

Last Updated: February 18, 2026
Introduction to Refsum Disease
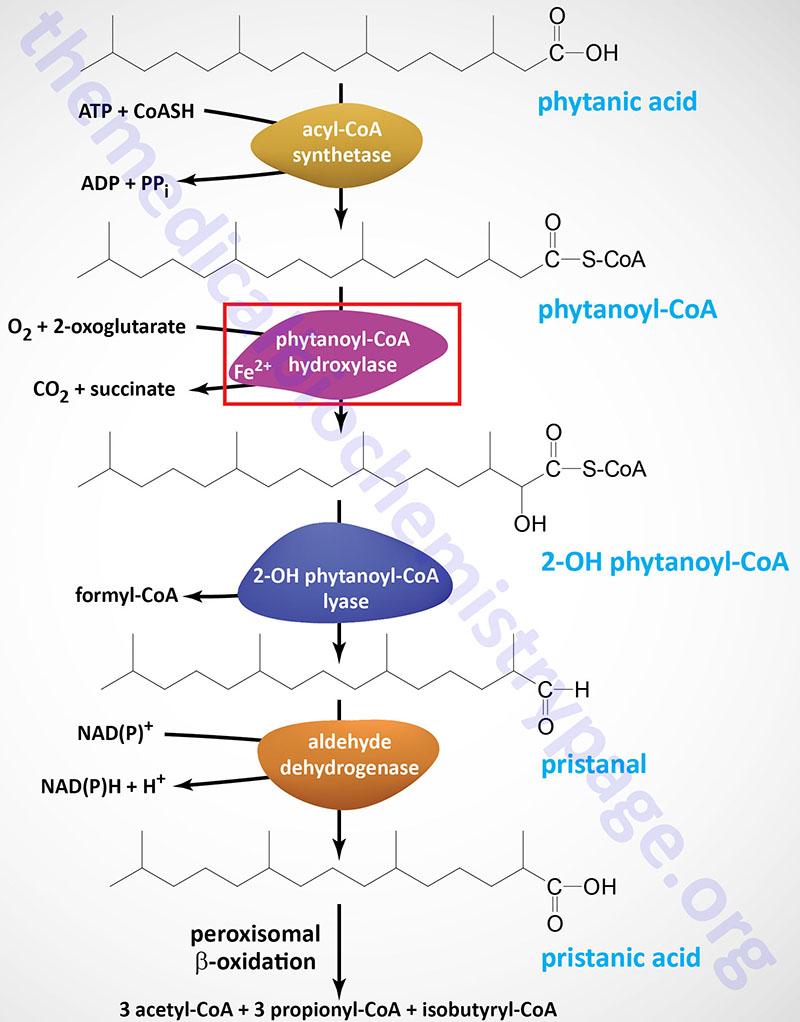
Refsum disease (heredopathia atactica polyneuritiformis) is an autosomal recessive disorder named for Sigvald Refsum who, in 1945, initially characterized the cardinal clinical features of this disease that results from defects in peroxisomal fatty acid metabolism. Specifically, the disorder is due to deficiencies in the peroxisomal enzyme responsible for one of the initial steps in the oxidation of phytanic acid, a 3-methyl substituted fatty acid derived from the chlorophyll, phytol.
Phytanoyl-CoA hydroxylase, PhyH (also called phytanoyl-CoA dioxygenase), carries out an initial α-oxidation reaction in the pathway resulting in the generation of the 19-carbon fatty acid, pristanic acid. Phytanoyl-CoA hydroxylase is a member of the large family of 2-oxoglutarate and Fe2+-dependent dioxygenases (2-OGDD).
Pristanic acid can then be oxidized by the remainder of the normal peroxisomal fatty acid β-oxidation pathway. Phytanic acid cannot be synthesized de novo in humans so dietary sources are the exclusive origin of this fatty acid. Dairy products, meat, ruminant fats, and fish are abundant sources of phytanic acid.
PhyH is localized to the peroxisomes and disorders in peroxisome biogenesis, the peroxisome biogenesis disorders (e.g. Zellweger syndrome), manifest with symptoms overlapping with PhyH deficiency.
Peroxisome Biogenesis and Function
The peroxisomes are a single membrane organelle, similar to lysosomes, present in virtually all eukaryotic cells. The peroxisome is a specialized enzyme “factory” that contains in excess of 50 different enzymes involved in a variety of metabolic processes including β-oxidation of very long chain fatty acids, α-oxidation of fatty acids and synthesis of ether-lipids. Proteins that are involved in and necessary for correct peroxisome biogenesis are called peroxins (PEX). At least 16 PEX genes have been identified in humans. Enzymes that are targeted to the peroxisomes contain either of two amino acid consensus elements called peroxisome targeting sequences (PTS).
The PTS1 is a C-terminal consensus sequence of –(S/A/C)(K/R/H)(L/M) referred to as the SKL motif. This sequence element is recognized by a cytosolic PTS1 receptor encoded by the PEX5 gene. There are two isoforms of PEX5 encoded proteins in humans identified as Pex5pS and Pex5pL (for Pex5 protein short and long forms, respectively). The Pex5pL protein has an internal 37 amino acid insertion, hence the “long” designation.
The PTS2 is an N-terminal consensus sequence of –(R/K)(L/V/I/Q)XX(L/V/I/H/Q)(L/S/G/A/K)X(H/Q)(L/A/F)–, (where X represents any amino acid). The PTS2 receptor is encoded by the PEX7 gene and the encoded protein is referred to as Pex7p. Proteins that are targeted to the membrane of the peroxisome (called peroxisome membrane proteins, PMP) contain a consensus sequence identified as the PEX19 binding site (PEX19BS) and this site is recognized by the membrane protein receptor encoded by the PEX19 gene.
Pex5pS, Pex5pL, and Pex7p interact with newly synthesized target proteins in the cytosol and direct them to the peroxisome. On the membrane of the peroxisome is a component of the protein import machinery encoded by the PEX14 gene called Pex14p. Following interaction of Pex5pS or Pex5pL, bound to a protein containing a PTS1 sequence, with Pex14p, the PTS1 containing protein is transferred into the peroxisome. The activity of Pex7p in peroxisome protein import actually requires Pex5pL as well. PTS2 containing proteins interact with Pex7p and then, in conjunction with Pex5pL, the complex interacts with Pex14p and the PTS2 containing protein is transferred into the peroxisome. Very few proteins contain a PTS2 sequence but phytanoyl-CoA hydroxylase (PHYH) is one such enzyme.
Molecular Biology of Refsum Disease
The gene (PHYH) encoding phytanoyl-CoA hydroxylase resides on chromosome 10p13 spanning 21 kb and composed of 10 exons that generate six alternatively spliced mRNAs that encode a total of five distinct protein isoforms. One of the mRNAs encodes the larger protein of 338 amino acids that contains a cleavable peroxisomal targeting signal type 2 (PTS2). The PTS2 signal is found at the N-terminus of the PhyH protein.
The PTS2 signal is recognized by a specific peroxisome protein called the peroxisomal PTS2 receptor or peroxisome biogenesis factor 7, PEX7 (also referred to as PAS7). Deficiencies in PEX7 result in rhizomelic chondrodyplasia punctata, type 1 (RCDP1) which manifests with an associated elevation in phytanic acid due to the inability to properly target PhyH to the peroxisome.
PhyH is an FK506-binding protein (FKBP)-associated protein, specifically FKBP52 (also called FKBP4). FK506 (also known as Tacrolimus or Fujimycin) is an immunosuppressant drug used in patients following allogenic organ transplantation. FK506-binding proteins were subsequently identified in complexes with FK506. The FK506-binding proteins are called immunophilins which are peptidylprolyl isomerases (PPIases) that catalyze isomerization of peptide bonds. Because of the interactions of PhyH and FKBP it is important to be aware of the potential side-effects, related to defective phytanic acid metabolism, in patients treated with immunosuppressants.
Clinical Features of Classic Refsum Disease
The cardinal clinical features of Refsum disease are retinitis pigmentosa, chronic polyneuropathy, cerebellar ataxia, and elevated protein levels in cerebrospinal fluid. Most Refsum patients have electrocardiographic changes, and some have sensorineural hearing loss and/or ichthyosis. Multiple epiphyseal dysplasia (skeletal malformations) is a conspicuous feature in some cases. Refsum disease is a slowly developing, progressive peripheral neuropathy. Progression of the disease results in severe motor weakness and muscle wasting, particularly in the lower extremities.