be in a specific orientation and at a relatively fixed position in order to properly function, whereas, enhancer elements can function in either orientation, relative to the transcriptional start site, and they can be displaced large distances relative to their naturally occurring locations, and yet will still function as cis-acting enhancer elements.
Eukaryotic mRNA Transcription Initiation
The process of eukaryotic mRNA transcriptional initiation is an extremely complex event. There are numerous protein factors controlling initiation, some of which are basal factors present in all cells and others that are specific to cell type and/or the differentiation state of the cell. Two basal promoter elements that are found in essentially all eukaryotic mRNA genes are the TATA-box and the CAAT-box. Many constitutively expressed mRNA genes (house-keeping genes) also contain a GC-box promoter element (generally GGGCGG). These elements are so called because of the DNA sequences that constitute the promoter element.
The TATA-box can be found approximately 25–100 bases upstream (written -25 to -100) of the start site for transcription and the CAAT-box is generally in the -70 to -150 position. The TATA-box sequences are found ONLY in the coding strand of the gene, i.e. the strand that has the sequences identical to the resulting mRNA (except where T residues in the coding strand would be U residues in the mRNA). The CAAT-box and GC-box sequences are most often found in the template strand but can also reside in the coding strand.
Many of the basal transcription factors were originally identified by the fact that they controlled the activity of RNA pol II. Thus, the original nomenclature of these proteins was TFII, for Transcription Factor of RNA pol II. These original designations are still in common use although the accepted nomenclature now uses the GTF acronym for General Transcription Factor.
The original characterizations of numerous general transcription factors assumed they were individual activities, however, subsequent studies demonstrated that many were in fact multisubunit complexes. For example TFIID, which is now designated GTF2D, was originally identified as the factor that binds to the TATA-box. GTF2D (TFIID) is actually a complex composed of 14 different proteins. The 14 proteins of the GTF2D complex are encoded by the TAF1–TAF13 (TATA-box binding protein associated factor 1–13) and the TBP (TATA-box binding protein) genes.
The interaction of the GTF2D complex is facilitated by another complex identified as GTF2A (TFIIA) which is also a multisubunit complex composed of three different proteins. The three subunits of the GTF2A complex are encoded by the GTF2A1, GTF2A1L, and GTF2A2 genes.
The interactions of the GTF2D and GTF2A complexes then facilitates the interactions of GTF2B (TFIIB). The binding of GTF2B results in the recruitment of RNA polymerase II to the promoter. Following RNA pol II binding the GTF2E (TFIIE) and GTF2H (TFIIH) complexes bind. The GTF2E complex is composed of two proteins and the GTF2H complex is composed of 10 subunits (details in next paragraph). The two subunits of the GTF2E complex are encoded by the GTF2E1 and GTF2E2 genes.
As indicated GTF2H (TFIIH) is in fact a complex of ten proteins and this complex is not only involved in transcription but also in certain steps of DNA damage repair. There is a core GTF2H complex of seven proteins. These seven proteins are encoded by the excision repair cross complementing 2 (ERCC2) gene [also known as the xeroderma pigmentosum D (XPD) locus], the ERCC3 gene, the general transcription factor IIH subunit 1 (GTF2H1) gene, and the GTF2H2, GTF2H3, GTF2H4, and GTF2H5 genes. The role of GTF2H in DNA repair can be seen as critical since defects in its function are responsible for certain forms of xeroderma pigmentosum.
In addition to the seven core proteins, GTF2H also contains three proteins that form the active kinase activity of the complex. The critical role of GTF2H in transcription initiation is in fact due to the presence of these three proteins which function to phosphorylate serine residues in the C-terminal domain (CTD) of the large subunit of RNA pol II. These three proteins are cyclin-dependent kinase 7 (encoded by the CDK7 gene), cyclin H (encoded by the CCNH gene), and the CDK-activating kinase assembly factor (encoded by the MNAT1 gene). The overall activity of CDK7 is regulated by interaction with cyclin H.
The CTD of the large subunit of RNA pol II (the actual RNA polymerizing activity; encoded by the POLR2A gene) contains a tandem repeat sequence that is composed of the consensus heptad of amino acids: Y1S2 P3T4 S5P6 S7 which can be repeated from 25 to 52 times. It is Ser5 and Ser7 that become phosphorylated during transcriptional initiation. These serine residues are different from the serine (Ser2) phosphorylated in the CTD by P-TEFb involved in the capping process as discussed below. After transcriptional initiation has commenced and RNA pol II moves down the DNA template while the GTF2A and GTF2D complexes remain on the promoter to allow for additional rounds of initiation to take place.
Mediator Complex in Transcriptional Initiation
In addition to the complexes formed by the TFII (GTF) family proteins, another multisubunit complex termed the Mediator (MED) complex plays a critical role as a coactivator for RNA polymerase II-mediated transcription of mRNAs. In the context of mRNA transcription, the MED complex binds to the C-terminal domain of the holoenzyme of RNA polymerase II.
The MED complex was originally identified as the vitamin D receptor interacting protein (DRIP) coactivator complex and as the thyroid hormone receptor-associated protein (TRAP) complex, sometimes referred to as the TRAP/DRIP complex.
The Mediator complex is an essential transcriptional co-regulator that functions to transmit signals from promoter-bound activators and repressors to the RNA polymerase II preinitiation complex. The Mediator complex has also been found to interact at specific non-promoter domains termed chromosomal interaction domain boundaries.
Humans express 31 distinct genes that encode proteins that can contribute to the formation of the MED complex. The genes encoding proteins of the MED complex all have the MED nomenclature, such as MED1, MED4, and MED6. The functional MED complex is composed of 26 subunit proteins. The MED complex can also associate with kinases forming what is referred to as the cyclin-dependent kinase (CDK)-Mediator complex, simply identified as CDK-Mediator.
Formation of the CDK-Mediator involves the interaction of the Mediator complex with the Mediator kinase module, MKM. The MKM is composed of at least four proteins that includes cyclin C, cyclin-dependent kinase 8 (encoded by the CDK8 gene), and the proteins encoded by the MED12 and MED13 genes. Related proteins have been identified that may be able to be incorporated into the MKM and include cyclin-dependent kinase 19 (encoded by the CDK19 gene), and the proteins encoded by the MED12L and MED13L genes.
The organization of the MED complex is such that three modules have been described that includes the head, middle, and tail modules. The head module consists of proteins encoded by the MED6, MED8, MED11, MED14, MED17, MED18, MED20, MED22, MED27, MED28, and MED30 genes. The middle module consists of proteins encoded by the MED1, MED4, MED7, MED9, MED10, MED19, MED21, MED26, and MED31 genes. The tail module consists of proteins encoded by the MED15, MED16, MED23, MED24, MED25, and MED29 genes.
Within the transcriptional pre-initiation complex, RNA pol II makes contacts with several of the subunit proteins of the MED complex as well as with proteins of the TFIIB, TFIIH, and TFIIE complexes.
Eukaryotic mRNA Transcription Elongation
Elongation involves the addition of the 5’–phosphate of ribonucleotides to the 3’–OH of the elongating RNA with the concomitant release of pyrophosphate. Nucleotide addition continues until specific termination signals are encountered. Following termination the core polymerase dissociates from the template. In prokaryotic transcription, the core and sigma subunit can then re-associate forming the holoenzyme again ready to initiate another round of transcription.
Eukaryotic mRNA Transcription Termination
Transcriptional termination of eukaryotic mRNA genes occurs when RNA pol II encounters the sequence, 3′-TTATTT-5′, in the template DNA which directs the incorporation of the termination and polyadenylation [poly(A)] signal, 5′-AAUAAA-3′ in the mRNA. The processes of mRNA 3′-end polyadenylation is described in detail below. Following incorporation of the AAUAAA element into the mRNA, the cleavage and polyadenylation specificity complex, which is associated with the RNA pol II complex, recruits other proteins to the site. The proteins that are recruited then cleave the mRNA freeing it from the transcription complex and transcription terminates. RNA pol II activity can be terminated by this process within 500–2,000 nucleotides of the AAUAAA element.
Termination of RNA pol I transcription requires an RNA pol I specific termination factor that is a DNA-binding proteins. Termination of RNA pol III transcription occurs following the incorporation of a series of U residues in the transcript.
Prokaryotic Transcription
E. coli RNA polymerase is composed of five distinct protein subunits. Association of several of these generates the RNA polymerase holoenzyme. The sigma (σ) subunit is only transiently associated with the holoenzyme. This subunit is required for accurate initiation of transcription by providing polymerase with the proper cues that a start site has been encountered. In both prokaryotic and eukaryotic transcription the first incorporated ribonucleotide is a purine and it is incorporated as a triphosphate. In E. coli several additional nucleotides are added before the sigma subunit dissociates.
In E. coli, transcriptional termination occurs by both factor-dependent and factor-independent means. Two structural features of all E. coli factor-independently terminating genes have been identified. One feature is the presence of two symmetrical GC-rich segments that are capable of forming a stem-loop structure in the RNA and the second is a downstream A rich sequence in the template. The formation of the stem-loop in the RNA destabilizes the association between polymerase and the DNA template. This is further destabilized by the weaker nature of the AU base pairs that are formed, between the template and the RNA, following the stem-loop. This leads to dissociation of polymerase and termination of transcription. Most genes in E. coli terminate by this method. Factor-dependent termination requires the recognition of termination sequences by the termination protein, rho (ρ). The rho factor recognizes and binds to sequences in the 3′ portion of the RNA. This binding destabilizes the polymerase-template interaction leading to dissociation of the polymerase and termination of transcription.
Co- and Post-transcriptional Processing of RNAs
When transcription of bacterial rRNAs and tRNAs is completed these molecules are immediately ready for use in translation. No additional processing takes place. Translation of bacterial mRNAs can begin even before transcription is completed due to the lack of the nuclear-cytoplasmic separation that exists in eukaryotes. The ability to initiate translation of prokaryotic RNAs while transcription is still in progress affords a unique opportunity for regulating the transcription of certain genes. An additional feature of bacterial mRNAs is that most are polycistronic. This means that multiple polypeptides can be synthesized from a single primary transcript. Polycistronic mRNAs are very rare in eukaryotic cells but have been identified. The mitochondrial genomes in mammals and the slime mold, Dictyostelium discoideum, encode polycistronic mRNAs that are processed into primarily mono-, di-, and tricistronic transcripts. In addition, several viruses encode polycistronic RNAs.
In contrast to bacterial transcripts, eukaryotic RNAs (all three classes) undergo significant processing, some of which occurs co-transcriptionally and some post-transcriptionally. All three classes of RNA are transcribed from genes that contain introns. The RNA sequences encoded by the intronic DNA must be removed from the primary transcript prior to the RNA being biologically active. The process of intron removal is called RNA splicing. Additional processing occurs to mRNAs that can alter the 5′- and 3′-ends of the transcripts.
mRNA 5′-End Capping
The 5′ ends of nearly all eukaryotic mRNAs are capped with a unique 5′ → 5′ linkage to a 7-methylguanosine residue. Synthesis of the mRNA cap structure is catalyzed by the bifunctional enzyme encoded by the RNGTT gene (RNA guanylyltransferase and 5′-phosphatase).
The RNGTT gene is located on chromosome 6q15 and is composed of 19 exons that generate three alternatively spliced mRNAs, each of which encode a distinct protein isoform.
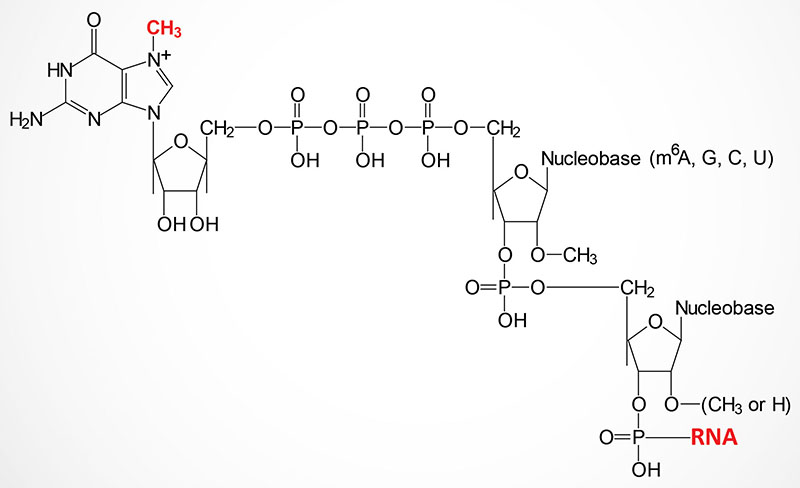
The RNGTT encoded enzymes possess mRNA 5′-triphosphatase activity in the N-terminal portion of the enzyme and mRNA guanylyltransferase activity in the C-terminal part. The mRNA 5′-triphosphatase activity of the enzyme hydrolyzes the 5′-triphosphate group of the 5′-nucleotide of the mRNA to generate a diphosphate-mRNA. The guanylyltransferase activity then adds GMP to the diphosphate–mRNA generating the 5′ → 5′ triphosphate linkage.
The guanine residue of the cap is then methylated by a second enzyme encoded by the RNMT gene (RNA guanine-7 methyltransferase). The RNMT encoded protein functions within a complex with the RAMAC (RNA guanine-7 methyltransferase activating subunit) encoded protein. The RAMAC encoded protein is often identified as RNMT-activating miniprotein (RAM). This complex is referred to as the RNMT-RAMAC complex, or often as the RNMT-RAM complex. The methylation of the N7 position of the guanosine in the mRNA cap structure requires S-adenosylmethionine (SAM) as is typical for most methyltransferase catalyzed reactions. The capped end of the mRNA is thus, protected from exonucleases and more importantly is recognized by specific proteins of the translational machinery.
The RNMT gene is located on chromosome 18p11.21 and is composed of 14 exons that generate two alternatively spliced mRNAs, each encoding distinct protein isoforms.
The RAMAC gene is located on chromosome 15q25.2 and is composed of 4 exons that encode a protein of 118 amino acids.
The capping process occurs after the newly synthesizing mRNA is around 20–30 bases long, at which point RNA pol II pauses. While RNA pol II is paused on the template, the kinase complex, known as positive transcription elongation factor b (P-TEFb), phosphorylates the catalytic enzyme of the RNA pol II complex (the POLR2A encoded enzyme) on the serine-2 residue (Ser2) in the repeat unit of the C-terminal domain (CTD). This pausing and regulatory phosphorylation event allows for the potential of attenuation in the rate of transcription.
The P-TEFb complex is composed of cyclin-dependent kinase 9 (CDK9) and either cyclin T1, T2, or K. The complex is also called C-terminal domain kinase 1 (CTDK1). There are two isoforms of the T2 cyclin identified as T2a and T2b. All four of these cyclins can associate with CDK9 resulting in the formation of multiple different forms of P-TEFb. The expression of the cyclin K gene (CCNK) is induced by the tumor suppressor, p53.
The methylation of the 2′-OH of the ribose of the nucleotide to which the cap structure is attached is catalyzed by the CMTR1 [cap-specific mRNA (nucleoside-2′-O-)-methyltransferase 1] encoded enzyme. Methylation of the 2′-OH of the ribose of the second nucleotide from the cap structure is catalyzed by the CMRT2 [cap-specific mRNA (nucleoside-2′-O-)-methyltransferase 2] encoded enzyme.
The CMTR1 gene is located on chromosome 6p21.2 and is composed of 27 exons that encode a protein of 837 amino acids.
The CMTR2 gene is located on chromosome 16q22.2 and is composed of 4 exons that generate six alternatively spliced mRNAs, all of which encode the same 770 amino acid protein.
mRNA 3′-End Polyadenylation
Almost all mammalian mRNAs are polyadenylated at the 3′-end. A specific sequence, AAUAAA, is the primary sequence recognized by one of several proteins and multiprotein complexes. In addition to the AAUAAA sequence element in the mRNA, an upstream UGUA sequence and a downstream GU-rich element act in cis to promote the recognition of the 3′-end of an mRNA by the cleavage and polyadenylation complexes. These protein complexes are responsible for recognizing the cis-acting signals in the mRNA and then catalyzing the mRNA cleavage and subsequent polyadenylation reactions.
In mammals the 3′-end cleavage and polyadenylation reactions are regulated by the interactions of at least four multiprotein complexes identified as the cleavage and polyadenylation specificity factor (CPSF), cleavage stimulatory factor (CSTF), cleavage factor I (CFI; more commonly identified as CFIm where the “m” refers to mRNA), and cleavage factor II (CFIIm).
In addition to these four complexes the actual polyadenylation reactions are catalyzed by poly(A) polymerases (PAP). Additional proteins required for mRNA polyadenylation are nuclear poly(A)-binding protein (encoded by the PABPN1 gene), symplekin, and the C-terminal domain (CTD) of the large subunit of RNA pol II.
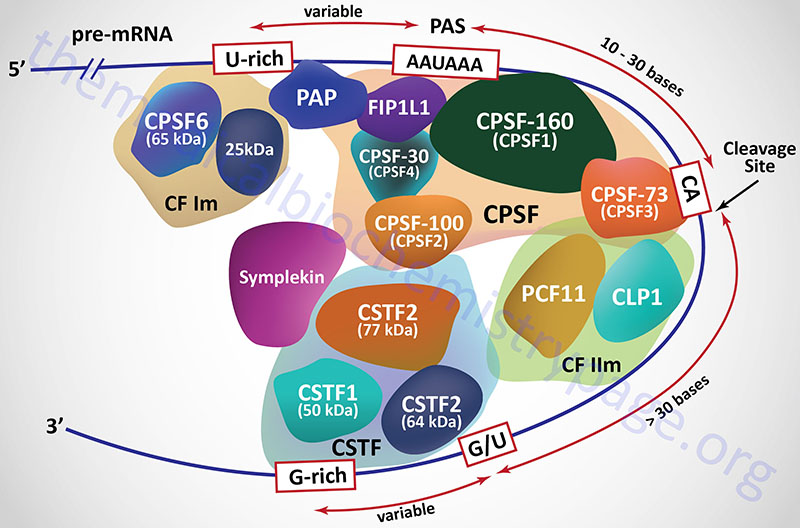
The CPSF is composed of at least seven distinct proteins, four of which were originally identified and named based upon their molecular weights. These four proteins are called CPSF-30, CPSF-73, CPSF-100, and CPSF-160 where the number represents the protein size in kDa. The CPSF-160 protein is encoded by the CPSF1 gene. The CPSF-160 protein physically binds to the AAUAAA sequence in the mRNA. The CPSF-100 protein is encoded by the CPSF2 gene. The CPSF-73 proteins is encoded by the CPSF3 gene. The CPSF-73 protein is a hydrolase that cleaves the mRNA downstream of the AAUAAA sequence element. The CPSF-30 protein in encoded by the CPSF4 gene. An additional protein that is found associated with the CPSF, that links the CPSF with poly(A) polymerases [specifically poly(A) polymerase alpha], is encoded by the FIP1L1 gene (factor interacting with PAPOLA and CPSF1). The FIP1L1 protein binds to U-rich sequences that reside upstream (5′) of the AAUAAA element and stimulates poly(A) polymerase activity.
Additional proteins that constitute the large CPSF complex include symplekin, FIP1, and WDR33. Symplekin is a scaffolding protein that not only functions in the nucleus in the process of mRNA polyadenylation, but also functions in tight junction homeostasis. The name symplekin is derived from the Greek language meaning “to tie together” or “weave”. The FIP1 protein physically interacts with poly(A) polymerases and as such the name is acronym for Factor Interacting with Poly(A) Polymerase 1. The WDR33 protein interacts with CPSF30 and this complex directly binds to an mRNA. The WDR33 protein is so-called because is is the 33rd WD repeat domain protein characterized.
The cleavage stimulatory factor (CSTF) is a complex composed of three distinct proteins. These proteins are identified as CSTF1 (50 kDa protein; also identified as CTSF50), CSTF2 (64 kDa protein; also identified as CTSF64), and CSTF3 (77 kDa protein; also identified as CTSF77) and each is encoded by a gene of the same name. The recruitment of the CSTF complex to the 3′-end of an mRNA is stimulated by the CPSF complex.
Cleavage factor I (CFIm) contains a 68 kDa protein encoded by the CPSF6 gene (cleavage and polyadenylation specific factor 6) and a smaller 25 kDa subunit. The binding of CFIm to the mRNA is facilitated by the RNA recognition motif in the N-terminus of the 68 kDa CPSF6 encoded protein. The primary function of CFIm is to recognize and bind the UGUA element in the mRNA. In addition to binding the UGUA element, CFIm has been shown to be involved in the regulation of alternative splicing. Functional CFIIm is a complex consisting of an essential component (identified as CFIIAm) and a stimulatory component (identified as CFIIBm). The CFIIAm component of the complex is composed of two proteins. These two proteins are encoded by the CLP1 gene (cleavage and polyadenylation factor I subunit 1) and the PCF11 gene (protein 1 of cleavage factor I). The CLP1 encoded protein of the CFIIm complex interacts with the CFIm complex and also with the CPSF complex.
Humans express a family of three polyadenylate polymerases (PAP), identified as poly(A) polymerase alpha (PAPOLA gene), poly(A) polymerase beta (PAPOLB gene), and poly(A) polymerase gamma (PAPOLG gene). These poly(A) polymerases possess both mRNA endonuclease activity and polyadenylate polymerase activity. The endonuclease activity cleaves the primary mRNA approximately 11–30 bases 3′ of the AAUAAA sequence element. A stretch of 20–250 adenosine residues is then added to the 3′-end by the non-template requiring polyadenylate polymerase activity of the enzymes.
Processing of tRNA and rRNA
In addition to intron removal in tRNAs, extra nucleotides at both the 5′ and 3′ ends are cleaved, the sequence 5’–CCA–3′ is added to the 3′ end of all tRNAs and several nucleotides undergo modification. There have been more than 60 different modified bases identified in tRNAs.
The post-transcriptional addition of the CCA sequences is carried out by an RNA polymerase identified as tRNA nucleotidyl transferase 1 that is encoded by the TRNT1 gene. The TRNT1 encoded enzyme is also referred to as ATP(CTP):tRNA nucleotidyltransferase or is simply called CCase. The TRNT1 gene is located on chromosome 3p26.2 and is composed of 11 exons that generates five alternatively spliced mRNAs that collectively encode two proteins of 434 amino acids (isoform 1) and 414 amino acids (isoform 2).
The TRNT1 encoded enzyme catalyzes the sequential addition of the CCA nucleotides in a template-independent but sequence-specific nucleotide polymerization reaction. This polymerase sequentially adds these three nucleotides to every tRNA transcript. The CCA terminus of all tRNAs is subjected to frequent turnover such that the TRNT1 encoded enzyme has the additional responsibility to regenerate the CCA terminus and thus, the maintenance of the proper tRNA terminus.
Both prokaryotic and eukaryotic rRNAs are synthesized as long precursors termed pre-ribosomal RNAs. In eukaryotes a 45S pre-ribosomal RNA serves as the precursor for the 18S, 28S and 5.8S rRNAs.
Splicing of RNA
Spliceosome-Mediated RNA Splicing
The removal of intronic RNA from precursor mRNA, tRNA, and rRNA molecules, in humans and other higher eukaryotes, requires a complex machinery termed the spliceosome which is composed of numerous small nuclear RNAs (snRNAs) and numerous proteins. The spliceosome catalyzes the reactions that result in intron removal and the joining together of the protein-coding exons. The spliceosome has been shown to be composed of as many as 300 distinct proteins and at least five non-coding RNAs. The five small nuclear RNAs (snRNAs) that constitute the spliceosome RNAs are identified as U1, U2, U4, U5, and U6. Each of these snRNAs is around 100–300 nucleotides in length and each are associated with several proteins forming individual small nuclear ribonucleoprotein (snRNP: pronounced “snurp”) complexes.
The composition of the U1 snRNP consists of the U1 snRNA and at least 10 proteins. The U1 RNA itself is 164 nucleotides in length. The U1 snRNP is the first complex to interact with pre-mRNAs and this interaction is key to the identification of potential intron boundaries. The U1 RNA base-pairs with the invariant consensus of nine nucleotides that resides at the exon-intron boundary. The proteins of the U1 snRNP are identified as Sm (for Smith antigen) splicesomal proteins of which there eight genes in humans. These proteins are also identified as small nuclear ribonucleoprotein polypeptides. There are also at lease three U1 specific small nuclear ribonucleoproteins encoded by the SNRNP70 encoding (U1-70K), SNRPA (encoding U1-A), and SNRPC (encoding U1-C) genes.
The composition of the U2 snRNP consists of the U2 snRNA and at least 19 proteins.
The composition of the U4/U6 snRNP consists of the U4 and U6 snRNAs and at least 12 proteins.
The composition of the U5 snRNP consists of the U5 snRNA and at least 15 proteins.
Several of the proteins present in the snRNP complexes are members of the DEAD-box helicase family of enzymes that are involved in numerous aspects of RNA metabolism. The original members of the DEAD-box helicase family were so-called because they all contained the four amino acid sequence: D-E-A-D (Asp-Glu-Ala-Asp). As a result of the isolation of variant family members, the family is more commonly referred to as the DExD/H-box protein family. Additional important protein components of the overall spliceosome are members of the SR protein family. These proteins get their name from the fact that they are enriched in Ser and Arg residues. At least 18 different SR protein encoding genes have been identified in the human genome. The activity of the SR proteins in the splicing process is controlled by their state of phosphorylation.
Introns in higher eukaryotic mRNAs can be of considerable length, in many cases spanning several thousands of bases and sometimes comprising up to 90% of the precursor mRNA. In addition, numerous precursor mRNAs undergo alternative exon splicing, a process controlled by many factors such as the cell type in which the mRNA gene is expressed. Indeed, as discussed below, the vast majority of eukaryotic mRNAs undergo some level of alternative splicing. The size and the number of introns in many mRNAs, in addition to the potential for alternative splicing, present an array of complexities that govern the control of, and catalytic processes of intron removal and exon joining.
The vast majority of eukaryotic mRNAs contain a highly conserved set of dinucleotides at the boundaries of every intron. These highly conserved sequences are GU at the 5′-end of the intron and AG at the 3′-end (shown in Figure below). In addition to these highly conserved cis-acting sequence elements there are several other important sequence elements in most introns that are necessary to control efficient and accurate splicing. Introns that contain the GU-AG consensus are spliced by the major U1, U2, U5, and U4/U6 snRNP containing spliceosomes. These introns are spliced by what is called the U2-type spliceosome. However, numerous introns have been characterized whose 5′-end and 3′-end consensus sequences are AT-AC instead of the more typical GU-AG. These second type of intron has been shown to be spliced by a spliceosome composed of a different set of snRNPs, specifically the U4/U6atac, U5, U11, and U12 snRNPs. The AT-AC introns are spliced by what is called the U12-type spliceosome. To date no precursor RNA has been identified that contains intronic RNA sequences that are spliced by both types of spliceosome. All spliced RNAs contain exclusively U2-type introns (the majority) or U12-type introns.
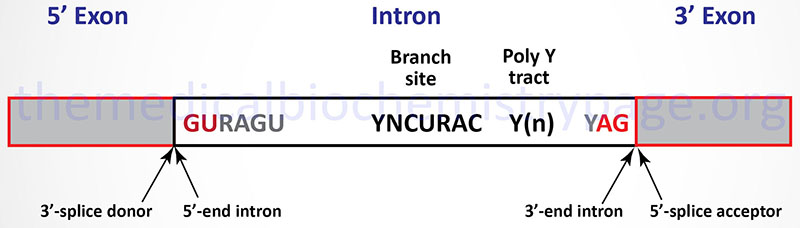
The first stage in U2-type intron splicing in mRNAs is recognition of the GU consensus element at the 5′-end of the intron by the U1 snRNP. The branch point sequence element is recognized by an additional factor called splicing factor 1, SF1 (also called the branch point binding protein, BBP). This is followed by recognition of the AG consensus element at the 3′-end of the intron and the poly(Y) tract by the U2 snRNP. Binding of the U2-snRNP results in displacement of the SF1. Once the U1 and U2 snRNP complexes are bound to the mRNA, the complex consisting of the U4/U6, and U5 snRNPs (called the tri-snRNP complex) binds to the mRNA. At this point the splicing complex is referred to as the pre-catalytic spliceosome complex. The next step involves release of the U1 and U4 snRNPs. The complex of mRNA, U2, U5, and U6 snRNP is now catalytically active and the intron is removed and the upstream and downstream exons are joined together.
The Exon Junction Complex, EJC
The exon junction complex (EJC) is a multiprotein complex that is deposited on spliced mRNAs at exon-exon junctions and functions in mRNA splicing, mRNA transport out of the nucleus, translation, and nonsense-mediated mRNA decay (NMD). The complexes of mRNA, splicesomes, and EJC packages the mRNAs into particles referred to as messenger ribonucleoprotein particles, mRNPs.
The EJC is a dynamic multiprotein complex whose core proteins are encoded by the EIF4A3, MAGOH, RBM8A, and CASC3 genes. The EIF4A3 encoded protein is a DEAD-box RNA helicase that is closely related to two translation initiation factors, eIF4A1 and eIF4A2, but it is not directly involved in the processes of translational initiation. The eIF4A3 protein functions as a clamp that binds to mRNAs in a sequence-nonspecific manner. The protein encoded by the MAGOH (mago homolog, exon junction complex subunit) gene and the protein encoded by the RBM8A (RNA binding motif protein 8A) gene form a heterodimer to lock eIF4A3 onto the mRNA. The RBM8A encoded protein is also identified as Y14. Humans express another gene that is closely related to MAGOH identified as MAGOHB whose encoded protein functions indistinguishably from the MAGOH encoded protein. The CASC3 (CASC3 exon junction complex subunit) encoded protein contacts eIF4A3 and the mRNA providing additional stability to the EJC. The protein encoded by the CASC3 is commonly identified as MLN51 (metastatic lymph node 51). The EIF4A3, MAGOH, and RBM8A encoded proteins are nuclear while the CACC3 encoded protein is mainly cytoplasmic. Nonetheless, all four core proteins do shuttle between the nucleus and the cytosol.
The EJC is not preassembled but is deposited onto mRNAs by the splicesomes and its assembly is closely linked to the process of splicing. The deposition of the EJC occurs, most often, 20-24 nucleotides upstream of spliced exon junctions during splicing. However, the EJC is not present at every exon-exon junction, and it does not always bind at the canonical position. The EJC remains associated with the mRNAs when they are transported from the nucleus to the cytoplasm. Within the cytosol the EJC serves as a platform to which numerous additional proteins to interact. There are at least 13 peripheral proteins known to interact with the EJC within the cytosol. During the first round of translation the EJC is removed and recycled back into the nucleus.
Alterations in the expression and function of EJC components is associated with several developmental defects and diseases. Complete loss of the MAGOH gene in mice is embryonic lethal. Disruptions of the activity of MAGOH and RBM8A have been associated with the loss of asymmetric cell division and anterior–posterior axis formation. Reduced expression of the EIF4A3 gene is associated with a hypopigmentation phenotype caused by mitotic arrest of melanoblasts. Mutations in the RBM8A gene are associated with thrombocytopenia with absent radii (TAR) syndrome. Expansion of an 18–20 nucleotide non-coding repeat in the 5′ untranslated region (UTR) of the EIF4A3 gene is associated with Richieri-Costa-Pereira syndrome which is characterized by craniofacial abnormalities and limb defects.
Alternative Splicing
The process of alternative splicing involves multiple interactions between splicing proteins and snRNPs that results in different patterns of exon joining from the same pre-mRNA in different cell types or under different stages of development and differentiation. Alternative splicing allows for the generation of protein isoforms that exhibit different biological properties, that differ in protein-protein interaction, that are localized to different subcellular locations, or that exhibit different catalytic activities and/or abilities. The process of alternative splicing has been identified to occur in the primary transcripts from at least 80% of all human protein coding genes.
The molecular decisions that control which exon(s) is removed and which exon(s) is included in a resultant mRNA involves both cis-acting RNA sequence elements and various protein regulators. The various cis-acting regulatory elements of an mRNA have been divided into four categories: exonic splicing enhancers (ESEs), exonic splicing silencers (ESSs), intronic splicing enhancers (ISEs) and intronic splicing silencers (ISSs). The ESEs are usually bound by members of the SR protein family which were described above. Proteins that are known to interact with the ISS and ESS sequences of the mRNA are members of the heterogeneous nuclear RNP (hnRNP) family. There are 14 known hnRNP encoding genes in the human genome. Several additional proteins are necessary for alternative splicing and these proteins (at least 18 characterized members) are expressed in a tissue-specific patterns. In addition to cis-acting sequence elements in the control of alternative splicing, secondary structure in the mRNA itself is known to regulate the alternative splicing process.
The overall process of alternative splicing requires that certain proteins are expressed that allow for splice site recognition and selection as well as expression of proteins that inhibit splice site recognition. In most cases of alternative splicing the regulation and specificity of which introns are removed and which exons are joined together is the result of a combinatorial interaction between both cis– and trans-acting activators and inhibitors.
Self-Splicing Introns
There are several different classes of reactions involved in intron removal. The two most common are the group 1 and group 2 introns. Group 1 introns are found in mRNA, tRNA, and rRNA molecules found in the chloroplasts and mitochondria of lower eukaryotic organisms as well as being found in bacterial RNA molecules. Group 2 introns are found in mRNA, tRNA, and rRNA molecules found in the chloroplasts and mitochondria of fungi, plants, and protists. The characteristic feature of both group 1 and group 2 introns is that they are self-splicing. The removal of these types of introns is catalyzed by the RNA itself via the ribozyme activity inherent in the RNA.
Group 1 introns require an external guanosine nucleotide as a cofactor. The 3’–OH of the guanosine nucleotide acts as a nucleophile to attack the 5’–phosphate of the 5′ nucleotide of the intron. The resultant 3’–OH at the 3′ end of the 5′ exon then attacks the 5′ nucleotide of the 3′ exon releasing the intron and covalently attaching the two exons together. The 3′ end of the 5′ exon is termed the splice donor site and the 5′ end of the 3′ exon is termed the splice acceptor site.
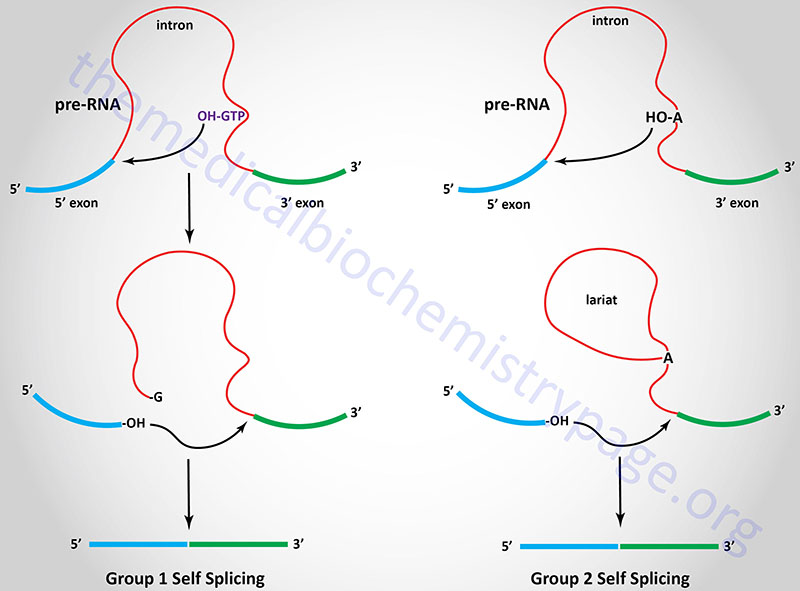
Group 2 introns are spliced similarly except that instead of an external nucleophile the 2’–OH of an adenine residue within the intron is the nucleophile. This residue attacks the 3′ nucleotide of the 5′ exon forming an internal loop called a lariat structure. The 3′ end of the 5′ exon then attacks the 5′ end of the 3′ exon as in group I splicing releasing the intron and covalently attaching the two exons together.
Clinical Significances of Alternative and Aberrant Splicing
The presence of introns in eukaryotic genes would appear to be an extreme waste of cellular energy when considering the number of nucleotides incorporated into the primary transcript only to be removed later, as well as the energy utilized in the synthesis of the splicing machinery. However, the presence of introns can protect the genetic makeup of an organism from genetic damage by outside influences such as chemical or radiation. An additionally important function of introns is to allow alternative splicing to occur, thereby, increasing the genetic diversity of the genome without increasing the overall number of genes. By altering the pattern of exons that are spliced together, from a single primary transcript, different proteins can arise from the processed mRNA from a single gene. Alternative splicing can occur either at specific developmental stages or in different cell types. As indicated earlier, the process of alternative splicing has been identified to occur in the primary transcripts from at least 80% of all human protein coding genes. One of the first clinically relevant examples of alternative splicing in humans involved the calcitonin gene (CALCA). Depending upon the site of transcription, the calcitonin gene yields an RNA that synthesizes calcitonin (thyroid) or calcitonin gene related peptide (CGRP, brain). Even more complex is the alternative splicing that occurs in the α-tropomyosin transcript. At least eight different alternatively spliced α-tropomyosin mRNAs have been identified.
Abnormalities in the splicing process can lead to various disease states. Diseases that have been identified as being due to alteration in, or the result of, alternative splicing are numerous. The causes of the alterations in the alternative splicing process are also numerous. There are diseases that are the result of mutations in splicing regulatory sequences in exons (e.g. the spinal muscular atrophies, SMA) resulting in inappropriate exon skipping. Alterations in alternative splicing can also lead to changes in protein isoform ratios that ultimately results in manifestation of disease (e.g. the diseases of the brain that result from abnormal accumulation of the tau protein). Mutations in sequences within introns can lead to the activation of cryptic splice sites resulting in abnormally spliced exons.
Numerous diseases are the result of mutations in either the 5′- or the 3′-splice sites such as various β-thalassemias. Diseases are also caused by mutations in genes the encode proteins of the spliceosomal machinery. Numerous human cancers are caused by mutations that alter splice site selection, particularly in tumor suppressor genes, or by mutations in genes encoding protein factors of the splicing machinery.
Patients suffering from a number of different connective tissue diseases exhibit humoral auto-antibodies that recognize small nuclear RNA-protein complexes (snRNPs). Patients suffering from systemic lupus erythematosis (SLE) have auto-antibodies (anti-nuclear antibodies) that recognize the U1 RNA of the spliceosome.
Nonsense-Mediated mRNA Decay: NMD
Nonsense-mediated mRNA decay (NMD) is a term that was originally used to define a control process that was identified as being responsible for the degradation of mRNAs containing truncated open reading frames, ORF. These truncated ORF were due to the presence of mutations in the encoding gene that resulted in the presence of premature termination codons (PTC) in the mRNA. However, subsequent studies found that numerous mRNAs that encoded full-length functional proteins were also degraded by the NMD machinery. It is now clear that NMD directly or indirectly influences the steady-state levels of approximately 10% of mRNAs in mammalian cells. The process of NMD, therefore, is a major contributor to overall processes of the posttranscriptional regulation of gene expression.
Specific determinants of the processes of NMD remain to be elucidated. However, in mRNAs harboring a PTC it has been determined that if the PTC is positioned more than 50 nucleotides upstream of an intron it will triggers efficient mRNA degradation. Another component of the NMD process is the exon junction complex.
Factors in the NMD Pathway
The initial identification of factors with roles in the NMD pathway was the result of studies in yeast (Saccharomyces cerevisiae) and the round worm (Caenorhabditis elegans). These studies led to the identification of seven genes in C. elegans that were identified as SMG1–SMG7 where SMG refers to suppressor with morphological effect on genitalia. These mutations were so-called because they resulted in abnormal morphogenesis of the male bursa and the hermaphrodite vulva. In studies with S. cerevisiae three genes were characterized and identified as UPF1–UPF3 for up–frameshift. These three yeast genes are the orthologues of the C. elegans SMG2, SMG3 and SMG4 genes, respectively. Subsequent homology searches identified orthologous genes in other species, including humans. In humans there are a total of 10 genes that encode proteins in the NMD pathway. These genes are identified as UPF1, UPF2, UPF3A, UPF3B, SMG1, SMG5, SMG6, SMG7, SMG8, and SMG9.
Many of the genes encoding NMD factors encode enzymes of the RNA helicase family. RNA helicases utilize the energy of ATP hydrolysis to translocate along nucleic acids. This translocation can either unwind secondary structures in the RNA thus, acting to remodel RNA-protein complexes, or the process may serve as a place marker where the helicase remains temporarily fixed in a defined position to signal to, or to directly recruit, the other components of the NMD pathway. In the case of serving as a place market the RNA helicases clamp the RNA in an ATP-dependent fashion providing nucleation centers upon which larger RNA-protein complexes can assemble.
The central factor in the NMD pathway in all organisms is UPF1. UPF1 functions as a monomeric RNA helicase. UPF1 is a member of the superfamily 1 helicase family of helicases. Superfamily 1 helicases are nucleic acid motor proteins that couple ATP hydrolysis to translocation along DNA or RNA with concomitant unwinding of secondary structures in the molecule. The central helicase domain of UPF1 is composed of two flexible RecA domains. The helicase domain of UPF1 binds single-stranded RNA and DNA and has been shown to be able to unwind long double-stranded structures. The central helicase of UPF1 is flanked by a conserved N-terminal domain rich in cysteine and histidine (CH) and a serine- and glutamine-rich (SQ) C-terminal domain. The C-terminal SQ domain of UPF1 is targeted for phosphorylation by SMG1 at multiple SQ motifs in the protein. The CH domain is the site of interaction between UPF1 and UPF2. When the helicase activity of UPF1 is activated the NMD complex translocates along the mRNA resolving secondary structure and clearing the proteins from the mRNA which allows access of the mRNA to nucleases.
UPF2 is the second core protein of the NMD pathway. The interaction of UPF2 with UPF1 induces a large conformational change in UPF1 that is necessary for its phosphorylation. This conformational change also promotes the ATPase and helicase activity of UPF1. UPF2 functions as a ring-like scaffold linking UPF1 and UPF3. Among the three core NMD factors UPF3 is the least conserved. In fact humans and other vertebrates contain two UPF3 genes identified as UPF3A and UPF3B. Both the UPF3A and UPF3B genes in humans encode alternatively spliced mRNAs. The N-terminus of the UPF3 proteins contain an RNA recognition motif that does not bind RNA but instead serves as an interaction surface for UPF2. A short motif in the C-terminus of vertebrate UPF3 interacts with several exon junction complex (EJC) proteins such as eIF4A3 and RNA binding motif protein 8A (RBM8A). UPF3 is found primarily in the nucleus where it associates with the EJCs that have been deposited on newly spliced mRNAs. Similar to the stimulatory activity of UPF2, UPF3 has also been shown to enhance the ATPase and helicase activity of UPF1. Once the complex of UPF1, UPF2, and UPF3b forms and the kinase activity of SMG1 is active, the factors SMG5, SMG6, and SMG7 interact with the phosphorylated UPF along with other general mRNA degradation factors.
The SMG1 protein of the NMD complex is a member of the phosphatidylinositol 3-kinase-related kinase family and its main function is to phosphorylate UPF1 as well as two regulatory factors, SMG8 and SMG9. SMG8 functions as a while SMG9 is a nucleotide triphosphatase (NTPase). Both SMG8 and SMG9 interact with the N-terminus of SMG with SMG9 interacting first followed by SMG8 binding to the SMG1-SMG9 complex. The interaction of SMG8 and SMG9 with SMG1 keeps the kinase domain of SMG1 in an inactive conformation. When SMG8 and SMG9 dissociate from SMG1, the kinase is activated. SMG5, SMG6, and SMG7 form a phosphate-binding complex that interacts with phosphorylated UPF1. SMG5 and SMG7 also interact with protein phosphatase 2A (PP2A) and this interaction is assumed to play a role in the dephosphorylation of UPF1. SMG5 and SMG7 function as a heterodimer and bind to phosphorylated UPF1. SMG6 appears to function as a monomer and it is an endonuclease involved in mRNA cleavage in the area of an mRNA where there are termination codons that trigger the NMD process.
In addition to the more well characterized NMD factors, a number of other proteins have been shown to be required for NMD in humans. These proteins include the RNA helicase encoded by the DHX34 (DExH-box helicase 34) gene, the DEAD-box RNA helicases encoded by the DDX5 and DDX17 genes, the NBAS (neuroblastoma amplified sequence) encoded protein, the GNL2 (G protein nucleolar 2) encoded protein, and the SEC13 (SEC13 homolog, nuclear pore and COPII coat complex component) encoded protein. This is of course, not intended to represent the full complexity of the NMD complexes that ultimately function to degrade various mRNAs, but is intended only to indicate that there are many factors required for the overall processes of NMD and its regulation.
The factors involved in NMD and an understanding of the process is undergoing rapid advancement but it still remains poorly understood how various NMD targets are selected in a global scale. Many examples of EJC-dependent and EJC-independent models for target recognition have been shown to be applicable but there are likely to be many mechanisms and target specific mechanisms functioning in the cell. This complexity can be appreciated in experiments that show that in cells that have been engineered to lack UPF1, many of the upregulated mRNAs lack previously characterized NMD recognition NMD features. The current state of NMD research suggests that it is most likely that no single NMD feature will be globally sufficient to trigger the NMD process. It is most likely that a combination of NMD-targeting and NMD-antagonizing processes function in the determining whether any given mRNA is susceptible to NMD.
Small Non-Coding RNAs (sncRNA)
It was believed that the only non-coding RNAs were the tRNAs and the rRNAs of the translational machinery. However, in a landmark study published in 1993 on the control of developmental timing in the roundworm, Caenorhabditis elegans, it was shown that the control of one gene was exerted by the small non-coding RNA (sncRNA) product of another gene. This regulatory gene is identified as lin-4 (lin-4 controls the activity of the lin-14 gene product) and it codes for two RNAs, one is approximately 22 nucleotides (nt) and the other is approximately 61 nt. Examination of the sequences of the larger RNA revealed that it could form a stem-loop structure which then serves as the precursor for the shorter RNA. The shorter lin-4 RNA is considered the founding member of class of small non-coding regulatory RNAs called microRNAs or miRNAs that consist, in their functional state, of approximately 22 nt.
It is estimated that the human genome contains at least 1000 miRNA genes and that the miRNA transcripts target over 60% of the rest of the genes in the human genome, either at the level of the mRNA products or the genes themselves. The majority of the miRNA genes in the human genome are transcribed via the activity of RNA polymerase II. The largest percentage of miRNAs are produced from independent genes that harbor their own promoter and regulatory elements. However, there are a small number of miRNAs that are derived from intronic sequences in canonical (mRNA) genes and as such are transcriptionally regulated by the “host” gene promoter and regulatory elements.
Following transcription the intronic miRNA is processed from the removed intronic RNA. miRNAs are found not only within the nucleus and the cytosol of the cell but they have also been discovered to function in various extracellular sites. In addition to miRNAs the sncRNA family includes small interfering RNAs (siRNAs) and the PIWI-interacting RNAs (piRNAs). The PIWI family represents a class of genes that were originally designated as such due to the founding member being a fruit fly (D. melanogaster) gene called P element Induced WImpy testes. The human PIWI genes encode proteins involved in stem cell differentiation and cell division in germ cells. The PIWI domain is now defined as a protein domain present in a large number of proteins that interact with nucleic acids, particularly proteins that bind and hydrolyze RNA.
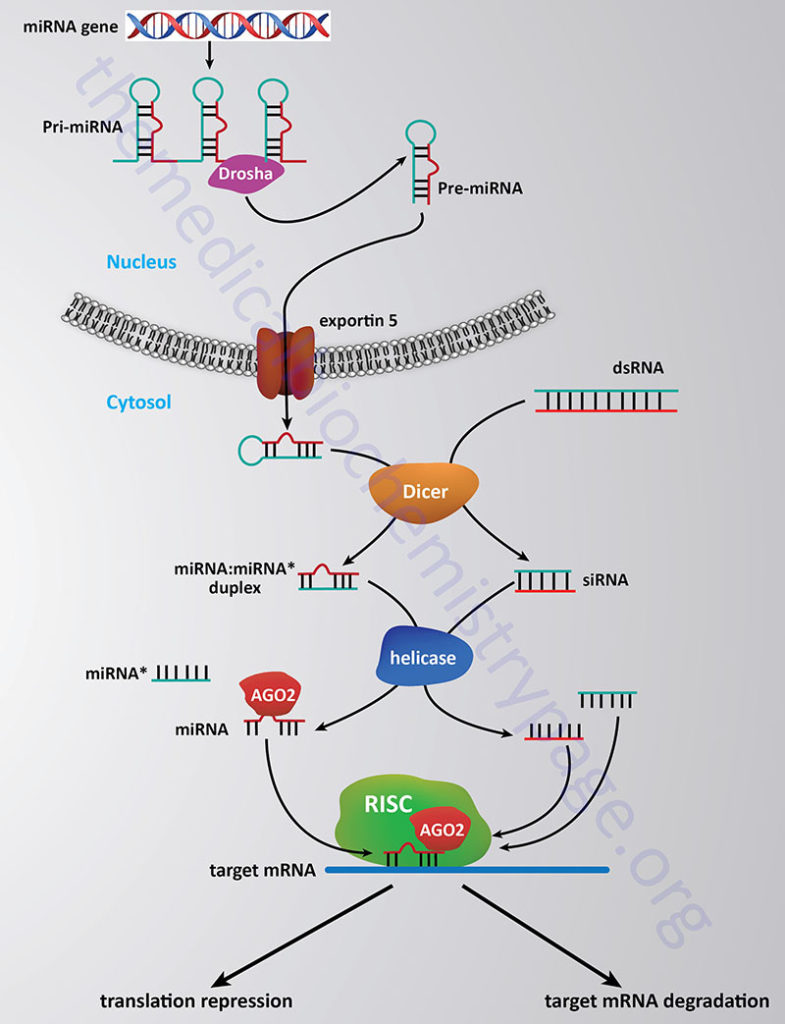
The processing and functioning of miRNAs is similar to that of the RNA silencing pathway identified in plants known as the post-transcriptional gene silencing (PTGS) pathway and the RNA inhibitory/interference (RNAi) pathway in mammals. For more details go to the Regulation of Gene Expression page. The RNAi pathway involves the enzymatic processing of double-stranded RNA into small interfering RNAs (siRNAs) of approximately 22–25 nt that may have evolved as a means to degrade the RNA genomes of RNA viruses such as retroviruses. The pathway of processing both miRNAs and siRNAs in diagrammed in the Figure below.
The stem-loop of the primary miRNA gene transcript (pri-miRNA) is first cleaved through the action of the RNase III-related activity called Drosha which takes place in the nucleus and generates the precursor miRNA (pre-miRNA). In the siRNA pathway the duplex RNAs are cleaved into 22–25 nt pieces through the action of the enzyme Dicer in the cytosol. Processed miRNA stem-loop structures are transported from the nucleus to the cytosol via the activity of exportin 5. In the cytosol the processed miRNA stem-loop is targeted by Dicer which removes the loop portion. The nomenclature of the mature miRNA duplex is miRNA:miRNA*, where the miRNA* strand is the non-functional half of the duplex (commonly referred to as the passenger strand).
Ultimately, fully processed miRNAs (commonly referred to as the guide strand) and siRNAs interact with proteins of the Argonaute (AGO) family. The active (guide) strand of RNA derived either from the miRNA or siRNA pathway is anti-sense to a region of a specific target mRNA. The role of the Argonaute proteins is to load the guide miRNA into the miRNA-induced silencing complex (miRISC) which then targets the miRNA to the correct mRNA and initiates silencing of the target mRNA. Humans express eight Argonaute encoding genes, however, in the RNAi-dependent gene silencing pathway it is exclusively the AGO2 protein that participates.
In addition to targeting mRNA stability and inhibition of protein synthesis as a means to change the level of gene expression, miRNAs have been shown to interfere with gene expression through alterations in the processes of histone modification and DNA methylation at promoter sites in target genes. These effects represent a form of epigenetic regulation of gene expression. The mechanisms by which miRNAs exert epigenetic regulation is by altering the level of DNA methyltransferases and histone deacetylases. Of profound clinical significance is that dysregulation of miRNA expression and regulation is associated with a contributory effect in the development of numerous human cancers. In addition, the activity of miRNAs appears to be associated with the oncogenic character of several genes as well at to be involved in the down-regulation of tumor suppressor genes in certain cancers. Many of the miRNAs whose activities have been shown to be deregulated in cancers have been shown to have a normal function that would exert tumor suppressive activity and/or to inhibit tumor metastasis. In addition to involvement in cancer, dysregulation or mutation in miRNA genes has been associated with numerous diseases in humans.
miRNAs and Disease
Given the critical roles of miRNAs in the regulation of diverse biological processes including cell growth and differentiation, metabolism, and apoptosis, it is not difficult to appreciate that dysfunction in miRNA regulated functions can, and indeed does, lead to pathophysiological states. Because there is a complex series of processes that must occur for a function miRNA to exerts its normal activity there are several different mechanisms that can be abnormal leading to disease pathology. Mutations in an miRNA gene or its associated regulatory elements can result in reduced or no expression or expression of a non-functional RNA product.
Mutations in the sequences of the target domain of an mRNA targeted by a miRNA can result in pathology due to loss of target mRNA regulation. Mutations in any of the genes that encode proteins required for processing of miRNAs as well as those required for target mRNA engagement can also lead to pathology. Several inherited diseases have been identified as being the result of mutations in miRNA genes. For example deletion of the miR-17-92 array is associated with growth defects and skeletal abnormalities.
Beckwith-Wiedemann syndrome, which is a classic imprinting defect related disease, has associated with the chromosome 11 deletion that results in the disease, the loss of the miR-675 gene. The most significant pathology that results from abnormal miRNA function is cancer.
With respect to cancer some miRNAs exhibit tumor suppressor activity while others can acquire oncogenic characteristics. Tumor suppressor miRNAs include miR-15a and miR-16-1. Downregulation, or loss, of either of these two miRNA genes is associated chronic lymphocytic leukemia, CLL. Oncogenic activity is associated with many miRNAs including miR-21 and miR-155 both of which are associated with CLL as well as many other cancers such as pancreatic, lung, and breast cancers.
Extracellular Small Non-Coding RNAs (RNAkines)
Several groundbreaking studies carried out in 2008 found that numerous miRNAs could be found in the blood and that several of these miRNAs were associated with disease states. Subsequently it has been shown that several secreted miRNAs, as well as other non-coding RNAs (ncRNA), exert effects in a manner classically defined for hormones. These extracellular functional ncRNA have been termed RNAkines.
The pancreas, liver, skeletal muscle, and adipose tissue have all been found to secrete RNAkines.
RNAkines that have been identified as being secreted by pancreatic β-cells includes members of the miR-29 family (miR29a, b, and c) and miR-26a. The secretion of miR-29 contributes to the recruitment of macrophages which in turn contributes to the promotion of inflammation typical of that associated with type 2 diabetes. Secreted miR-26a acts on the liver to promote insulin sensitivity. Indeed, in individuals with type 2 diabetes there is an associated decrease in the level of circulating miR-26a.
RNAkines identified as being secreted by the liver include miR-107, miR-192, and miR-1297, each of which promotes hepatic stem cell proliferation. Additionally, miR-122, miR-122-5p, and miR-192-5p have been shown to be secreted by hepatocytes and to act on immune cells of the macrophage monocyte lineage to activate inflammatory processes.
Skeletal muscle secrete miR-133a and miR-133b which act on the liver to promote insulin sensitivity.
Adipose tissue secretes both small non-coding RNAs and long non-coding RNAs (lncRNA). The secretion of miR-34a functions to promote the role of macrophages in obesity-induced inflammation in adipose tissue. The secretion of the lncRNA, lncSNHG9, by adipose tissue acts on endothelial cells of the vasculature to prevent dysfunction which in turn contributes to an inhibition of intravascular inflammation.
Cell Surface Glycosylated Small Non-Coding RNAs
Numerous extracellular molecules, either soluble or tethered to the surface of cells, are modified by the addition of carbohydrates. The typical molecules are glycolipids and glycoproteins. However, recent evidence (2021) has demonstrated that small non-coding RNAs are tethered to the exterior surface of cells and that these RNAs are modified by complex sialic acid containing carbohydrate structures. These modified RNAs have been termed glycoRNAs.
GlycoRNAs are primarily of the Y RNA family of sncRNAs and the SNO (small nucleolar) family of sncRNAs. The Y RNAs were originally identified as components of ribonucleoprotein (RNP) complexes. Humans express four Y RNA genes identified as Y1, Y3, Y4, and Y5. These Y RNA genes encode RNAs that are transcribed by RNA polymerase III and that range in size from 84 to 113 nucleotides.
The SNO family of sncRNAs are all designated by the nomenclature snoRNA. The snoRNA family, as the name implies, are predominantly found in the nucleoli. The snoRNA family of RNAs are primarily encoded within the introns of both protein coding and non-protein coding genes. The snoRNAs range in size from 60 to 300 nucleotides. The primary function of snoRNAs is in the processing of rRNAs.
Glycosylation of sncRNAs involves many of the enzymatic processes required for N-glycosylation of proteins and requires the endoplasmic reticulum (ER)/Golgi machinery for processing and cell surface presentation. However, the precise mechanism by which glycoRNAs are trafficked to and stably presented on the surface of cells is yet to be determined. Nonetheless, when present on the surface of cells the glycoRNAs have been shown to be recognized by anti-RNA antibodies as well as by the Siglec (I-type lectins) family of cell surface receptors.
Long Non-Coding RNAs (lncRNA)
Another biologically significant class of non-coding RNAs are termed the long non-coding RNAs, designated lncRNA. Like the majority of the small non-coding RNAs of the miRNA family, the vast majority of lncRNAs are transcribed by RNA polymerase II. The distinction for the term long non-coding RNA is that these RNA molecules are greater than 200 nucleotides in length.
Most of the lncRNAs are post-transcriptionally processed like mRNAs, the other major RNA polymerase II derived transcripts. Most of the lncRNAs are capped, polyadenylated, and spliced, yet unlike mRNAs the lncRNAs are not translated into protein. In addition, unlike mRNAs which are only functional in the cytoplasm, the subcellular localization of lncRNAs are diverse including nuclear, cytoplasmic, and extracellular. In addition to diverse localization, the functions of this class of RNA are also highly diverse.
According to the lncRNA database, LNCipedia, there are over 320,000 transcripts generated from over 95,000 genes that constitute the number of lncRNA in humans. Currently, less than 1% of the annotated lncRNAs have been associated with a defined functional role.
Most of the lncRNAs (representing the two major classes of lncRNA) are transcribed from either intergenic regions (i.e. between mRNA genes) or from the opposite strand of protein coding mRNA genes. The intergenic lncRNAs are referred to as large intergenic non-coding RNAs and given the designation, lincRNA. The lncRNAs that are transcribed across protein coding mRNA genes but in the opposite direction utilizing the opposite strand of DNA are referred to as natural antisense transcripts and given the designation, NAT. Of the two major lncRNA classes the lincRNA class is by far the largest with over 10,000 identified transcripts.
Although there are two major classifications for lncRNAs, there are a number of other types of functional lncRNAs and the processes by which a fully functional lncRNA are derived are also quite diverse. The next most abundant classes of lncRNAs are those that originate within enhancer elements (called eRNAs) and those that originate from promoter elements (called PROMPTs). Dependent on the mechanism for processing the 3′-end of certain lncRNAs, another class derived from intergenic regions, as for the lincRNAs, contain a 3′ triple helical domain. Another class of lncRNA molecules contain small nucleolar RNA (snoRNA) structures at the 5′- and 3′-ends (called sno-lncRNAs). Another class of lncRNA is derived from intronic sequences and when fully processed are circular RNAs whose ends are connected via a 2′,5′-phosphodiester linkage (called ciRNAs) or via a 3′,5′-phosphodiester linkage (called circRNAs).
Accumulating evidence has demonstrated that lncRNAs, like the miRNAs, have important roles in the regulation of gene expression at both the transcriptional and post-transcriptional levels in diverse cellular contexts and a variety of biological processes. The lncRNAs that remain in the nucleus have been shown to play roles in the integrity of the structure of the nucleus and in the regulation of expression of nearby genes. These lncRNA effects are referred to as cis-acting effects. Nuclear lncRNAs can also exert transcriptional effects via trans-acting effects through interactions with other proteins (e.g. transcription factors or RNA-binding proteins), RNAs (e.g. miRNAs), or DNA. The lncRNAs localized to the cytosol can also exert trans-acting effects on gene expression by interacting with proteins and RNAs.
Based upon observation of lncRNA localization and function three primary classifications of these RNAs have been designated. One class are those lncRNAs that are absolutely nuclear and exert their effects in cis, another are those lncRNAs that are mainly nuclear localized and exert their effects in trans, and lastly those lncRNAs that primarily localized to the cytoplasm.
Nuclear lncRNAs that exert their effects in cis can carry out these effects in numerous ways. The lncRNAs can form DNA-RNA triple helical structures that anchor the lncRNA to the promoter regions of targeted genes. The nuclear lncRNAs can also recruit transcription factors or chromatin modifiers to the local regions of the chromosome where the lncRNA gene resides and, thereby, affect local transcriptional events. An important example of this cis-acting effect of lncRNA is the regulated expression of the gene encoding the transcription factor MYC. MYC is a critical transcription factor regulating the expression of hundreds of genes whose encoded proteins control cell growth and differentiation events. The lncRNA identified as CCAT1-L (colon cancer associated transcript 1) is transcribed from the upstream super enhancer region of the MYC gene. The accumulation of the CCAT1-L RNA with this enhancer results in the recruitment of the insulator protein CTCF (a chromatin organizer) resulting in enhanced transcription of the MYC gene. The CTCF protein is a major DNA-binding protein that regulates gene expression through its ability to modulate transcription factor interactions with DNA and to modulate the activity of chromatin remodeling complexes.
In addition to its role in controlling MYC gene transcription CTCF is involved in the pattern of imprinting at the IGF-2 locus and is involved in the regulated transcription of the XIST gene which is required for X chromosome inactivation. The XIST gene encodes a lnRNA, the expression of which is absolutely require for X chromosome inactivation in females. In addition to XIST, there are several other genes that are critical to the process of X chromosome inactivation that encode lncRNAs including the TSIX, FTX, and JPX genes. Imprinting effects are also exerted by lncRNAs as evidenced by the effects of the NAT encoded from the antisense strand of the AIR (acute insulin response) gene, referred to as the Airn lncRNA. The Airn RNA recruits the histone methyltransferase encoded by the EHMT2 gene (also known as KMT1C) to the locus of the IGF-2 receptor gene (IGF2R) to maintain the imprinted status of that locus which contains several other imprinted genes. Cytoplasmic lncRNAs exert their effects on gene expression as well. The mechanisms include interference with post-translational protein modifications, directly interfering with mRNA translation, activating mRNA decay processes, and acting as decoy targets for miRNAs.
lncRNAs in Oxidative Stress Responses
Many lncRNA are involved in the responses of cells to oxidative stress and hypoxia. Several of the oxygen-sensitive lncRNA include lincNORS (Noncoding Oxygen-Sensitive Regulator of Sterol Homeostasis), NEAT1 (Nuclear Enriched Abundant Transcript 1), MALAT1 (Metastasis Associated Lung Adenocarcinoma Transcript 1; MALAT1 is also known as NEAT2), MEG3 (Maternally Expressed Gene 3), H19, NLUCAT1 (Nuclear LUng Cancer Associated Transcript 1), HOTAIR (HOX Antisense Intergenic RNA), HIF1A-AS2 (HIF1A–AntiSense RNA 2), and MIR210HG. Although these lncRNA are associated with effects that are exerted during oxygen deprivation, they are also involved in a wide array of other biochemical processes.
The lincNORS RNA is derived from the locus identified as MIR193BHG which encodes a tandem array of two miRNA genes, miRNA193b and miRNA365a. The NEAT1 lncRNA is derived from the NEAT1 gene. The MALAT1 lncRNA is derived from the MALAT1 gene. The MEG3 lncRNA is derived from the MEG3 gene. The NLUCAT1 lncRNA represents a nuclear localized lncRNA that is derived from the LUCAT1/lnc-ARRDC3-1 locus. The HOTAIR lncRNA is derived from HOTAIR gene which is expressed in the antisense direction to the HOXC11 gene and as such is also referred to as HOXC11-AS1. The HIF1A-AS2 lncRNA is expressed from the 3′-end of the HIF1A gene in the anti-sense direction. The MIR210HG lncRNA is encoded by the MIR210HG gene.
lncRNAs and Lipid Metabolism
The role of lncRNAs in the regulation of lipid homeostasis is an emerging area of research. Several lncRNAs have been found to regulate sterol (cholesterol) synthesis, triglyceride synthesis, and/or lipoprotein biogenesis.
MALAT1 has been shown to regulate hepatic lipid accumulation by increasing the stability of SREBP-1c, a major transcriptional regulator of the expression of several genes whose encoded proteins are required for lipid biosynthesis. Palmitic acid levels are associated with increases in the expression of MALAT1 which in turn is correlated to increased levels of SREBP-1c. MALAT1 binds to SREBP-1c in the nucleus preventing the transcription factor from being ubiquitylated. The significance of the role of MALAT1 in hepatic lipid homeostasis has been shown by the fact that if MALAT1 is knocked-out in mice the levels of hepatic lipids are significantly reduced.
H19 has also been shown to affect SREBP-1c levels by stabilizing the mRNA encoding the protein.
In contrast to the positive effects of MALAT1 and H19 on lipid biosynthesis, the lncRNA identified as lncHR1 (lncRNA HCV regulated 1; where HCV is hepatitis C virus) represses the expression of SPREB-1c resulting in reduced hepatic lipid content as well as reduced circulating triglycerides.
Another hepatic lncRNA, identified as LeXis (liver-expressed liver X receptor-induced sequence), represses the expression of several genes encoding enzymes involved in cholesterol biosynthesis. Expression of LeXis increases in the liver in response to consumption of a high-fat diet. The reduced hepatic and serum cholesterol levels that are found in response to the activity of LeXis involves the SREBP family member, SREBP-2.
Table of Several lncRNA Involved in Regulation of Hepatic Lipid Homeostasis
| lncRNA Name | Gene | Functions / Comments |
| lincNORS | MIR193BHG | nuclear-localized lncRNA that is induced under conditions of hypoxia; expression is regulated by HIF-2α; functions with RNA-binding protein RALY to regulate expression of numerous genes involved in the synthesis of cholesterol including MVK (mevalonate kinase), MVD (diphosphomevalonate decarboxylase), FDPS: (farnesyl diphosphate synthase), SQLE 9squalene epoxidase; also called squalene monooxygenase), and DHCR7 (7-dehydrocholesterol reductase) |
| H19 | H19 | promotes hepatic lipogenesis; stabilizes both the SREBP-1c protein and the mRNA encoding SREBP-1c |
| metastasis associated lung adenocarcinoma transcript 1 | MALAT1 | promotes hepatic lipogenesis; interacts with SREBP-1c and prevents its ubiquitylation, thereby enhancing the levels of functional SREBP-1c |
| LeXis: liver-expressed liver X receptor-induced sequence | CT70 | interacts with SREBP-2 and HMG-CoA reductase; reduces the synthesis of cholesterol and other sterols by blocking the expression of genes (e.g. HMGCR) encoding cholesterol synthesizing enzymes; also blocks expression of SREBP2 gene |
| lncARSR: lncRNA regulator of Akt signaling associated with HCC and RCC | LNCARSR | interacts with SREBP-2 and HMG-CoA reductase; enhances cholesterol synthesis |
| lncHR1: lncRNA HCV regulated 1 | LNCHR1 | reduces the level of active SRFEBP-1c resulting in a decrease in hepatic triglycerides and, consequently, serum triglycerides |
| APOA1-AS | APOA1-AS | regulation of the levels of the mRNAs that encode apo-A1, apo-C3, and apo-A4; inhibits expression of the APOA1 gene; reduces the formation of HDL; interacts with proteins involved in chromatin silencing |
| APOA4-AS | APOA4-AS | increases the expression of APOA4 gene; stabilizes the mRNA encoding apo-A4 |
| lncHC | MN026163 | interacts with PPARγ and ABCA1; promotes cholesterol accumulation in hepatocytes; degrades the mRNAs encoding CYP7A1 and ABCA1 |
| lncLSTR | prevents transcription factor binding to the promoter of the CYP8B1 gene that encodes an enzyme in bile acid synthesis | |
| HULC: hepatocellular carcinoma up-regulated long non-coding RNA | HULC | enhances triglyceride and cholesterol accumulation by increasing the level of acyl-CoA synthetase long chain family member 1 (ACSL1) |
lncRNAs and Disease
Given that the evidence is clear that lncRNAs exert numerous important effects on the regulation of expression of numerous genes, it is not surprising that mutations in lncRNA genes, as well as dysregulation in lncRNA functions, have been correlated to numerous disease states in humans. Indeed the progression of diabetes, breast cancer, ovarian cancer, prostate cancer, hepatocellular cancer, colon cancer, lung cancer, and bladder cancer has been associated with abnormal lncRNA activity. Indeed, more than 200 human diseases have been shown to be associated with lncRNA activity.
The H19 gene encodes a lncRNA whose expression is regulated by imprinting. The H19 gene is only expressed from the maternal allele. Overexpression of H19 is associated with the development of breast cancers. Several lncRNAs function as tumor-suppressor non-coding RNAs while other lncRNAs function as oncogenic non-coding RNAs.
The MALAT1 lncRNA is overexpressed in a number of different types of lung, cervical, hepatocellular, and colorectal cancers. The normal function of the MALAT1 lncRNA is the regulation of alternative splicing and, therefore, it is suspected that overexpression leads to aberrant splicing events resulting in loss of synthesis of important regulatory proteins. As indicated above, MALAT1 is also involved in cellular responses to oxidative stress such as in the case of hypoxia.
RNA Editing
RNA editing was a term first used to describe an unusual form of post-transcriptional processing involving the insertion of uridine (U) residues into a mitochondrial mRNA found in Trypanosoma brucei. This particular form of editing was then found to occur in many eukaryotic mRNAs. The process of RNA editing is now known to encompass a wide variety of mechanistically unrelated processes that change the nucleotide sequence of an RNA species relative to that directed by the encoding DNA. Currently RNA editing systems are divided into two general classes: substitution and insertion/deletion. In the first class, the coding sequences of a mature RNA and its gene are co-linear as they contain the same number of nucleotides but differ in nucleotide sequence where editing has occurred. In the second class, the nucleotide sequence of the mature RNA product is not co-linear with that of its DNA coding sequence since the final RNA product contains extra nucleotides relative to the encoding gene. All of the major types of cellular RNA (mRNA, rRNA, and tRNA) have been shown to be subject to editing in different organisms.
The term “RNA editing” is not used to refer to RNA modifications such as 5′-capping, splicing, and 3′-polyadenylation, nor to the formation of modified nucleosides in RNA (as is typical in tRNAs). However, it is important to keep sight of the fact that the distinctions between “RNA editing” and “RNA modification” can be less than obvious. To illustrate this fact, consider that there are instances of RNA editing involving deamination of A residues forming I (inosine) residues (see next section). If this editing occurs in the coding region of an mRNA, the edited site (I) is recognized as G during translation. However, it is also known that A residues in the wobble position of tRNA anticodons (the 5′-nucleotide) undergo deamination (by an evolutionarily related enzyme) to I, which similarly results in a change in the anticodon pairing properties. Thus, under these circumstances editing and modification can result in the same effects at the level of the resultant protein.
RNA editing systems have been identified that result in changes in A residues to I residues, referred to as A-to-I editing systems, or changes in C residues to U residues, referred to as C-to-U editing systems. The enzymes that catalyze the A-to-I edits are members of a family of adenosine deaminases that act on RNA (ADAR). This distinguishes these enzymes from the adenosine deaminase involved in the catabolism and salvage of purine nucleotides. The enzymes that catalyze C-to-U edits are called cytosine deaminases that act on RNA (CDAR). A sequence comparative analysis of ADAR and CDAR sequences demonstrated that they all belong to a superfamily of RNA-dependent deaminases that also includes tRNA-specific deaminases (ADAT). A common feature of ADAR, CDAR, and ADAT is the presence in the deaminase domain of conserved residues that are essential for catalysis. All three types of deaminases likely arose from an ancestral cytidine deaminase via the acquisition of RNA-binding domains.
The clinical significances of the editing of human RNAs is demonstrated by the observations that mutations in the ADAR1 gene are associated with rare autosomal skin pigmentation disorder (dyschromatosis symmetrica hereditaria, DSH) and with Aicardi-Goutières syndrome (AGS), an early-onset encephalopathy that often results in severe and permanent neurological damage. Defective RNA editing is also associated with a number of neurological diseases including suicidal depression, epilepsy, schizophrenia, and amyotrophic lateral sclerosis (Lou Gehrig disease).
A-to-I Editing
The process of A-to-I editing occurs on nuclear transcripts and is catalyzed by a family of enzymes referred to as ADAR. ADAR activity was initially characterized as a double-stranded RNA (dsRNA) unwinding activity and as such, these observations emphasize that ADAR are dsRNA-binding proteins and that their catalytic activity is directed toward duplex regions in RNA. Although the most biologically significant functions of ADAR is site-specific deamination in mRNA, it is known that RNA duplex regions in several types of non-coding RNAs, including microRNAs (miRNA) and small interfering RNAs (siRNA), as well as some viral RNAs are also substrates for ADAR.
Three mammalian ADAR genes give rise to four known isoforms: ADAR1p150 (interferon-inducible), ADAR1p110, ADAR2 and ADAR3. The ADAR1 proteins are encoded by the ADAR gene encoded mRNAs. Alternative promoter usage within the ADAR gene generates the full length (ADAR1p150) isoform and an N-terminally truncated (ADAR1p110) isoform. Both ADAR1 isoforms contain three dsRNA-binding domains and the deaminase domain. The ADAR1 variants and ADAR2 are expressed in many tissues, whereas the ADAR3 protein is only expressed in the brain. Although ADAR3 is catalytically inactive, it competes with ADAR1 and ADAR2 enzymes for RNA binding substrates, thereby, altering the overall profile of edited RNAs via that mechanism. The protein identified as ADAR2 is encoded by the ADARB1 gene and the ADAR3 protein is encoded by the ADARB2 gene.
The vast majority of A residues that are targets for editing are localized near splice junctions in the pre-mRNA. The formation of a dsRNA-ADAR substrates in intronic sequences could, therefore, obscure splice sites from the splicing machinery resulting in alternative splicing events. In addition, the editing of select A residues could lead to the creation or elimination of splicing sites which also could result in alternative splicing events.
A-to-I editing occurs in more RNAs than does C-to-U editing. By far, most of the mammalian mRNAs found to undergo A-to-I editing are expressed in the nervous system. Physiologically significant examples are transcripts of the ionotropic glutamate receptor (GluR) family and the serotonin receptor family. In both cases the deamination of exonic A residues leads to single amino acid changes in the resulting proteins.
Editing of glutamate receptor mRNA occurs specifically in the mRNA encoding the GluA2 (also identified as GluR2 and GluRB; encoded by the GRIA1 gene) subunit of the AMPA (2-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptors. Editing of the GluA2 mRNA is catalyzed by both the ADAR gene and the ADARB1 gene encoded enzymes (ADAR1 and ADAR2, respectively) and these edits occur at two non-synonymous sites termed the Q/R and R/G sites. These sites are so-called because the editing results in the change of a glutamine (Q) residue for an arginine (R) residue in the first site and a change of arginine (R) for glycine (G) in the second. The Q/R site is encoded by exon 11 and resides within the second transmembrane domain (TMII) of the protein. The R/G site is located just one nucleotide from the boundary between exon 13 and the downstream intron. When this site is edited, splicing favors inclusion of exon 15 over that of exon 14.
With respect to the Q/R site, editing has a profound effect on the calcium permeability of the resulting AMPA receptor. Calcium permeability of all AMPA receptor isoforms is controlled by the GluA2 subunit. In the proteins derived from the unedited GluA2 mRNA the presence of the Q residue allows Ca2+ permeability whereas the edited amino acid (R) does not. Almost all of the GluA2 present in the human brain is edited. The importance of GluA2 mRNA editing can be demonstrated by the phenotype of ADARB1 (ADAR2) knockout mice. These mice have significantly reduced editing of the Q/R site which causes them to be highly seizure-prone, and they die within 3 weeks of birth.
Editing of serotonin receptor mRNA occurs specifically in the mRNA encoding the 5-HT2C subtype within the cells of the prefrontal cortex. This mRNA, encoded by the HTR2C gene, contains five sites that are A-to-I edited. These sites are referred to as A, B, C’ (E), C, and D. The editing of the HTR2C encoded mRNA is catalyzed by both the ADAR gene and the ADARB1 gene encoded enzymes (ADAR1 and ADAR2, respectively). The most commonly detected edited 5-HT2C mRNAs are edited at the AC’C, ABD, and ABCD combination sites. There is a strong correlation to severe psychiatric behaviors and 5-HT2C mRNA editing combinations. In victims of suicide, who had been diagnosed with a history of major depression, the level of C’ editing is much higher and the level of D editing is significantly decreased when compared in unaffected individuals. Interestingly, when mice are treated with the antidepressant, fluoxetine, the pattern of C, C’, and D editing in the 5-HT2C mRNA is the exact opposite to that observed in victims of suicide.
A-to-I editing also occurs in the non-coding region of the ADAR2 (ADARB1 encoded) pre-mRNAs. The consequence of ADAR2 editing its own mRNA is the generation of an alternative splice acceptor site in intron 1, resulting in an alternative splicing event that creates a nonfunctional ADAR2 protein.
The A-to-I editing process also influences the biogenesis and target recognition of siRNAs involved in the RNAi pathway (see above). siRNA biogenesis requires processing of long dsRNA precursors into 21- to 23-nucleotide RNA duplexes which ultimately initiate transcriptional and post-transcriptional sequence-specific silencing. For details on the processing of siRNA (and miRNA) go to the Regulation of Gene Expression page. The RNA editing and RNAi pathways both involve dsRNA, therefore, editing could potentially antagonize the RNAi pathway. A-to-I edits could potentially alter the required dsRNA structures of siRNAs (and miRNAs) leading to reduced processing and thus, decreased functional siRNAs. In addition, editing of siRNAs and miRNAs could change their proper targeting to sequence-specific silencing sites in target mRNAs.
C-to-U Editing
The first reported instance of C-to-U editing was within the mRNA encoding apolipoprotein B (apoB). Editing of the apoB mRNA changes a CAA codon (at amino acid 2180) to a UAA translational stop codon leading to premature termination of protein synthesis. When the APOB gene is transcribed within hepatocytes the mRNA is not edited and a full-length apoB protein is generated called apoB-100. This apolipoprotein (apoB-100) is found exclusively with the VLDL particles produced and secreted by the liver. Within intestinal enterocytes, the APOB mRNA is edited resulting in the generation of a smaller protein called apoB-48. This apolipoprotein (apoB-48) is found exclusively associated with chylomicrons, the lipoprotein particles produced by the intestines and released to the lymphatic system.
C-to-U editing of the APOB mRNA requires a single-stranded RNA template with well defined characteristics in the immediate vicinity of the edited base, as well as protein cofactors that assemble into a functional complex referred to as a holoenzyme or editosome. This functional complex includes a minimal core composed of apolipoprotein B mRNA editing enzyme, catalytic polypeptide 1 (APOBEC-1; the catalytic deaminase) and a competence factor, APOBEC-1 complementation factor (A1CF). The function of A1CF is to act as an adaptor protein by binding both the APOBEC-1 enzyme and the mRNA substrate.
Another example of C-to-U mRNA editing involves site-specific deamination of a CGA to UGA codon in the neurofibromatosis type 1 (NF1) mRNA. The NF1 mRNA encodes a protein identified as neurofibromin 1. The editing of the NF1 mRNA introduces a translational stop codon at position 3916 that results in a truncation of the neurofibromin 1 protein in a critical domain involved in GTPase activation. Although no demonstration of a truncated NF1 protein has been shown, the editing of the NF1 mRNA has been demonstrated in peripheral nerve sheath tumors from patients with type 1 neurofibromatosis.
A third C-to-U edited mRNA encodes eukaryotic initiation factor 4, gamma 2, eIF-4G2 (also identified as p97, DAP5, and NAT1) which is a translational repressor that may be involved in repression of global translation. The editing of the eIF-4G2 mRNA was identified in studies that demonstrated the oncogenic potential of APOBEC-1 when it was overexpressed in experimental animals. In these studies it was found that the eIF-4G2 mRNA underwent C-to-U editing at multiple sites, creating of stop codons that in turn reduced the abundance of the eIF-4G2 protein. The eIF-4G2 protein has a crucial role in early embryogenesis since eIF-4G2-negative embryos die during gastrulation. Although the precise mechanism through which elevated APOBEC-1 activity leads to dysplasia and cancer is not yet defined, host adaptations have been shown to modulate the expression of APOBEC-1 in sporadic human colorectal cancers.
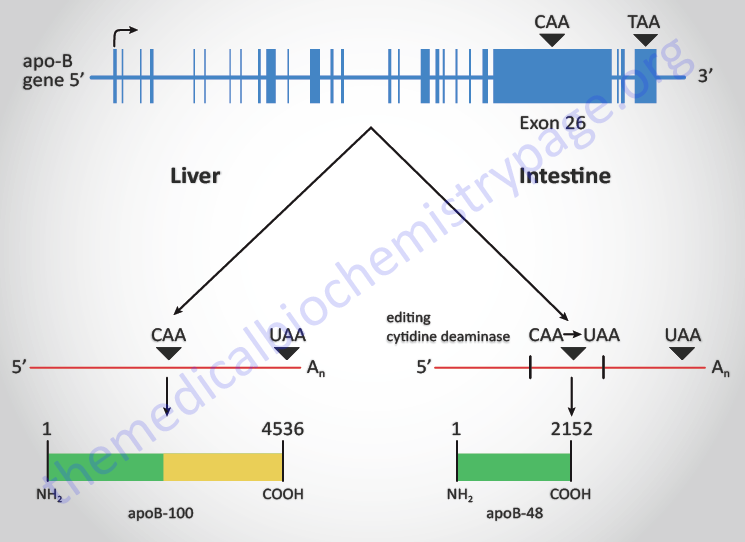
The APOBEC-1 deaminase is encoded by the APOBEC1 gene located on chromosome 12p13.1 and is composed of 6 exons that generate three alternatively spliced mRNAs that encode two distinct protein isoforms. The APOBEC1 gene is a member of a large cytidine deaminase gene family but is the only member of the family that encodes an mRNA-specific editing enzyme. All the other members of the family function primarily to edit cytidine residues in different types DNA molecules.
The other members of the family include APOBEC2, APOBEC3A, APOBEC3B, APOBEC3C, APOBEC3D, APOBEC3F, APOBEC3G, APOBEC3H, APOBEC4, and activation-induced cytidine deaminase (AICDA). Although the APOBEC3A encoded protein functions principally to deaminate cytidines of single-stranded DNA and to inhibit viruses and retrotransposons, it is also known to deaminate cytidines in mRNAs in monocytes and macrophages in response to hypoxia.
The enzymes encoded by the APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H genes function as anti-retroviral enzymes and have been shown to restrict HIV infection. Each of these four enzymes gets assembled into infectious virion particles where they deaminate cytidine residues in the viral cDNA resulting in reduced progression of reverse transcription. The resulting uracil residues induce G-to-A hypermutations in the HIV-1 genome since A base pairs with U during DNA replication.
RNA Methylation and Demethylation
Modified nucleotides, that serve distinctive functional purposes, have been known to exist in tRNA molecules from invertebrates and vertebrates for many years, where up to 25% of the nucleotides have been identified as being modified. Indeed, more than 160 distinct modifications have been characterized in all types of RNAs including tRNAs and other non-coding RNAs as well as rRNAs and mRNAs. The modifications of mRNA nucleotides, although widespread, are sparse in comparison to their density in tRNAs when comparing the level of nucleotide modification in a single tRNA to those in a single mRNA.
Although there have been numerous modified nucleotides identified in mRNAs, the modifications in mRNAs that are likely to be the most functionally significant include N6-methyladenosine (m6A, m6A, or 6mA), 5-methylcytosine (m5C, m5C, or 5mC), and pseudouridine (Ψ; 5-ribosyl isomer of uridine). The majority of m6A sites in mRNA are found in long exons, near stop codons, and in 3′ untranslated regions (3′ UTR). In addition to being found in mRNA, m6A is found in rRNA and both short and long non-coding RNAs (sncRNA and lncRNA, respectively).
Most recently N1-methyladenosine (m1A, m1A, or 1mA) has been identified as a modified nucleotide in mRNAs but little is currently understood regarding the incorporation and removal of this particular modification. The m1A was initially identified in rRNA and tRNA and this modification is involved in the tertiary structures of these RNA molecules. The incorporation of m1A in rRNA and tRNA is catalyzed by the tRNA methyltransferase 6/tRNA methyltransferase 61A (TRMT6/TRMT61A) complex. The TRMT6 encoded protein is the non-catalytic subunit of the TRMT6/TRMT61A complex, and the TRMT61A encoded protein is the catalytic subunit of the heterodimeric complex. Removal of m1A methylation is catalyzed by the ALKBH3 demethylase.
The other identified methylated sites in the nucleotides of mRNAs include the N3 position of cytidine, the N7 position of guanosine, and the 2′-OH of the ribose of adenosine. The latter methylation is most often observed in the context of N6-methyladenosine (m6A).
More data have been gathered on the mechanisms of incorporation and removal of the m6A modification and the consequences of these changes than those related to the m5C and Ψ modifications. Given that there is clear evidence demonstrating that these mRNA modifications exert functional consequences on the mRNA, these dynamic mRNA marks have been collectively termed the epitranscriptome similar to DNA methylation and histone modifications being defined as the epigenome.
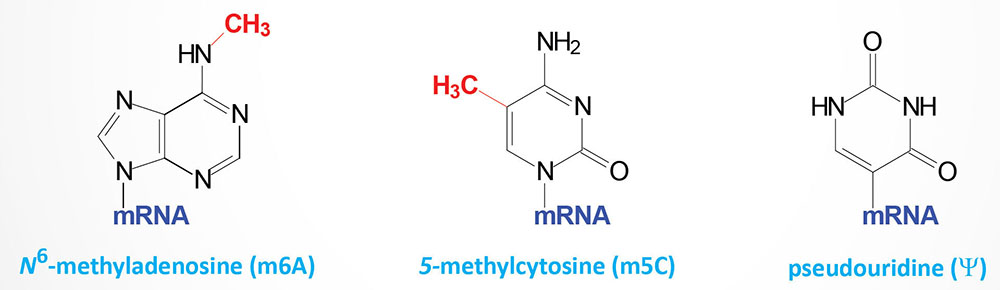
With respect to mRNA m6A modification there are enzymes that incorporate the methyl group (referred to as writers), and enzymes that remove the methyl group (referred to as erasers) and proteins that recognize the m6A structure to effect functional consequences of the modification (referred to as readers). mRNA methylation readers have been found in both the nucleus and the cytoplasm. The m6A modification has been found on adenosine residues in intronic regions of pre-mRNAs indicating that this methylation occurs co-transcriptionally or relatively soon after transcription prior to intron splicing.
Methylation of adenosine residues generating the m6A modification is restricted to adenosines residing in the context of the motif identified as R-A*–C where R=G or A; A*=methylatable A; C=cytidine. Transcriptome-wide analysis studies extended the motif in which methylated adenosines could be found to be N1-R-A*-C-N2 where N1 is A or G in 90% of the cases and N2 is very rarely a G. These transcriptome-wide m6A mapping approaches allowed for a broader consensus to be defined and has since been termed the DRACH motif where D=A, G or U; R=G or A; A=methylatable A; C=cytidine; H=A, C or U.
Formation of m6A residues in DRACH motifs has been shown to occur by default. However, if a DRACH motif resides within approximately 100 nucleotides of a splice junction the methylation of the adenosine is excluded. This exclusion of methylation is mediated by the exon junction complex (EJC; see the Splicing of RNA section above).
mRNA Methylation “Writers”
In mammalian mRNA, the m6A modification is primarily produced by a complex composed of the methyltransferases encoded by the METTL3 and METTL14 (METTL: methyltransferase like) genes. An additional methyltransferase, encoded by the METTL4 gene, may also be involved in the generation of m6A residues in mRNAs. The METTL3 protein of the complex is the subunit that binds the S-adenosylmethionine (SAM or AdoMet) that serves as the methyl donor for the methylation reaction. The role of the METTL14 subunit of the complex is the scaffold that binds the mRNA and enhances the catalytic activity of METTL3. Similar to the function of METTL3, the METTL16 methyltransferase also catalyzes the m6A modification.
The significance of the METTL3 and METTL14 enzymes was demonstrated by knocking the genes out in mice and showing that up to 99% the m6A sites were lost. In addition, the loss of the METTL3 gene in mice results in embryonic lethality demonstrating that RNA methylation is required for viability.
There are several additional proteins that are required for the activity of the methylases that generate the m6A modification. The association of all these additional factors form what is referred to as the MACOM (m6A-METTL-associated complex) complex.
Addition of the methyl group, catalyzed by the METTL3 and METTL14 complex requires the regulatory protein encoded by the WTAP (Wilms tumor 1-associating protein) gene. Another critical regulatory protein is encoded by the RBM15 (RNA-binding motif protein 15) gene. The RBM15 protein binds to m6A-methylation complexes and recruits the complex to specific sites in RNA which leads to the methylation of adenosine residues in adjacent m6A consensus motifs. Additional components of the MACOM complex include the VIRMA gene encoded protein (vir like m6A methyltransferase associated), the CBLL1 gene encoded protein (Cbl proto-oncogene like 1), and the ZC3H13 encoded protein (zinc finger CCCH-type containing 13).
The VIRMA gene is the human homolog of the Drosophila gene called Virilizer. The presence of VIRMA stimulates the METTL3/METTL14 mediated incorporation of m6A in the 3′ UTR and around the stop codon, thereby mediating alternative polyadenylation. The CBLL1 encoded protein is a member of the RING-finger type E3 ubiquitin ligase family. The CBLL1 encoded protein is often identified as Hakai which is derived from the Japanese word for “destruction” or “disruption”. The ZC3H13 encoded protein serves as the anchor for the formation of the MACOM complex. The WTAP encoded protein binds to the METTL3/METTL14 heteromeric enzyme thereby functioning as a regulator of the methyltransferase activity. The ZC3H13 encoded protein also functions as a regulator of the METTL3/METTL14 methyltransferase.
Methylated mRNA “Erasers”
As indicated, removal of the methylation in m6A residues is catalyzed by specific demethylases referred to as erasers. The fat mass and obesity-associated protein encoded by the FTO gene (also known as the ALKBH9 gene: alkB homolog 9), was the first mammalian RNA demethylase shown to catalyze m6A demethylation. Another demethylase, ALKBH5 (alkB homolog 5), is a conserved eraser of m6A methylation. The ALKBH5 gene is highly expressed in the testes and has been shown to be required for spermatogenesis and fertility in mice. The alkB gene is a bacterial gene responsible for DNA damage repair in response to alkylation damage. Mammalian homologs of the gene are, therefore, identified as ALKBH genes with there being nine identified members.
The ALKBH genes encode enzymes that are members of the large family of 2-oxoglutarate and Fe2+-dependent dioxygenases (2OG-oxygenases). Whereas the FTO and ALKBH5 enzymes have specificity for m6A residues in RNA, several of the ALKBH enzymes function in the demethylation of DNA. For instance ALKBH2 and ALKBH3 have been shown to demethylate N1-methyladenosine (1mA or m1A) and N3-methylcytosine (3mC or m3C) residues in DNA. Indeed, the ALKBH2 protein is the primary enzyme responsible for demethylation repair of alkylated DNA. The ALKBH4 gene encodes a lysine demethylase. The ALKBH8 gene encodes a tRNA methyltransferase. During the process of nucleotide demethylation, of both DNA and RNA, functional intermediates have been shown to exist. The intermediates generated by the ten eleven translocation (TET) family of DNA demethylases are discussed in the DNA: Chromatin Structure, Replication, and DNA Damage Repair page. During FTO-mediated m6A demethylation the intermediates 6-hydroxymethyladenosine (6hmA) and 6-formyladenosine (6fA) are generated and these have both been shown to exist stably in mRNA molecules.
Methylated mRNA “Readers”
The proteins that recognize the various mRNA methylation marks are responsible for the actual decoding of this information. These methylated mRNA recognition proteins, broadly categorized as RNA binding proteins (RBP) are referred to as the readers.
Humans express at least five m6A reader genes, all of which are members of the YTH (YT homology) domain family of m6A-binding proteins. The YTH domain refers to the fact that this domain was found in proteins shown to have homology to the Drosophila RNA splicing factor protein identified as YT521-B.
The five human YTH domain genes are identified as YTHDC1 (YTH m6A RBP C1), YTHDC2, YTHDF1, YTHDF2, and YTHDF3. The YTHDC1 and YTHDC2 proteins are localized to the nucleus, whereas the YTHDF1, YTHDF2, and YTHDF3 proteins are localized to the cytoplasm. Studies on the activity of these proteins have shown that the cytoplasmic readers appear to be highly specific for binding to m6A residues in mRNA. The YTHDF1 protein promotes mRNA translation while the YTHDF2 protein promotes mRNA decay. The affinity of the YTHDC2 protein for m6A sites in mRNA is weak suggesting that the function of this RBP may not rely on m6A sites.
The functions of the YTHDF proteins have been found to regulated by post-translational modifications (PTM) and subcellular localization as well as by competition for binding partners. The major PTM include O-GlcNAcylation of YTHDF1 and YTHDF3 and phosphorylation and SUMOylation of YTHDF2.
RNA reader proteins dedicated to the recognition of the m5C or Ψ modified nucleotides have yet to be characterized.
In addition to the YTH family of m6A readers there are proteins of the heterogeneous nuclear ribonucleoprotein (hnRNP) family that m6A readers. The protein encoded by the HNRNPA2B1 gene is involved in miRNA processing and the protein encoded by the HNRNPC gene is involved in mRNA splicing. Proteins of the insulin like growth factor 2 mRNA binding protein (IGF2BP) family have also been shown to be m6A readers. In this family it is the IGFBP1, IGFBP2, and IGFBP3 encoded proteins that are members of the m6A reader family. By binding to methylated sites in target mRNAs, the IGFBP proteins enhance mRNA stability.
The methylation of mRNA has the potential to affect most of the posttranscriptional steps in the processes of regulated gene expression. Indeed, evidence has demonstrated that nuclear mRNA methylation readers are involved in the control of mRNA stability and splicing, and micro-RNA (miRNA) processing, whereas cytoplasmic readers are known to be involved in the regulation of mRNA translation. The mechanisms of mRNA methylation-mediated effects are clearly the result of interactions with the m6A reader proteins, and may very well involve additional RNA binding proteins since, as indicated, readers for m5C and Ψ are yet to be characterized. Clearly defined functions for mRNA methylation in the control of splicing, stability, and translation have been identified.
However, additional important consequences of these modifications are speculated. Synthesis of truncated proteins could result as a consequence of site-specific ribosome stalling at modified codons. One of the most significant consequences of mRNA methylation may be the possibility for altered gene function as a result of regulated rewiring of the genetic code. Although the data for this latter possibility are limited in humans, there are examples in bacteria showing that the insertion of m5C leads to recoding of proline as leucine. Given that both m5C and Ψ have been shown to affect tRNA and rRNA tertiary structure, these same modifications in mRNA may also affect mRNA structures resulting in altered accessibility of binding sites for regulatory factors.
Comparatively little is known about the mechanisms of m5C production in mRNA even though m5C is common in noncoding RNAs from all domains of life. Nonetheless, the significance of m5C in human cells is evidenced from the fact that over 8,000 such sites have been identified in human mRNAs. In human cells, the methyltransferases DNMT2 (a known DNA CpG dinucleotide methyltransferase) and NSUN2 have been shown to modify certain mRNAs. There are seven humans genes encoding proteins of the NSUN methyltransferase family.
Like the potential for further chemical modification of Ψ sites in mRNAs, the m5C modification can also undergo further chemical modification. The enzymes known to carry out these m5C modifications are the same as those responsible for the step-wise removal of m5C in DNA. These enzymes are members of the ten eleven translocation (TET) gene family of demethylases. As for m5C modification by TET enzymes in DNA, the m5C in mRNAs can be modified to 5-hydroxymethylcytidine (5hmC), 5-formylcytidine (5fC), and 5-carboxylcytidine (5caC).
Clinical Implications of mRNA Nucleotide Modifications
The FTO gene (also identified as the ALKBH9 gene) was the first gene to be shown to play a role in common obesity in humans. In this original association it was found that a single nucleotide polymorphism (SNP) in the first intron of the FTO gene was correlated to increased fat mass. In animal studies it was discovered that overexpression of the FTO gene resulted in increased fat mass and body weight, whereas knocking the gene out in mice resulted in reduced fat mass and body weight. The results of numerous studies on the FTO gene have demonstrated that its role in tuning the status of m6A methylation in mRNA plays a direct role in the regulation of fat mass and obesity.
The FTO demethylase has also been shown to play a role in the regulation of dopamine signaling in the brain. Maintenance of an appropriate m6A status in a specific subset of mRNAs encoding components of the neuronal dopamine signaling pathway has been shown to be important in the overall regulation of dopamine signaling. These observations have led to the suggestion that the function of FTO may be important in the onset and progression of Parkinson disease. Indeed, malfunction in FTO has been associated with reduced brain volume in healthy elderly individuals, in individuals with attention deficit disorders, and in the propensity for addictive behaviors. Alterations in both the methylation writers, METTL3, METTL14, and WTAP, and the FTO demethylase eraser have been associated with numerous forms of cancer. These results clearly indicate that maintenance of the methylation status of the mRNA pool within numerous different cell types is critical for normal cellular function.
Aberrant m6A modification has been shown to be associated with various types of human cancers. Several proteins that were found to be associated with acute myelocytic leukemia (AML) were subsequently shown to have roles in m6A methylation of mRNAs. The WTAP, which is the regulatory protein in the METTL3/METTL14 complex, is highly expressed in AML and the level of its expression correlates with poor prognosis. The METTL3 gene is also highly expressed in AML and this results in elevated levels of m6A methylation in the BCL-2 and PTEN mRNAs. BCL-2 is an anti-apoptotic protein. PTEN is a phosphatidylinositol-3-phosphate phosphatase that was originally identified as a tumor suppressor located on chromosome 10q23. The m6A modified BCL-2 and PTEN mRNAs exhibit a higher level of translation than the unmodified mRNAs and this results in activation of AKT/PKB-mediated signal transduction.
Elevated expression of the eraser, FTO demethylase, has also been identified in AML. Overexpression of the FTO gene results in decreased levels of global mRNA m6A modification. This decrease in m6A level promotes cell proliferation and viability, while decreasing apoptosis. Expression of another m6A eraser, ALKBH5, is also elevated in AML and this increased expression correlates poor prognosis in AML patients.
Methylated RNA Readers and Malignancy
Recent studies have identified a critical role for the YTHDF2 protein in the process of immune invasion by B cell cancers. Indeed, YTHDF2 is classified as an oncogene responsible for several types of B cell malignancies by functioning as a dual methylated mRNA reader to stabilize mRNAs harboring m5C modifications while destabilizing m6A-modified mRNAs.
YTHDF2 binds to m5C-modified mRNAs that encode protein components of the mitochondrial ATP synthase (complex V). These mRNAs are encoded by the ATP5PB (also known as ), the ATP5MF (also known as ), and the ATP5MG (also known as ) genes. The ability of the YTHDF2 protein to stabilize these three mRNAs involves recruitment of the protein encoded by the PABPC1 [poly(A) binding protein cytoplasmic 1] gene. The PABPC1 encoded protein is the principal cytoplasmic polyA-binding protein of mRNA translation. This protein binds to the polyA tail of mRNAs and provides a link to the translation initiation factor, eIF-4G. The result of the YTHDF2-mediated stabilization of these mRNAs encoding components of ATP synthase is that cells are able to increase the synthesis of ATP which in turn promotes malignant cell proliferation.
The m6A-modified mRNAs that are destabilized by YTHDF2 binding are encoded by the CD19, HLA-DMA (major histocompatibility complex, class II, DM alpha), and HLA-DMB (major histocompatibility complex, class II, DM beta) genes. The CD19 gene is expressed exclusively in B cells and the encoded protein is a member of the immunoglobulin gene superfamily. The CD19 protein is the target of chimeric antigen receptor (CAR) T-cells that are used in the treatment of lymphoblastic leukemias. The HLA-DMA and HLA-DMB encoded proteins are involved in the presentation of non-self peptides to cells of the immune system. By destabilizing these mRNAs, YTHDF2 function contributes to the ability of malignant cells to evade immune surveillance.
Pseudouridine in mRNAs
Within the context of non-coding RNAs, such as tRNAs and rRNAs, the nucleotide pseudouridine (Ψ) is the most abundant non-standard nucleotide. Pseudouridine is the 5-ribosyl isomer of uridine that is generated via a 180 degree rotation of the uridine base such that the base is attached to the 1′ carbon of the ribose via a carbon-carbon glycosidic bond instead of the normal nitrogen-carbon glycosidic bond. The estimate for the level of Ψ in non-coding RNAs is on the order of 7%–10% of all the uridine. Within mRNA the level of Ψ is significantly lower and is also much less abundant than m6A. The primary human enzyme that is responsible for the conversion of uridine to pseudouridine (Ψ) is encoded by the PUS1 (pseudouridylate synthase 1) gene. Several PUS genes (at least 13) have been identified in the human genome with four (PUS1, PUS7, TRUB1, and DKC1) being identified as capable of synthesizing pseudouridine from uridine in mRNAs. Unlike the m6A modification, pseudouridylation of mRNA is believed to be irreversible.
Additionally, the presence of the Ψ residue may mediate additional chemical modifications at those sites. For example, Ψ can be further modified by N1 methylation. In addition to the normal patterns of mRNA pseudouridylation that have been detected and identified, numerous additional sites of pseudouridylation are found in cells in response to stress related stimuli such as heat shock and increased production of reactive oxygen species (ROS).
The significance of pseudouridylation, whether it be in non-coding RNAs or in mRNAs, can be evidenced from the fact that mutations in several of the pseudouridine synthase genes have been identified in various disorders. Mutations in the PUS1 gene are associated with a form of mitochondrial myopathy identified as MLASA (myopathy, lactic acidosis and sideroblastic anemia). Mutations in the dyskerin pseudouridine synthase 1 (DKC1) gene result in the multisystem disorder known as X-linked dyskeratosis congenita (X-DC). The DKC1 encoded enzyme is responsible for pseudouridylation of rRNA (as well as being a component of the telomerase complex), yet the loss of this modification leads to impaired translation of mRNAs that contain IRES elements. Internal ribosome entry sites (IRES) are used for the translation of mRNAs lacking a 5′-cap structure as well as for the translation of mRNAs under conditions where cap-dependent translation is impaired, such as in nutrient deprivation. Numerous anti-apoptotic protein coding mRNAs and tumor suppressor encoding mRNAs contain IRES elements, thus a loss of their translation, due to mutations in the DKC1 gene, can lead to development of cancers.
Catalytically Active RNAs: Ribozymes
Ribozymes represent a special class of RNA molecules that possess catalytic activity. Ribozyme are composed of well-defined tertiary structures that impart the RNAs with their unique biological activity as nucleic acid enzymes. Ribozymes have been identified in a wide range of genomes from viruses to mammals. To date, eight naturally occurring classes of ribozyme have been defined, all of which catalyze cleavage or ligation of the RNA backbone by trans-esterification or hydrolysis of phosphate groups. The catalytic properties of ribozymes are exclusively due to the capacity of these RNA molecules to assume particular structures. RNA molecules have the capacity to fold into several distinct structures which can enable a single RNA to perform more than one function.
RNA-mediated catalysis was first demonstrated in the process of intron splicing (group I and II introns). Subsequently, numerous RNAs harboring catalytic activity have been described. Ribozymes have been shown to be involved in tRNA processing (RNaseP), phosphoryl transfer reactions catalyzing the cleavage or ligation of the RNA phosphodiester backbone, in protein synthesis (peptidyltransferase) and in the regulation of gene expression. Despite the similarity of the chemistry of the reactions catalyzed by ribozymes, each molecule possesses a completely unique sequence, tertiary structure, and a specific catalytic mechanism, which reflects the diversity of catalytic strategies of ribozymes. Peptidyltransferase activity of the ribosome represents a distinct ribozyme structure and activity.
The enzymatic activity of ribozymes depends on the capacity of the RNA to fold into specific structures that impart catalytic specificity. The possibility, for a single RNA molecule, to fold into more than one structure, implies that a single RNA polymer could have more than one function. This means the RNA molecules could perform more than one task resulting in a single sequence (the genotype) manifesting multiple phenotypes. That this is indeed the case has been demonstrated for short (25-34 nucleotides) RNA sequences which exhibit the ability to bind two different ligands such as GMP and L-arginine. In addition, another experiment, designed to select for a ribozyme that catalyzed the ligation of two RNA substrates, discovered that the RNA molecule could also undergo a separate self-cleavage reaction. These two distinct catalytic reactions, ligation and cleavage, were imparted by two distinct sites of the RNA molecule. Multiple bifunctional ribozymes have been identified.
Group I introns are considerably larger and more structurally complex than any of the self-cleaving RNAs. This class of ribozyme is found in precursor mRNA, tRNA, and rRNA transcripts from a variety of organisms. The catalytic reaction carried out by group I intron ribozymes occurs in two steps. The reactions result in the ligation of flanking 5′ and 3′ exons to yield the mature RNA. Several hundred examples of this class of ribozyme have been identified. All of them share a common secondary structure and most likely a similar reaction mechanism. The Tetrahymena thermophila rRNA intron was the first group I self-splicing intron discovered (see section above). The ribozyme derived from this intron is 421 nucleotides long and is composed of a conserved catalytic core of roughly 200 nucleotides. This ribozyme catalyzes the first step of intron self-splicing using an oligonucleotide to mimic the 5′-exon. The 3′ oxygen of an exogenous guanosine serves as the nucleophile for this reaction (see Figure in the Splicing of RNA section above).
The most recently discovered functional class of ribozymes include those that are involved in the regulation of protein synthesis. Two of these newly identified ribozymes are the mammalian cytoplasmic polyadenylation element-binding protein 3 (CPEB3) ribozyme and a variant hammerhead ribozyme embedded in mammalian mRNAs. Hammerhead ribozymes are so-called because of the secondary structure evident in the active ribozyme. The hammerhead, hepatitis delta virus (HDV), hairpin, Neurospora Varkud satellite (VS), and glmS ribozymes are a class of small RNAs (50–150 nucleotides) that catalyze site-specific self-cleavage and were originally characterized in viral, virusoid, bacterial, or satellite RNA genomes.
The glmS ribozyme is a ribozyme found in Gram-positive bacteria. It is considered a metabolite-responsive ribozyme since it was originally discovered by its ability to catalyze site-specific RNA cleavage in the presence of glucosamine-6-phosphate (GlcN6P). The glmS ribozyme was originally identified in the 5′-untranslated region of the GLMS gene which is involved in the synthesis of GlcN6P. The glmS ribozyme is also considered a riboswitch since it is involved in the regulation of gene expression in response to changing concentrations of a metabolite.
The CPEB3 ribozyme is a self-cleaving non-coding RNA located in the second intron of the CPEB3 (cytoplasmic polyadenylation element-binding protein 3) gene, which belongs to a family of genes regulating the translation of mRNAs. In the case of CPEB3, neurotransmitter receptor mRNA translation is regulated. A 72 nucleotide core of the CPEB3 ribozyme sequence is sufficient to carry out self-cleavage. The cleavage activity of the CPEB3 ribozyme is slow which, under normal conditions, allows normal splicing of the CPEB3 pre-mRNA to occur. A trans-acting factor is known to interact with the ribozyme cleavage site thereby, regulating the rate of ribozyme self-cleavage. When self-cleavage is increased, the level of truncated CPEB3 pre-mRNAs increases resulting in degradation of the cleaved RNA fragments. This process may serve as a switch to turn off the synthesis of the CPEB3 protein.
Extracellular Cell Surface Associated RNAs
Extracellular RNAs were originally identified as components of ribonucleoprotein complexes that were released from dying cells. These RNAs often become autoantigens such as is the case for the anti-nuclear antigen associated with many cases of systemic lupus erythematosus (SLE). Only recently have experiments demonstrated that nuclear encoded RNAs can be found tethered to the extracellular surface of cells. These RNAs have been termed membrane-associated extracellular RNAs (maxRNA).