
Last Updated: September 15, 2022
Introduction to Pseudo-Hurler Polydystrophy
Pseudo-Hurler polydystrophy (also called mucolipidosis III α/β, ML-IIIα/β) is an autosomal recessive disorder that results as a consequence of defective targeting of lysosomal hydrolases to the lysosomes. The disorder is related to I-cell disease (mucolipidosis II α/β, ML-IIα/β) which was first described in 1967 and originally thought to be a variant of Hurler syndrome. However, I-cell disease patients present with symptoms earlier than do Hurler syndrome patients and in addition, I-cell patients do not exhibit mucopolysacchariduria.
I-cell disease is so called because fibroblasts from afflicted patients contain numerous phase-dense inclusion bodies in the cytosol. The inclusion bodies seen in I-cell disease are also observed in pseudo-Hurler polydystrophy. The symptoms associated with pseudo-Hurler polydystrophy are less severe than those of I-cell disease and present later in life. In addition, whereas, I-cell disease is a fatal disorder, patients with pseudo-Hurler polydystrophy survive into adulthood. The use of the term mucolipidosis for these related disorders is to denote diseases whose combined clinical manifestations are common to both the mucopolysaccharidoses and the sphingolipodoses.
Lysosomal Enzyme Targeting
The targeting of lysosomal enzymes to lysosomes is mediated by receptors that bind mannose 6-phosphate recognition markers on the enzymes (see Glycoproteins page). The recognition marker is synthesized in a two-step reaction in the Golgi complex. The enzyme that catalyzes the first step in this process is UDP-N-acetylglucosamine:lysosomal-enzyme N-acetylglucosaminyl-1-phosphotransferase (GlcNAc-phosphotransferase). The phosphotransferase is a low abundance membrane-associated enzyme possessing two activities. One activity is responsible for the recognition of enzymes that need to be targeted to the lysosomal compartment and the other activity is the catalytic component.
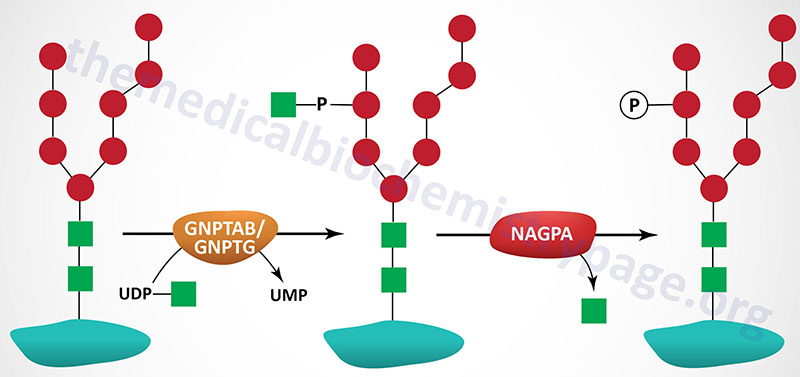
Molecular Biology of Pseudo-Hurler Polydystrophy
GlcNAc-phosphotransferase is an α2β2γ2 hexameric complex whose protein subunits are encoded by two genes. The α- and β-subunits of the phosphotransferase are encoded by the GNPTAB gene and the γ-subunits are encoded by the GNPTG gene.
The GNPTAB gene is located on chromosome 12q23.2 and is composed of 23 exons that encode a 1256 amino acid precursor protein. The protein is proteolytically processed generating a 928 amino acid fragment from the N-terminus (the α-subunit) and a 328 amino acid fragment from the C-terminus (the β-subunit).
Both pseudo-Hurler polydystrophy and I-cell disease result from defects in the GNPTAB gene. However, whereas the mutations in the GNPTAB gene that cause I-cell disease are null mutations (i.e. no enzyme activity is present) the mutations that cause Pseudo-Hurler polydystrophy reduce the activity of the encoded enzyme but do not eliminate activity. A variant disorder called mucolipidosis III γ (ML-IIIγ) results from defects in the GNPTG gene.
Clinical Features of Pseudo-Hurler Polydystrophy
The symptoms of pseudo-Hurler polydystrophy are similar to those seen in I-cell disease but are much less severe and manifest later in development, usually appearing around 2 to 4 years of age. The clinical features of pseudo-Hurler polydystrophy also overlap those seen in the mild to severe forms of mucoplosaccharidosis type I (Hurler syndrome) and type IV (Morquio syndrome). However, unlike the mucopolysaccharidotic diseases, pseudo-Hurler polydystrophy does not present with mucopolysacchariduria. This latter clinical observation is the same as for I-cell disease.
One of the most common symptoms of the disease is stiffness of the hands and shoulders similar to that seen with rheumatoid arthritis. The joint stiffness makes it difficult for pseudo-Hurler polydystrophy patients to dress themselves without assistance. By the time afflicted individuals reach 4 to 6 years of age most will manifest with claw-hand deformities, scoliosis, and short stature. A progressive destruction of the hip joints represents one of the most disabling features of this disease, often leading to a characteristic waddling gait. Mild coarsening of the facial features and thickening of the skin will be apparent in patients by the age of 6. Pelvic and vertebral abnormalities that are characteristic of pseudo-Hurler polydystrophy include flattening of the proximal femoral epiphyses, underdevelopment of the posterior parts of the vertebral bodies of the dorsal spine, hypoplasia of the anterior third of the vertebral bodies of the lumbar spine, and low iliac wings with hypoplastic bodies. Although intellectual impairment in pseudo-Hurler polydystrophy is milder than that seen in I-cell disease, at least 50% of patients will manifest with some level of learning disability or intellectual impairment.