
Last Updated: July 9, 2026
Introduction to Translation
Translation is the RNA directed synthesis of proteins. This process requires all three classes of RNA. Although the chemistry of peptide bond formation is relatively simple, the processes leading to the ability to form a peptide bond are exceedingly complex. The template for correct addition of individual amino acids is the mRNA, yet both tRNAs and rRNAs are involved in the process. The tRNAs carry activated amino acids into the ribosome which is composed of rRNA and ribosomal proteins. The ribosome is associated with the mRNA ensuring correct access of activated tRNAs and containing the necessary enzymatic activities to catalyze peptide bond formation.
Historical Perspectives
Early genetic experiments demonstrated:
- The co-linearity between the DNA and protein encoded by the DNA. Yanofsky showed that the order of observed mutations in the E. coli tryptophan synthetase gene was the same as the corresponding amino acid changes in the protein
- Crick and Brenner demonstrated, from a large series of double mutants of the bacteriophage T4, that the genetic code is read in a sequential manner starting from a fixed point in the gene, the code was most likely a triplet and that all 64 possible combinations of the 4 nucleotides code for amino acids, i.e. the code is degenerate since there are only 20 amino acids
The above mentioned experiments only indicated deductive correlation’s regarding the genetic code. The precise dictionary of the genetic code was originally determined by the use of in vitro translation systems derived from E. coli cells. Synthetic polyribonucleotides were added to these translation system along with all twenty amino acids. One amino acid at a time was radiolabeled. The first demonstration of the dictionary of the genetic code was with the use of poly(U). This synthetic polyribonucleotide encoded the amino acid phenylalanine, i.e. the resulting polypeptide was poly(F).
The utilization of a variety of repeating di- tri- and tetra polyribonucleotides established the entire genetic code. These results of these experiments confirmed that some amino acids are encoded for by more than one triplet codon, hence the degeneracy of the genetic code. These experiments also established the identity of translational termination codons.
An additional important point to come from these early experiments was that the 5′ end of the RNA corresponded to the amino terminus of the polypeptide. This was important since previous labeling experiments had demonstrated that the N-terminus is the beginning of the elongating polypeptide. Therefore, in vitro translation experiments established that the RNA is read in the 5′ to 3′ direction.
Crick first postulated that translation of the genetic code would be carried out through mediation of adapter molecules. Each adapter was postulated to carry a specific amino acid and to recognize the corresponding codon. He suggested that the adapters contain RNA because codon recognition could then occur by complementarity to the sequences of the codons in the mRNA.
During the course of in vitro protein synthesis and labeling experiments it was shown that the amino acids became transiently bound to a low molecular weight mass fraction of RNA. This fraction of RNAs have been termed transfer RNAs (tRNAs) since they transfer amino acids to the elongating polypeptide. These results indicate that accurate translation requires two equally important recognition steps:
- The correct choice of amino acid needs to be made for attachment to the correspondingly correct tRNA
- Selection of the correct amino acid-charged tRNA by the mRNA. This process is facilitated by the ribosomes which we will discuss below
Summary of Experiments to Determine the Genetic Code
1. The genetic code is read in a sequential manner starting near the 5′ end of the mRNA. This means that translation proceeds along the mRNA in the 5′ → 3′ direction which corresponds to the N-terminal to C-terminal direction of the amino acid sequences within proteins.
2. The code is composed of a triplet of nucleotides.
3. That all 64 possible combinations of the 4 nucleotides code for amino acids, i.e. the code is degenerate since there are only 20 amino acids.
The precise dictionary of the genetic code was determined with the use of in vitro translation systems and polyribonucleotides. The results of these experiments confirmed that some amino acids are encoded by more than one triplet codon, hence the degeneracy of the genetic code. These experiments also established the identity of translational termination codons.
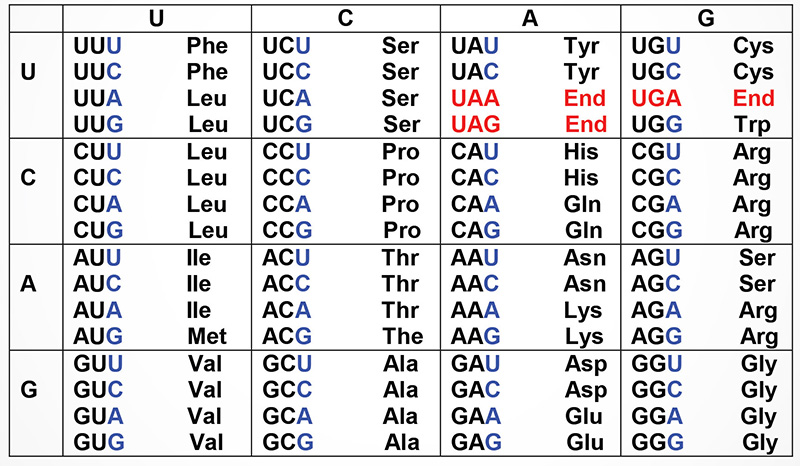
The Genetic Code
Shown below are the triplets that are used for each of the 20 amino acids found in eukaryotic proteins. The row on the left side indicates the first nucleotide of each triplet and the row across the top represents the second nucleotide. The wobble position nucleotides are indicated in blue. The three stop codons are highlighted in red.
Characteristics of tRNAs
More than 300 different tRNAs have been sequenced, either directly or from their corresponding DNA sequences. tRNAs vary in length from 60–95 nucleotides (18–28 kD). The majority contain 76 nucleotides. Evidence has shown that the role of tRNAs in translation is to carry activated amino acids to the elongating polypeptide chain. All tRNAs:
1. Exhibit a cloverleaf-like secondary structure.
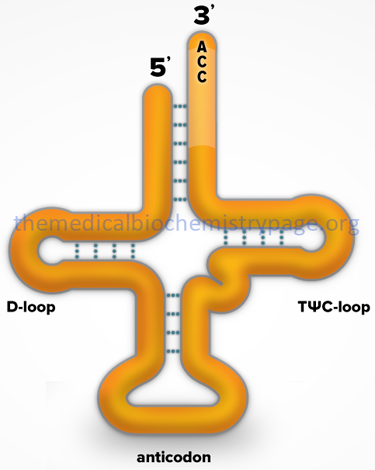
2. Have a 5′-terminal phosphate.
3. Have a 7 bp stem that includes the 5′-terminal nucleotide and may contain non-Watson-Crick base pairs, e.g. 5′-GU-3′. This portion of the tRNA is called the acceptor since the amino acid is carried by the tRNA while attached to the 3′-terminal OH group.
4. Have a D loop and a TΨC loop.

5. Have an anticodon loop.
6. Terminate at the 3′-end with the sequence 5’–CCA–3′.
7. Contain 13 invariant positions and 8 semi-variant positions.
8. Contain numerous modified nucleotide bases (see Biochemistry of Nucleic Acids for structures of several modified nucleotides in tRNAs).
Activation of Amino Acids
Activation of amino acids is carried out by a two step process catalyzed by aminoacyl-tRNA synthetases. Humans express both nuclear genome and mitochondrial genome encoded aminoacyl-tRNA synthetases. The aminoacyl-tRNA synthetases are divided into two classes: class I and class II.
The class I enzymes catalyze addition of the amino acid to the 2′-OH of a target tRNA and do so as either monomeric or dimeric enzymes. Humans express 19 class I aminoacyl-tRNA synthetases of which eight function in the translation of mitochondrial genome (mtDNA) encoded RNAs.
The class II enzymes catalyze addition of the amino acid to the 3′-OH of a target tRNA and do so as dimeric or tetrameric enzymes. Humans express 19 class II aminoacyl-tRNA synthetase genes which includes two genes that encode the two subunits (α and β) of a phenylalanine-tRNA synthetase and eight enzymes that function in the translation of mitochondrial genome (mtDNA) encoded RNAs.
Two of the class II aminoacyl-tRNA synthetases, encoded by the AARS1 and AARS2 genes, have been shown to function not only in the process of amino acid activation for translation, but also in the process of protein lysine lactylation. The AARS1 gene encodes the cytoplasmic alanyl-tRNA synthetase 1, whereas the AARS2 gene encodes the mitochondria-localized alanyl-tRNA synthetase 2. The ability of these two enzymes to function as lactylation writers stems, in part, from the fact that alanine and lactate are structurally very similar.
Activation of amino acids requires energy in the form of ATP and occurs in a two step reaction catalyzed by the aminoacyl-tRNA synthetases. First the enzyme attaches the amino acid to the α-phosphate of ATP with the concomitant release of pyrophosphate (PPi). This is termed an aminoacyl-adenylate (aminoacyl-AMP) intermediate. In the second step the enzyme catalyzes transfer of the amino acid to either the 2’–OH (class I enzymes) or 3’–OH (class II enzymes) of the ribose portion of the 3′-terminal adenosine residue of the tRNA generating the activated aminoacyl-tRNA. Although these reactions are freely reversible, the forward reaction is favored by the coupled hydrolysis of PPi.
Accurate recognition of the correct amino acid as well as the correct tRNA is different for each aminoacyl-tRNA synthetase. Since the different amino acids have different R groups, the enzyme for each amino acid has a different binding pocket for its specific amino acid. It is not the anticodon that determines the tRNA utilized by the synthetases. Although the exact mechanism is not known for all synthetases, it is likely to be a combination of the presence of specific modified bases and the secondary structure of the tRNA that is correctly recognized by the synthetases.
It is absolutely necessary that the discrimination of correct amino acid and correct tRNA be made by a given synthetase prior to release of the aminoacyl-tRNA from the enzyme. Once the product is released there is no further way to proof-read whether a given tRNA is coupled to its corresponding amino acid. Erroneous coupling would lead to the wrong amino acid being incorporated into the polypeptide since the discrimination of amino acid during protein synthesis comes from the recognition of the anticodon of a tRNA by the codon of the mRNA and not by recognition of the amino acid. This was demonstrated by reductive desulfuration of cys-tRNAcys with Raney nickel generating ala-tRNAcys. Alanine was then incorporated into an elongating polypeptide where cysteine should have been.
The Wobble Hypothesis
As discussed above, 3 of the possible 64 triplet codons are recognized as translational termination codons. The remaining 61 codons might be considered as being recognized by individual tRNAs. Most cells contain isoaccepting tRNAs, different tRNAs that are specific for the same amino acid, however, many tRNAs bind to two or three codons specifying their cognate amino acids. As an example yeast tRNAphe has the anticodon 5’–GmAA–3′ and can recognize the codons 5’–UUC–3′ and 5’–UUU–3′. It is, therefore, possible for non-Watson-Crick base pairing to occur at the third codon position, i.e. the 3′ nucleotide of the mRNA codon and the 5′ nucleotide of the tRNA anticodon. This has phenomenon been termed the wobble hypothesis

Once there is a sufficient pool of charged aminoacyl-tRNAs, and the mRNAs whose nucleotide sequences can be converted to amino acid sequences, the two RNAs need to brought together accurately and efficiently. This is the job of the ribosomes and numerous proteins of the translation machinery. Ribosomes themselves are complexes composed of ribosomal proteins and rRNAs.
All living organisms need to synthesis proteins and all cells of an organism need to synthesize proteins, therefore, it is not hard to imagine that ribosomes are a major constituent of all cells of all organisms. The make up of the ribosomes, both rRNA and associated proteins are slightly different between prokaryotes and eukaryotes.
Order of Events in Translation
The ability to begin to identify the roles of the various ribosomal proteins in the processes of ribosome assembly and translation was aided by the discovery that the ribosomal subunits will self assemble in vitro from their constituent parts.
Following assembly of both the small and large subunits onto the mRNA, and given the presence of charged tRNAs, protein synthesis can take place. To reiterate the process of protein synthesis:
- Synthesis proceeds from the N-terminus to the C-terminus of the protein
- The ribosomes “read” the mRNA in the 5′ to 3′ direction
- Active translation occurs on polyribosomes (also termed polysomes). This means that more than one ribosome can be bound to and translate a given mRNA at any one time
- Active translation occurs on polyribosomes (also termed polysomes). This means that more than one ribosome can be bound to and translate a given mRNA at any one time
Translation proceeds in an ordered process. First accurate and efficient initiation occurs, then chain elongation, and finally accurate and efficient termination must occur. All three of these processes require specific proteins, some of which are ribosome associated and some of which are separate from the ribosome, but may be temporarily associated with it.
Initiation
Initiation of translation in both prokaryotes and eukaryotes requires a specific initiator tRNA, tRNAimet, that is used to incorporate the initial methionine residue into all proteins. In E. coli a specific version of tRNAimet is required to initiate translation, [tRNAifmet]. The methionine attached to this initiator tRNA is formylated. Formylation requires N10-formy-THF and is carried out after the methionine is attached to the tRNA. The fmet-tRNAifmet still recognizes the same codon, AUG, as regular tRNAmet. Although tRNAimet is specific for initiation in eukaryotes it is not a formylated tRNAmet.
The initiation of translation requires recognition of an AUG codon. In the polycistronic prokaryotic RNAs this AUG codon is located adjacent to a Shine-Dalgarno element in the mRNA. The Shine-Dalgarno element is recognized by complimentary sequences in the small subunit rRNA (16S in E. coli).
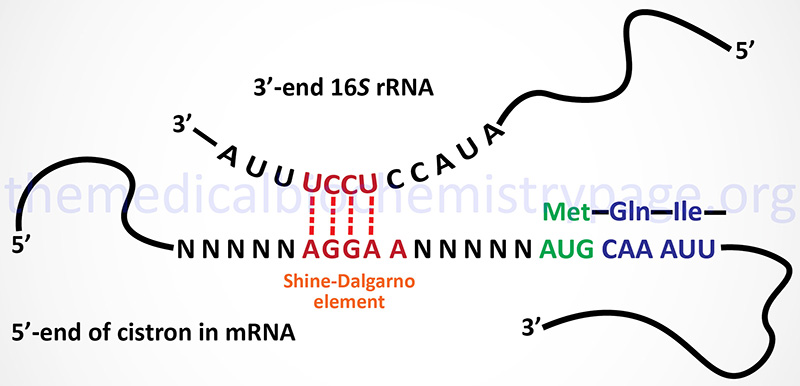
The Shine-Dalgarno element is found at the 5′ side of each initiator AUG codon in prokaryotic polycistronic mRNAs. This element is complementary to sequences present near the 3′-end of the 16S rRNA of the prokaryotic ribosome.
In eukaryotes, initiator AUGs are generally, but not always, the first encountered by the translational machinery. A specific sequence context, surrounding the initiator AUG, aids ribosomal discrimination. This context is A/GCCA/GCCAUGA/G in most mRNAs and is referred to as the Kozak consensus sequence.
Eukaryotic Initiation Factors and Their Functions
The specific non-ribosome-associated proteins required for accurate translational initiation are termed initiation factors. In E. coli they are IFs in eukaryotes they are eIFs. Numerous eIFs have been identified. Current research has shown that the mammalian eIF family of protein complexes consists of at least 12 members that together consist of at least 29 distinct proteins. By far the largest eIF complex is the eIF-3 complex (described in the next section) which contains 13 protein subunits and has an overall mass of >800 kDa.
Table of Eukaryotic Initiation Factors
| Initiation Factor | Activity |
| eIF-1 | repositioning of met-tRNA to facilitate mRNA binding; encoded by the EIF1 gene |
| eIF-1AX | X-linked eIF-1A encoded by the EIF1AX gene; required for the binding of the complex composed of 40S subunit, eIF2-GTP + initiator methionine tRNA (met-tRNAimet), and eIF-3 to the 5′ end of capped RNA; a Y-linked gene (EIF1AY) encodes a protein that may also perform similar function |
| eIF-2 | heterotrimeric G-protein composed of α, β, and γ subunits; formation of ternary complex consisting of eIF2-GTP + initiator methionine tRNA (met-tRNAimet); AUG-dependent met-tRNAimet binding to 40S ribosome; α-subunit encoded by the EIF2S1 gene, β-subunit encoded by the EIF2S2 gene, γ-subunit encoded by the EIF2S3 gene |
| eIF-2A | this is not the α-subunit of the heterotrimeric eIF-2 protein; encoded by the EIF2A gene; eIF-2A functions in the binding of the initiator tRNA to the 40S ribosomal subunit but does so in a codon-dependent process |
| eIF-2B | composed of five subunits: α, β, γ, δ, ε; functions in GTP/GDP exchange during eIF-2 recycling; genes encoding the subunits identified as EIF2B1–EIF2B5, mutations in any one of which causes the severe autosomal recessive neurodegenerative disorder called leukoencephalopathy with vanishing white matter (VWM) |
| eIF-3, composed of 13 subunits (see below) | ribosome subunit anti-association by binding to 40S subunit; eIF-3E and eIF-3I subunits transform normal cells when over-expressed, eIF-3A (also called eIF3 p170) over-expression has been shown to be associated with several human cancers |
| Initiation factor complex often referred to as eIF-4F composed of 3 primary subunits: eIF-4E, eIF-4A, eIF-4G and at least 2 additional factors: polyA-binding protein, Mnk1 (or Mnk2) | mRNA binding to 40S subunit, ATPase-dependent RNA helicase activity, interaction between polyA tail and cap structure; eIF-4A encoded by one of three genes (EIF4A1, EIF4A2, or EIF4A3); eIF-4E encoded by EIF4E gene; eIF-4G encoded by one of three genes (EIF4G1, EIF4G2, or EIF4G3) |
| polyA-binding protein C1 (PABPC1) | principal cytoplasmic polyA-binding protein of mRNA translation; binds to the polyA tail of mRNAs and provides a link to eIF-4G; encoded by the PABPC1 gene; humans express several cytoplasmic (PABPC) and at least one nuclear (PABPN) polyA-binding protein; the PABPN1 encoded protein is involved in polymerization of the polyA tail |
| Mnk1 and Mnk2 are eIF-4E kinases | phosphorylate eIF-4E increasing association with cap structure; Mnk1 is encoded by the MKNK1 (MAPK interacting serine/threonine kinase 1) gene; Mnk2 is encoded by the MKNK2 gene |
| eIF-4A | ATPase-dependent RNA helicase; encoded by one of three genes (EIF4A1, EIF4A2, or EIF4A3) |
| eIF-4E (see below) | 5′ cap recognition; encoded by the EIF4E gene; frequently found over-expressed in human cancers, inhibition of eIF4E is currently a target for anti-cancer therapies |
| 4E-BP | humans express 9 eIF-4 binding protein genes (EIF4BP1–EIF4BP9); original eIF-4BP (EIF4BP1) was called phosphorylated heat- and acid-stable protein 1 (PHAS1); when de-phosphorylated 4E-BP binds eIF-4E and represses its’ activity, phosphorylation of 4E-BP occurs in response to many growth stimuli leading to release of eIF-4E and increased translational initiation |
| eIF-4G | encoded by one of three genes (EIF4G1, EIF4G2, or EIF4G3); acts as a scaffold for the assembly of eIF-4E and -4A in the eIF-4F complex; interaction with PABP family member proteins allows 5′-end and 3′-ends of mRNAs to interact |
| eIF-4B | stimulates helicase, binds simultaneously with eIF-4F |
| eIF-5 | humans express several members of the eIF-5 family: EIF5, EIF5A, EIF5A2, EIF5B |
| eIF-5A | binds to the ribosome between the P-site and the tRNA exit site (E-site); promotes peptide bond formation by preventing ribosome stalling in mRNAs containing proline repeats as well as many other ribosome stalling sites; also enhances translation termination; only human protein that contains a modified lysine residue called hypusine (derived from hydroxyputrescine and lysine); the hypusine residue is catalyzed in a two-step reaction involving the polyamine, spermidine |
| eIF-5B | the EIF5B encoded protein is the human homolog of prokaryotic IF2; eIF-5B is a ribosome-dependent GTPase activating protein; interacts with the 40S initiation complex to promote hydrolysis of bound GTP with concomitant joining of the 60S ribosomal subunit to the 40S initiation complex; interacts with eIF-1A to position the initiation methionyl-tRNA on the start codon of the mRNA |
| eIF-6 | encoded by the EIF6 gene; contains two fibronectin type III domains that bind to the fibronectin type III domains of integrin beta-4 subunit (ITGB4) of intermediate filaments; functions as a metabolism-mediated translation initiation factor via ability to regulate the availability of 60S subunits for translation; translational regulation involves enhancement of the translation of mRNAs encoding transcription factors that regulate the expression of genes encoding lipogenic enzymes; the function of eIF-6 in the translation of these mRNAs involves its ability to allow the bypassing of inhibitory upstream open reading frames (uORF) in these mRNAs |
Activities of eIF-3
The eIF-3 complex is composed of 13 different subunits whose sizes, nomenclature and functions are described in the Table below. Although the protein complexity of the eIF-3 complex has been discerned, the functions of all of the proteins in the complex are not fully characterized. The importance of the eIF-3 complex in translation initiation is demonstrated by the fact that assembly of the eIF-2-GTP-met-tRNAimet (the ternary complex), binding of the ternary complex and other components of the 43S pre-initiation complex (PIC) to the ribosome 40S subunit, recruitment of the mRNA to the 43S PIC, and scanning of the mRNA for the initiator AUG codon recognition are all dependent on eIF-3 complex activity. Therefore, primary function of the components of eIF-3 is to act as a scaffold for the assembly of the PIC and this assembled complex is referred to as the multi-initiation factor complex (MFC).
Table of Proteins in the eIF-3 Complex
| Nomenclature | Human subunit designation | Function(s) / Comments |
| eIF3A | p170 | binds 40S subunit, binds eIF-4B, involved in formation of MFC, recruitment of mRNA and the ternary complex |
| eIF3B | p116 | binds 40S subunit, involved in formation of MFC, recruitment and scanning of mRNA, recruitment of ternary complex |
| eIF3C | p110 | binds 40S subunit, involved in formation of MFC, recruitment and scanning of mRNA, recruitment of ternary complex, recognition of the initiator AUG |
| eIF3D | p66 | major mRNA binding subunit |
| eIF3E | p48 | associates with eIF3A subunit; contains a PCI domain (Proteasome component, COP9, Initiation factor 3) |
| eIF3F | p47 | proposed to be the binding site for mTOR and p70S6K (see regulation of eIF-4E activity below) |
| eIF3G | p44 | binding of eIF-4B |
| eIF3H | p40 | aids the targeting of specific mRNAs to ribosomes especially during early embryogenesis |
| eIF3I | p36 | |
| eIF3J | p35 | binds 40S subunit, involved in formation of the MFC |
| eIF3K | p28 | |
| eIF3L | p67 | |
| eIF3M | GA17 | elevated levels of protein found in several types of cancer; contains a PCI domain (Proteasome component, COP9, Initiation factor 3) |
Specific Steps in Translational Initiation
Initiation of translation requires 4 specific steps:
1. A ribosome must dissociate into its’ 40S and 60S subunits.
2. A ternary complex termed the pre-initiation complex is formed consisting of the initiator, GTP, eIF-2 and the 40S subunit.
3. The mRNA is bound to the pre-initiation complex.
4. The 60S subunit associates with the pre-initiation complex to form the 80S initiation complex.
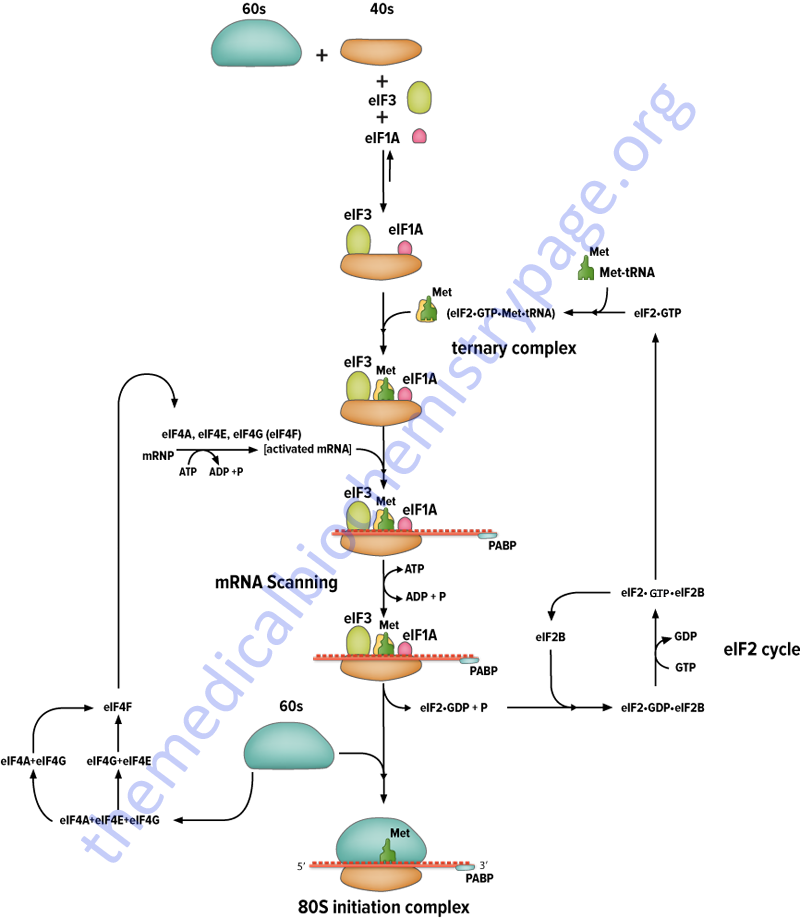
The initiation factors eIF-1 and eIF-3 bind to the 40S ribosomal subunit favoring anti-association to the 60S subunit. The prevention of subunit re-association allows the pre-initiation complex to form.
The first step in the formation of the pre-initiation complex is the binding of GTP to eIF-2 to form a binary complex. eIF-2 is composed of three subunits, α, β and γ. The binary complex then binds to the activated initiator tRNA, met-tRNAimet forming a ternary complex that then binds to the 40S subunit forming the 43S pre-initiation complex. The pre-initiation complex is stabilized by the earlier association of eIF-3 and eIF-1 to the 40S subunit.
The cap structure of eukaryotic mRNAs is bound by specific eIFs prior to association with the pre-initiation complex. Cap binding is accomplished by the initiation factor eIF-4F. This factor is actually a complex of 3 proteins; eIF-4E, A and G. The protein eIF-4E is a 24 kDa protein which physically recognizes and binds to the cap structure. eIF-4A is a 46 kDa protein which binds and hydrolyzes ATP and exhibits RNA helicase activity. Unwinding of mRNA secondary structure is necessary to allow access of the ribosomal subunits. eIF-4G aids in binding of the mRNA to the 43S pre-initiation complex.
The eIF-2 Cycle
Once the mRNA is properly aligned onto the pre-initiation complex and the initiator met-tRNAimet is bound to the initiator AUG codon (a process facilitated by eIF-1) the 60S subunit associates with the complex. The association of the 60S subunit requires the activity of eIF-5 which has first bound to the pre-initiation complex. The energy needed to stimulate the formation of the 80S initiation complex comes from the hydrolysis of the GTP bound to eIF-2. The GDP bound form of eIF-2 then binds to eIF-2B which stimulates the exchange of GTP for GDP on eIF-2.
The activity called eIF-2B is actually a complex of five subunits identified as α, β, γ, δ, and ε. The GDP for GTP exchange reaction is catalyzed by a sub-complex of the γ and ε subunits. The α, β, and γ subunits form another sub-complex that binds phosphorylated eIF-2α resulting in reduced nucleotide exchange. The phosphorylation of eIF-2α is discussed below. Thus, under conditions where eIF-2α is not phosphorylated is serves as a substrate for eIF-2B but when phosphorylated eIF-2α acts as a competitive inhibitor to eIF-2B activity. When GTP is exchanged eIF-2B dissociates from eIF-2.
The overall process of eIF-2B-mediated GTP for GDP exchange in eIF-2 is termed the eIF-2 cycle as shown in the Figure below. This cycle is absolutely required in order for eukaryotic translational initiation to occur.
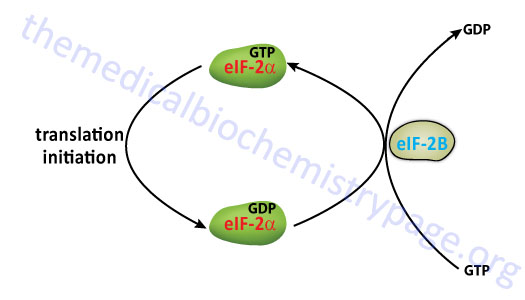
At this stage the initiator met-tRNAimet is bound to the mRNA within a site of the ribosome termed the P-site, for peptide site. The other site within the ribosome to which incoming charged tRNAs bind is termed the A-site, for amino acid site.
Role of the mRNA Poly(A) Tail in Initiation
As discussed in the RNA: Transcription and Processing page, most eukaryotic mRNAs possess a polyadenylated [poly(A)] tail at their 3’ end. One major function of the poly(A) tail is to protect the mRNA from exonuclease degradation. However, an equally significant function is its role in translational initiation. The poly(A) tail has been shown to interact with the 5′-cap structure to synergistically stimulate translation. This interaction is accomplished by the presence of specific RNA-binding proteins called poly(A)-binding proteins (PABPs). Multiple copies of these proteins are found associated with the poly(A) tail of most mRNAs. The PABPs interact with the scaffolding protein, eIF4G, resulting in a circularization of the mRNA leading to juxtaposition of the 5’ and 3’ ends.
Humans express four distinct cytoplasmic PABPs identified as PABPC1, PABPC3, PABPC4, and PABPC5. The “C” in the acronym stands for cytoplasmic. The PABPC1 protein is derived from the PABPC1 gene located on chromosome 8q22.2–q23 which is composed of 15 exons the encode a 636 amino acid protein. PABPC1 moves between the cytosol and the nucleus and binds the poly(A) tail of mRNAs in both compartments. PABPC1 is the major poly(A)-binding protein involved in the regulation of translational initiation. The PABPC3 gene is located on chromosome 13q12–q13 and is an intronless gene encoding a protein of 631 amino acids. The PABPC4 protein was originally isolated as an inducible RNA-binding protein in T cells and called inducible PABP (IPABP). The function of PABPC4 is thought to be necessary for stabilization of labile mRNAs in induced T cells. The PABPC4 gene is located on chromosome 1p34.2 and is composed of 16 exons that generate three alternatively spliced mRNAs. The PABPC5 gene is an X-linked gene (Xq21.3) and is composed of 2 exons that encode a 382 amino acid protein.
It is believed that this closed-loop of mRNA improves translational efficiency. One possible mechanism for the improved translation is that the circularized mRNA by facilitates the utilization and/or recycling of 40S ribosomal subunits. Another possible reason for enhanced translation is that only intact mRNAs are efficiently translated thus preventing the generation of potentially dominant-negative forms of a given protein from being translated. Another possible benefit to this closed-loop is that PABP can now participate in the promotion of the association of the 60S ribosomal subunit with the pre-initiation complex. It is also possible that the interaction of PABP with eIF4G might render changes to the overall activity of the eIF4F complex bound to the cap structure.
Recent evidence has demonstrated that mRNA circularization, effected by PABP, is indeed a key step in translation initiation and that this process represents a means to exert control over translation. Two PABP-binding proteins have recently been characterized and called PABP interacting protein 1 and 2 (Paip1 and Paip2). Paip1 interacts with eIF4A and has been shown to enhance translation. On the other hand, Paip2 inhibits the formation of the 80S initiation complex and thus, inhibits translation. Paip2 also competes with Paip1 for binding to PABP and is capable of displacing PABP from the poly(A) tail. Paip2 also competes with eIF4G binding to PABP due to an overlap in the binding sites for these two proteins on PABP. The PAIP1 gene is located on chromosome 5p12 and is composed of 13 exons that generate three alternatively spliced mRNAs. The PAIP2 gene is located on chromosome 5q31.2 and is composed of 6 exons that generate two alternatively spliced mRNAs, both of which encode the same 127 amino acid protein.
Humans also express a nuclear-localized poly(A)-binding protein identified by the acronym, PABPN1. This protein was identified after the identification of the original cytoplasmic PABP and was, therefore, originally referred to as PABP2. The PABPN1 gene is located on chromosome 14q11.2 and is composed of 7 exons that encode a protein of 306 amino acids. The primary function of PABPN1 is to control the efficient polymerization of the poly(A) tail ensuring a length of up to 250 nucleotides results. Clinical significance is associated with PABPN1 since the gene belongs to the family of the trinucleotide repeat disorder genes, specifically the polyalanine repeat diseases. The PABPN1 gene contains a GCG (encodes alanine) repeat in the N-terminal end of the coding region. The normal repeat length is 6 copies and expansion results in 8-13 copies. Individuals harboring an expanded repeat suffer from a disorder referred to as oculopharyngeal muscular dystrophy, OPMD. OPMD is an autosomal dominant disorder that manifests later in life with a characteristic dysphagia (difficulty swallowing) and progressive ptosis (drooping) of the eyelids.
Cap-Independent (IRES-Mediated) Translational Initiation
Cap-dependent translational initiation represents the primary mechanism for the translation of the vast majority of eukaryotic mRNAs. As discussed above, this process involves recognition of the cap structure by the eIF4F cap-binding complex (composed of eIF4A, eIF4E, and eIF4G). Binding of the cap structures is the function of eIF4E, while eIF4G is a scaffolding protein that binds eIF4E, eIF4A and the mRNA. The 40S ribosomal subunit binds a protein complex that includes the ternary complex (eIF2-GTP-tRNAi-met). The assembled protein complex then binds the mRNA at the cap structure and then scans along the mRNA until an AUG start codon is recognized in an appropriate sequence context.
During studies of the picornaviruses (including poliovirus) it was discovered that these viruses could initiate translation of their own RNAs, using the host translational machinery, via an internal ribosome entry site (IRES). An IRES is an RNA structure that directly recruits the 40S ribosomal subunits via a mechanism that is independent of both a cap structure and the extreme 5’ end of the viral mRNAs. Subsequently a number of eukaryotic viral mRNAs were shown to utilize cap-independent translational initiation mediated by IRES elements in their mRNAs. Picornaviruses encode a protease that hydrolyzes the eIF-4E binding site on eIF-4G which leads to impairment of global cap-dependent host translation but IRES-mediated translation remains intact. The hydrolyzed eIF-4G protein binds to IRES elements in the viral mRNAs allowing the picornavirus protein synthesis by the host translational machinery.
IRES elements were found to consist of long (300-500 nucleotides) stretches of highly structured sequences. However, the precise mechanisms by which the various IRES elements recruit the ribosomal machinery is not completely understood. Regardless, the viral IRES elements have been classified into four major structural groups, epitomized by poliovirus (PV; Type 1), encephalomyocarditis virus (EMCV; Type 2), hepatitis C virus (HCV; Type 3) and cricket paralysis virus (CrPV; Type 4). Subsequent to their identification in viral mRNAs, IRES elements, and cap-independent translational initiation, have been discovered in numerous mammalian mRNAs.
The ability of IRES-mediated translation initiation to occur is strictly dependent on the overall structure of the IRES. Single nucleotide substitutions, as well as small deletions or insertions, can either reduce or enhance the activity of a particular IRES. Given these requirements, it is likely that the IRES functionality may change under differing physiological and/or pathological conditions due to changes in the IRES structure under these differing conditions. Another contributor to IRES function, in some mRNAs (e.g. Myc and BiP) is the poly(A) tail despite being independent of the cap structure. Go to the section above to see the role of the poly(A) tail in cap-dependent translation initiation. In addition, many of the IRES elements function via interactions with several of the initiation factors (eIFs) required for cap-dependent initiation, such as eIF4G.
Several IRES elements require, not only a subset of eIFs, but also certain RNA binding proteins in order to facilitate cap-independent translational initiation. These non-eIF factors are called IRES trans-activating factors (ITAF). Several ITAF have been identified including polypyrimidine tract binding protein (PTB, also called hnRNP I), poly(rC) binding protein 2 (PCBP2, also called hnRNP E), hnRNP C1/C2, hnRNP D, upstream of N-ras (Unr), ITAF45, and the lupus autoantigen (La). It is potentially significant that some of these ITAF are hnRNPs (heterogeneous nuclear ribonucleoproteins) since these are a family of proteins involved in pre-mRNA processing, mRNA export, localization, and stability in addition to their roles in translation.
Several human genes encoding proto-oncogenes, growth factors, proteins involved in the regulation of programmed cell death (apoptosis), cell cycle progression and stress response have been shown to express mRNA that contain IRES elements in their 5’ untranslated regions (UTRs). The presence of IRES elements in these mRNAs, most of which also harbor a cap structure, allows translation to proceed under conditions where cap-dependent translational initiation is repressed. Thus, the hallmark of IRES-mediated translation is that it allows for enhanced or continued gene expression (at the level of protein synthesis) under conditions where normal, cap-dependent translation is shut-off or compromised.
Indeed, IRES elements have been shown to be active during and/or following irradiation, hypoxia, angiogenesis, apoptosis, and nutrient (amino acid) deprivation. Thus, IRES-mediated translation initiation represents a regulatory mechanism that allows cells to respond to, and cope with, various transient stress-related circumstances. Also IRES elements in certain mRNAs are likely to be important for the maintenance of normal physiological processes as well.
Table of Several Examples of Mammalian IRES-Containing mRNAs
| Encoded Protein | Protein Class / Activating Conditions | Known ITAF |
| eIF4G | translation | |
| Apoptotic protease activating factor 1, Apaf1 | apoptosis activator; IRES utilization during apoptosis | PTB, Unr, DAP5, hnRNPA1 |
| X-linked inhibitor of apoptosis protein, XIAP | apoptosis activator; IRES utilization during apoptosis | La, DAP5, hnRNPA1, hnRNPC1/C2, PTB |
| Bcl-2 | apoptosis activator; IRES utilization during apoptosis | DAP5 |
| NF-κB repressing factor, NKRF | transcription factor, blocks transcription of NF-κB- responsive genes | |
| FGF-1 | growth factor | |
| FGF-2 | growth factor | hnRNPA1 |
| lymphoid enhancer-binding factor 1, LEF-1 | transcription factor; IRES utilization during oncogenesis | |
| vascular endothelial growth factor, VEGF | growth factor, stimulates angiogenesis; IRES utilization during hypoxia | PTB |
| hypoxia-inducible factor 1α, HIF-1α | transcription factor, regulates expression of genes involved in energy metabolism, angiogenesis, and apoptosis; IRES utilization during hypoxia | PTB |
| Hsp70 | chaperone, IRES utilization during heat shock and apoptosis activator | |
| glucose-regulated protein 78-kDa, GRP78; also called Binding immunoglobulin heavy chain Protein, BiP; also identified as heat-shock 70kDa protein 5, HSPA5 | chaperone, IRES utilization during heat shock | La, NSAP1 |
Regulation of Translation Initiation: The eIF-2α Kinases
Regulation of translational initiation in eukaryotes is primarily controlled by phosphorylation of Ser (S) residues in the α-subunit of eIF-2. Phosphorylated eIF-2α, in the absence of eIF-2B, is just as active an initiator as non-phosphorylated eIF-2α. However, when eIF-2α is phosphorylated the GDP-bound complex is stabilized and exchange for GTP is inhibited. The exchange of GDP for GTP is mediated by eIF-2B (see section above). When the eIF-2 complex is phosphorylated on the α-subunit of the heterotrimeric complex (α, β, and γ subunits) eIF-2B binds more tightly slowing the rate of nucleotide exchange. It is this inhibited exchange that affects the rate of initiation.
Regulation of translational initiation, via phosphorylation of eIF-2α, occurs under a number of different conditions, particularly conditions of stress, that include endoplasmic reticulum (ER) stress (also referred to as the unfolded protein response, UPR), mitochondrial stress, nutrient stress (deprivation or restriction), viral infection, and in erythrocytes as a consequence of limiting concentrations of heme. Each of these distinct regulatory pathways involves an eIF-2α kinase, and there are four of these related kinases known to exist in mammalian cells. Each eIF-2α kinase contains unique regulatory domains that interact with various inducing agents in response to different stress-related conditions.
Because these four eIF-2α kinases are involved in the responses of cells to a variety of stressful conditions, their activation, and subsequent effects, are collectively referred to as the integrated stress response (ISR). A distinguishing feature of the ISR, that is not associated with the UPR, is that there is regulation of both translation and transcription as a result of the activation of the ISR.
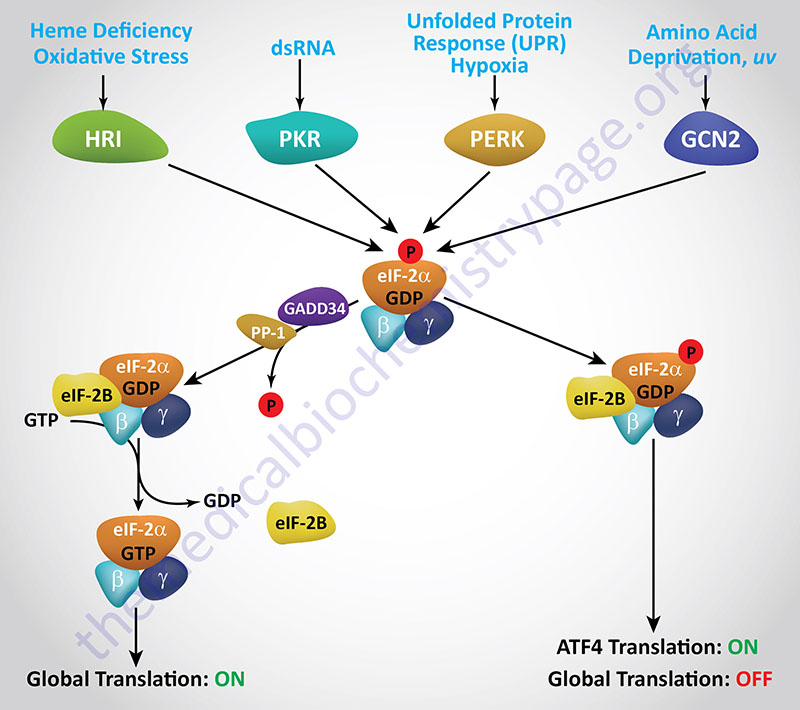
The first eIF-2α kinase identified and characterized was isolated from reticulocyte lysates and shown to be involved in the control of globin mRNA translation in response to deficiency in heme. This kinase (discussed in detail below) is called the heme regulated inhibitor (HRI). The protein is also called the heme controlled repressor (HCR) or the heme controlled inhibitor (HCI). The gene encoding HRI is identified as EIF2AK1.
Viral infection involving double-stranded RNA viruses activates the expression of the interferons which in turn activate the eIF-2α kinase known as PKR (RNA-dependent kinase). PKR is encoded by the EIF2AK2 gene and details of this kinase are described below in the section on Interferon Control of Translation.
Proteins that are destined for secretory vesicles are translated while associated with endoplasmic reticulum (ER) membranes. During translation secreted protein are transported into the lumen of the ER in an unfolded state. Proper folding occurs prior to the transfer of the protein to the Golgi apparatus. Under certain conditions the secretory responses of a cell can exceed the capacity of the folding processes of the ER. Accumulation of unfolded proteins triggers the unfolded protein response (UPR) which is a form of ER stress. The UPR involves different protein effectors allowing the cell to respond appropriately and enhance the processing, assembly, and transport of secreted proteins.
Among the proteins activated by the UPR is an eIF-2α kinase termed PERK which is a transmembrane protein kinase associated with the ER. PERK is RNA-dependent protein kinase (PKR)-like ER Kinase. PERK, also known as PEK (pancreatic eIF-2α kinase), is encoded by the EIF2AK3 gene. The aim of PERK-mediated phosphorylation of eIF-2α is to reduce global protein synthesis allowing the cell time to correct the impaired process of protein folding.
Deficiencies in EIF2AK3 gene which encodes PERK result in the extremely rare autosomal recessive disorder known as Wolcott-Rallison syndrome (WRS). WRS is characterized by dysfunction in specific secretory tissues including the pancreas. The defect in insulin secretion in WRS results in a specific form of neonatal diabetes.
Nutritional deprivation, in particular amino acid deficiency, results in the activation of the fourth eIF-2α kinase known as GCN2. GCN2 is the mammalian homolog of the General Control Non-derepressible-2 gene first identified in yeast. Mammalian GCN2 is encoded by the EIF2AK4 gene. Activation of GCN2, in response to amino acid deprivation, is the consequence of uncharged tRNAs binding to GCN2 and activating its kinase activity. In addition to nutritional deprivation, GCN2 is induced by UV irradiation, inhibition of proteasome function, and infection by certain viruses. With respect to amino acid deficiency, the activation of GCN2 occurs via the binding of uncharged tRNAs to the regulatory domain of the enzyme.
Evidence shows that the activities of PERK and GCN2 work in a concerted fashion to ensure that during periods of extended stress, global protein synthesis is inhibited. Under these types of conditions PERK serves a primary inhibitory function while GCN2 serves a secondary inhibitory role allowing for cell cycle arrest in response to ER stress.
The key protein whose translation is activated in response to stress-mediated phosphorylation of eIF-2α is the transcription factor ATF4 (activating transcription factor 4; also known as CREB2: cAMP response element-binding protein 2). In response to ER stress the levels of ATF4 mRNA do not change but the level of the protein increases dramatically, indicative of induced translation. The ATF4 mRNA contains two upstream open reading frames (uORFs) identified as uORF1 and uORF2. uORF1 encodes only three amino acids while uORF2 encodes 59 amino acids and overlaps the first 83 amino acids of the functional ATF4 protein.
Examination of the role of these two uORFs in ATF4 translational control using in vitro assays demonstrated that uORF1 serves a positive role in translational control allowing ribosomes to overcome the inhibitory role of uORF2 in response to eIF-2α phosphorylation. Under non-stressed conditions when eIF-2α phosphorylation is low the ribosomes rapidly scan to uORF2 and translate a non-functional protein thus, reducing the overall level of functional ATF4 protein. However, in the stressed condition there is less eIF-2-GTP which slows translational initiation allowing ribosomes that have engaged the limited eIF-2-GTP to scan to the correct ATF translational initiation site allowing for increases in functional ATF4 protein.
The function of ATF4 is to induce the expression of genes that allow the cell to respond to the stress conditions such as the transcription factors FOS, JUN, and C/EBP (CAAT-box/enhancer binding protein). ATF4 translation also activates a feedback regulatory mechanism to control the level of eIF-2α phosphorylation via increased expression of the GADD34 gene (growth arrest and DNA damage-inducible protein 34). GADD34 is a regulatory subunit of protein phosphatase-1 (PP-1) and when the GADD34 gene is activated by ATF4 there is a resultant increase in PP-1-mediated phosphate removal from eIF-2α due to increased GADD34 protein.
Elongation
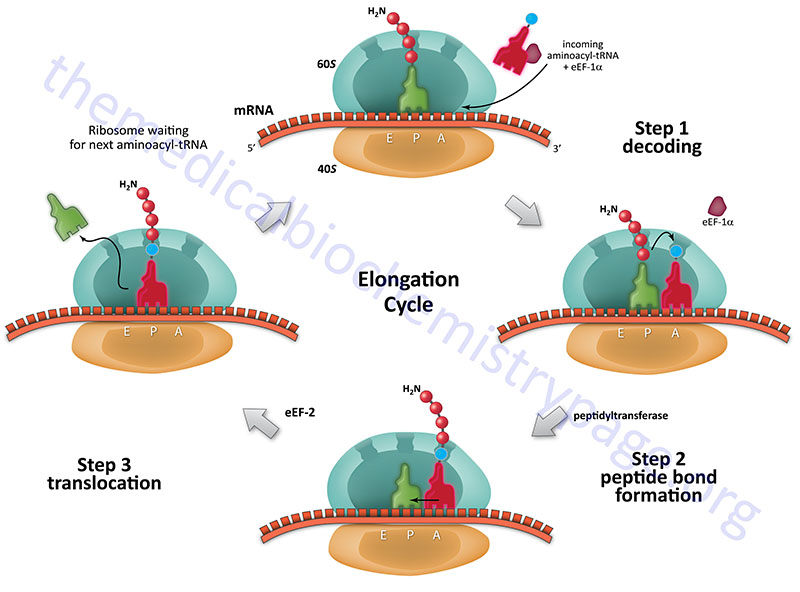
The process of elongation, like that of initiation requires specific non-ribosomal proteins. In prokaryotes these are designated EF and in eukaryotes, eEF. Elongation of polypeptides occurs in a cyclic manner such that at the end of one complete round of amino acid addition the A site will be empty and ready to accept the incoming aminoacyl-tRNA dictated by the next codon of the mRNA. This means that not only does the incoming amino acid need to be attached to the peptide chain but the ribosome must move down the mRNA to the next codon.
eEF-1 Complex Proteins
Similar to eIF-2, functional eEF-1 is a heterotrimeric G-protein. The eEF-1 complex also consists of several regulator proteins. Humans express at least six genes encoding proteins that can form the eEF-1 complex as well as a selenocysteine specific eEF encoded by the EEFSEC gene.
The EEF1A1 and EEF1A2 genes encode eEF-1α proteins. The EEF1A1 gene is located on chromosome 6q13 and is composed of 8 exons that encode a 462 amino acid protein. The EEF1A2 gene is located on chromosome 20q13.33 and is composed of 8 exons that encode a 463 amino acid protein. Expression of the EEF1A1 gene is ubiquitous whereas expression of the EEF1A2 gene predominates in the heart and brain.
The EEF1B2 gene encodes the β-subunit. The EEF1B2 encoded β-subunit possesses the guanine nucleotide exchange factor (GEF) activity of the overall eEF-1 complex. The EEF1B2 gene is located on chromosome 2q33.3 and is composed of 7 exons that generate three alternatively spliced mRNAs, all of which encode the same 225 amino acid protein.
The EEF1G gene encodes the γ-subunit. The EEF1G gene is located on chromosome 11q12.3 and is composed of 10 exons that encode a 437 amino acid protein.
The EEF1D gene encodes the eEF-1δ subunit that functions in the delivery of the aminoacyl-tRNAs to the ribosome and also possesses GEF activity. The EEF1D gene is located on chromosome 8q24.3 and is composed of 12 exons that generate 10 alternatively spliced mRNAs that collectively encode four protein isoforms.
The EEF1E1 gene encodes the eEF1epsilon1 (eEF-1ε1) protein which is a multifunctional protein localized to both the cytosol and the nucleus. Within the cytosol the eEF-1ε1 protein functions as a component of aminoacyl-tRNA synthetase complexes. Within the nucleus the eEF-1ε1 protein has been shown to be involved in the ATM-mediated activation of p53. The EEF1E1 gene is located on chromosome 6p24.3 and is composed of 5 exons that generate two alternatively spliced mRNAs encoding proteins of 174 amino acids (isoform 1) and 139 amino acids (isoform 2).
Steps of Elongation
Each incoming aminoacyl-tRNA is brought to the ribosome by an eEF-1α-GTP complex. When the correct tRNA is deposited into the A site the GTP is hydrolyzed and the eEF-1α-GDP complex dissociates. In order for additional translocation events the GDP must be exchanged for GTP. This is carried out by eEF-1βγ similarly to the GTP exchange that occurs with eIF-2 catalyzed by eIF-2B.
Humans express two genes encoding eEF-1α subunits of the eEF-1 complex, EEF1A1 and EEF1A2. The EEF1A1 gene is expressed in brain, placenta, lung, liver, kidney, and pancreas while the EEF1A2 gene is expressed in brain, heart, and skeletal muscle.
The peptide attached to the tRNA in the P site is transferred to the amino group at the aminoacyl-tRNA in the A site forming a new peptide bond. This reaction is catalyzed by peptidyltransferase activity which resides in what is termed the peptidyltransferase center (PTC) of the large ribosomal subunit (60S subunit). This enzymatic process is termed transpeptidation. This enzymatic activity is not mediated by any ribosomal proteins but instead by the large ribosomal RNA (28S) contained in the 60S subunit. This RNA encoded enzymatic activity is referred to as a ribozyme.
The elongated peptide now resides on a tRNA in the A site. The A site needs to be freed in order to accept the next aminoacyl-tRNA. The process of moving the peptidyl-tRNA from the A site to the P site is termed, translocation. Translocation is catalyzed by eEF-2 coupled to GTP hydrolysis. In the process of translocation the ribosome is moved along the mRNA such that the next codon of the mRNA resides under the A site. Following translocation eEF-2 is released from the ribosome. The cycle can now begin again.
The ability of eEF-2 to carry out translocation is regulated by the state of phosphorylation of the enzyme. When eEF-2 is phosphorylated its activity is inhibited. Phosphorylation of eEF-2 is catalyzed by the enzyme eEF2 kinase (eEF-2K; encoded by the EEF2K gene). Regulation of eEF-2K activity is normally under the control of insulin and Ca2+ fluxes. The Ca2+-mediated effects are the result of calmodulin interaction with eEF-2K. The association of a calmodulin subunit with eEF-2K is denoted by the fact that the enzyme is also referred to as Ca2+/calmodulin-dependent kinase III (CaMKIII). Activation of eEF-2K in skeletal muscle by Ca2+ is important to reduce consumption of ATP in the process of protein synthesis during periods of exertion which will lead to release of intracellular Ca2+ stores. eEF-2K itself is also regulated by phosphorylation and one of the kinases that phosphorylates the enzyme is regulated by the mTOR complex 1 (mTORC1; see Regulation of eIF-4E below). In addition, the master metabolic regulatory kinase, AMP-activated protein kinase (AMPK) will phosphorylate and activate eEF-2K leading to inhibition of eEF-2 activity.
Termination
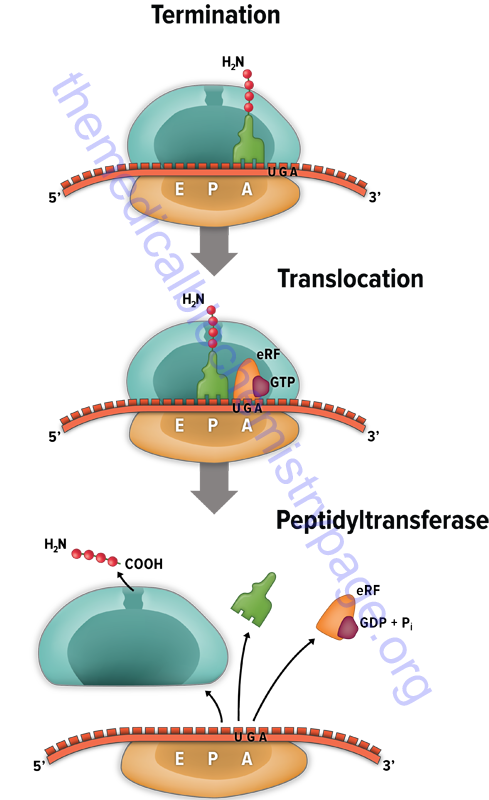
The signals for termination are the same in both prokaryotes and eukaryotes. These signals are termination codons present in the mRNA. There are three termination codons in mammalian mRNAs, UAG, UAA and UGA.
Like initiation and elongation, translational termination requires specific protein factors identified as releasing factors (RF) in prokaryotes and eRF in eukaryotes. In prokaryotes there are two distinct termination factors identified as RF-1 and RF-2. The termination codons UAA and UAG are recognized by RF-1, whereas RF-2 recognizes the termination codons UAA and UGA.
The translational termination process in humans involves a complex of eRF1, eRF3, and GTP. The eRF1 protein is encoded by the ETF1(eukaryotic translation release factor 1) gene. The eRF3 protein is encoded by the HBS1L (HBS1 like translational GTPase) gene.
The eRF3 protein hydrolyzes GTP which changes the conformation of eRF3 allowing it to dissociate and subsequently allowing the eRF1 protein to fully occupy the A-site of the ribosome. The ABCE1 encoded ATPase then interacts with eRF1 stimulating the peptidyltransferase activity to transfer the peptidyl group to water instead of an aminoacyl-tRNA. The resulting uncharged tRNA left in the P site is expelled with concomitant hydrolysis of GTP. The inactive ribosome then releases its mRNA and the 80S complex dissociates into the 40S and 60S subunits ready for another round of translation.
Selenocysteine Incorporation
Selenium is a trace element and is found as a component of several prokaryotic and eukaryotic enzymes that are involved in red-ox reactions. The selenium in these selenoproteins is incorporated as a unique amino acid, selenocysteine (Sec), during translation. Selenocysteine is often referred to as the 21st proteinogenic amino acid. Humans express several genes whose encoded proteins contain a selenocysteine residue. A particularly important group of eukaryotic selenoenzymes are the glutathione peroxidases (GPx) which are critically important anti-oxidant enzymes. The GPx enzymes utilize glutathione in the context of the reduction of hydrogen peroxide (H2O2) and other organic hydroperoxides, thereby detoxifying reactive oxygen species, ROS.
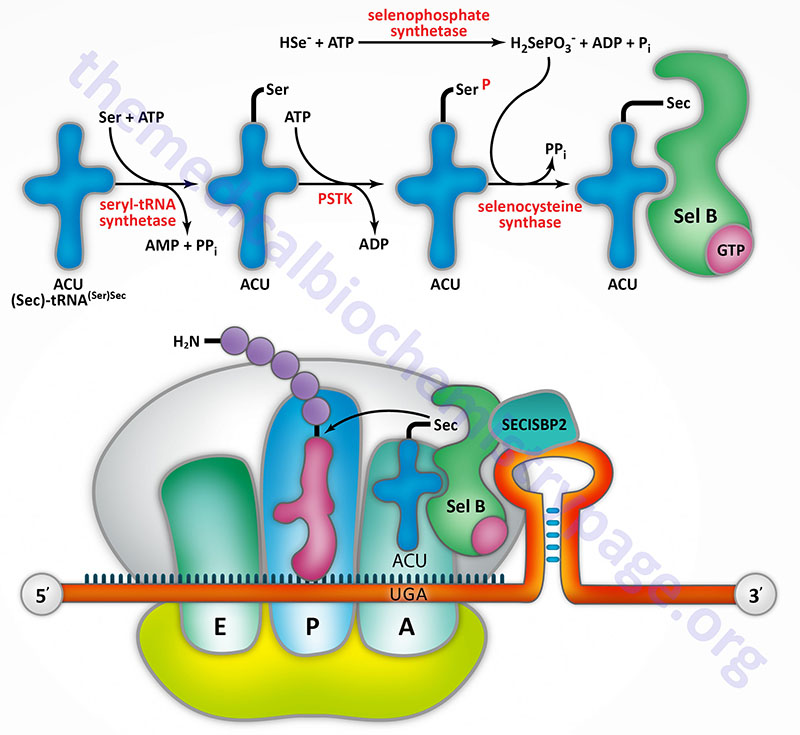
Selenocysteine incorporation in eukaryotic proteins occurs co-transcriptionally at UGA codons (normally stop codons) via the interactions of a number of specialized proteins and protein complexes. In addition, there are specific secondary structures in the 3′ untranslated regions of selenoprotein mRNAs, termed selenocysteine insertion sequence (SECIS) elements, that are required for selenocysteine insertion into the elongating protein. One of the complexes required for this important modification is comprised of a selenocysteinyl tRNA Sec-tRNA(Ser)Sec and its specific elongation factor identified as selenoprotein translation factor B (SelB). Human SelB is more commonly called eukaryotic elongation factor, selenocysteine-tRNA-specific and it is encoded by the EEFSEC gene.
The protein that is involved in the interaction of the SECIS element with the Sec-tRNA(Ser)Sec is referred to as SECIS binding protein 2 (SBP2) which is encoded by the SECISBP2 gene. Human Sec-tRNA(Ser)Sec is encoded by the TRU-TCA1-1 gene which is more commonly called TRSP. There are two isoforms of the Sec-tRNA(Ser)Sec in humans. These two Sec-tRNA(Ser)Sec isoforms differ from each other by a single 2′-O-methylribose which is located at position 34 and designated as Um34. Within the Sec-tRNA(Ser)Sec the 2′-O-methylribose is present in as 5-methoxycarbonylmethyluracil-2′-O-methylribose designated mcm5Um, whereas the Sec-tRNA(Ser)Sec that lacks the 2′-O-methylribose is designated mcm5U (5-methoxycarbonylmethyluracil).
The synthesis of the Um34 residue is regulated by the overall selenium status, being enriched when selenium is at adequate levels and being less abundant under conditions of selenium deficiency. On the other hand, the Sec-tRNA(Ser)Sec isoform that lacks the Um34 residue predominates under conditions of selenium deficiency. The two different Sec-tRNA(Ser)Sec isoforms have different roles in the synthesis of the repertoire of human selenoproteins. The mcm5U Sec-tRNA(Ser)Sec is involved in the synthesis of selenoproteins considered as housekeeping enzymes. The mcm5Um Sec-tRNA(Ser)Sec is active in the synthesis of both housekeeping selenoproteins and selenoproteins involved in cellular responses to stress such as the GPx isoforms.
Additional proteins involved in the selenoprotein synthesis pathway include two selenophosphate synthetases, SEPHS1 and SEPHS2, selenocysteine synthase (SEPSECS), ribosomal protein L30, SECp43 (encoded by the tRNA selenocysteine 1 associated protein 1 gene: TRNAU1AP), nucleolin, and eIF-4A3.
Humans express 25 genes that encode proteins that contain selenocysteine residues, several of which have no defined function at this time. The majority of these selenoproteins carry out oxidation-reduction reactions. Two critical redox enzyme families that require selenocysteine residues are the glutathione peroxidase and thioredoxin reductase families. Glutathione peroxidase is a critical enzyme involved in the protection of red blood cells from reactive oxygen species (ROS). This enzyme is a component of a redox system that also involves the enzyme glutathione reductase and NADPH as the terminal electron donor. This system is required for the continued reduction of oxidized glutathione (GSSG) and represents the single most significant system requiring continued glucose metabolism via the Pentose Phosphate Pathway in erythrocytes as the means for the production of the NADPH. Glutathione (GSH) becomes oxidized in the context of reducing various ROS and peroxides and to continue in this capacity the oxidized form needs to be continuously reduced.
Glutathione Peroxidases
Humans express eight different glutathione peroxidase genes identified as GPX1 through GPX8 with five of these enzymes (GPX1, GPX2, GPX3, GPX4, and GPX6) having been demonstrated to harbor selenocysteine residues. The enzyme encoded by the GPX1 gene (GPx1) is found in the cytosol of nearly all cell types in humans.
- The GPX1 gene is located on chromosome 3p21.3 and is composed of 2 exons that generate five alternatively spliced mRNAs via the use of alternative splice donor or splice acceptor sequences. GPx1 functions almost exclusively to reduce hydrogen peroxide (H2O2) to water. The GPX1 coding region contains a polyalanine tract in the N-terminal region of the protein. There are several alleles of this gene that have five, six, or seven alanine repeats. The allele with five alanine repeats has been shown to be highly correlated to increased risk for development of breast cancer.
- The GPX2 gene is located on chromosome 14q23.3 and is composed of 4 exons that encode a 190 amino acid protein.
- The GPX3 gene is located on chromosome 5q33.1 and is composed of 6 exons that generate two alternatively spliced mRNAs, each of which encode distinct proteins isoforms. Expression of the GPX3 gene is highest in the kidney. The protein encoded by the GPX3 gene, GPx3, is an extracellular enzyme found primarily in the plasma.
- The GPX4 gene is located on chromosome 19p13.3 and is composed of 8 exons that generate four alternatively spliced mRNAs. The GPx4 isoform A encoding mRNA generates a 197 amino acid precursor protein which represents the predominant form of GPx4 protein. Although ubiquitously expressed, expression of the GPX4 gene is highest in adipose tissue and the testis. The GPX4 encoded enzyme, GPx4, is an extracellular enzyme as well.
- The GPX5 gene is located on chromosome 6p22.1 and is composed of 6 exons that generate two alternatively spliced mRNAs, each of which encode distinct protein isoforms. The resultant GPX5 mRNA does not contain the canonical selenocysteine codon (UGA) and thus, the resulting protein does not contain a selenocysteine residue. Expression of the GPX5 gene is regulated by androgens and the gene is expressed exclusively in the epididymis in the male reproductive tract where the expressed protein, GPx5, is involved in protecting spermatozoa membranes from the damaging effects of lipid peroxidation.
- The GPX6 gene is located on chromosome 6p22.1 and is composed of 5 exons that encode a 221 amino acid precursor protein. GPX6 expression is restricted to embryonic tissues and the adult olfactory system.
- The GPX7 gene is located on chromosome 1p32.3 and is composed of 3 exons that encode a 187 amino acid precursor protein.
- The GPX8 gene is located on chromosome 5q11.2 and is composed of 4 exons that generate four alternatively spliced mRNAs, each of which encodes a unique protein isoform.
Thioredoxin Reductases
The thioredoxin reductases are involved in the reduction of thioredoxin which itself is principally involved in the reduction of oxidized disulfide bonds in proteins. The reduction of these disulfide bonds results in oxidation of thioredoxin which then is reduced by thioredoxin reductase. The overall process, like the glutathione peroxidase system, requires NADPH as the terminal electron donor for the reduction process. A critically important reaction that is coupled to the thioredoxin system is the formation of deoxynucleotides.
Humans contain three thioredoxin reductase genes that encode three distinct enzymes identified as TrxR1, TrxR2, and TrxR3. The TrxR1 enzyme is functional in the cytosol and is primarily involved in the maintenance of the ribonucleotide reductase system. The TrxR1 enzyme is encoded by the TXNRD1 gene located on chromosome 12q23.3 and is composed of 18 exons that generate seven alternatively spliced mRNAs encoding five different isoforms of TrxR1.
The TrxR2 enzyme is functional in the mitochondria where it is principally involved in the detoxification of reactive oxygen species (ROS) produced in this organelle. The TrxR2 enzyme is encoded by the TXNRD2 gene located on chromosome 22q11.21 and is composed of 22 exons that generate six alternatively spliced mRNAs resulting in six different isoforms of TrxR2.
TrxR3 is a testes-specific isoform of the enzyme. The TrxR3 enzyme is encoded by the TXNRD3 gene located on chromosome 3q21.3 and is composed of 16 exons that generate two alternatively spliced mRNAs resulting in two different isoforms of TrxR3. The mRNA encoding the TrxR3 isoform 1 protein utilizes a non-AUG codon (CUG) to initiate translation.
Thyroid Deiodinases
The enzymes of the deiodinase family are also important selenocysteine-containing enzymes. Clinically relevant enzymes in this family are the thyroid deiodinases that are critical for the maturation and catabolism of the thyroid hormones. Humans express three different thyroid deiodinase genes identified as DIO1, DIO2, and DIO3.
The enzyme encoded by the DIO1 gene, thryroxine deiodinase type I (also called iodothyronine deiodinase type I) is involved in the peripheral tissue conversion of thyroxine (T4) to the bioactive form of thyroid hormone, tri-iodothyronine (T3). In addition to its role in the generation of T3, thyroxine deiodinase I is involved in the catabolism of thyroid hormones. The DIO1 gene is located on chromosome 1p32.3 and is composed of 5 exons that generate five alternatively spliced mRNAs, each of which encodes a distinct protein isoform.
The enzyme encoded by the DIO2 gene, iodothyronine deiodinase type II, is also involved in the conversion of T4 to T3 but does so within the thyroid gland itself. The activity of iodothyronine deiodinase II has been associated with the thyrotoxicosis of Graves disease. The DIO2 gene is located on chromosome 14q31.1 and is composed of 7 exons that generate three different mRNAs through alternative promoter usage and alternative splicing. The DIO2 isoform a mRNA encodes two distinct protein isoforms via the use of alternative in-frame translation termination codons.
The enzyme encoded by the DIO3 gene is involved only in the inactivation (catabolism) of T3 and T4. Expression of the DIO3 gene is highest the female uterus during pregnancy and in fetal and neonatal tissue suggesting a role for this enzyme in the regulation of thyroid hormone levels and functions during early development. The DIO3 gene is located on chromosome 14q32.31 and is an intronless gene (is a single exon gene) that encodes a protein of 304 amino acids.
Selenoprotein P
One human selenocysteine containing protein, identified as selenoprotein P, contains ten selenocysteine residues. Selenoprotein P is encoded by the SELENOP gene which is located on chromosome 5p12 and is composed of 7 exons that generate three alternatively spliced mRNAs, two of which encode the same 381 amino acid protein. This form of selenoprotein P represents the major form of the protein. Selenoprotein P is synthesized primarily by the liver and secreted into the plasma. The primary function of selenoprotein P is to serve as a selenium transporter to deliver selenium to other organs.
Regulation of eIF-4E Activity
The cellular levels of eIF-4E are the lowest of all eukaryotic initiation factors which makes this factor a prime target for regulation. Indeed, at least 3 distinct mechanisms are known to exist that regulate the level and activity of eIF-4E. These include regulation of the level of transcription of the eIF-4E gene, post-translational modification via phosphorylation and inhibition by interaction with binding proteins.
Although the exact mechanisms used to upregulate the transcription of the eIF-4E gene are not yet well understood, it is known that exposure of cells to growth factors as well as activation of T cells leads to increased expression of eIF-4E. The proto-oncogene MYC is believed to play a role in the transcriptional activation of eIF-4E as 2 functional MYC-binding sites have been found in the promoter region of the eIF-4E gene. Of significant note is the finding that cells that are stably over-expressing the MYC gene also have enhanced levels of eIF-4E. Quite strikingly it has been shown that promiscuous elevation in the levels of eIF-4E lead to tumorigenesis placing this translation factor in the category of proto-oncogene.
Numerous extracellular stimuli (e.g. insulin, EGF, angiotensin II and gastrin) that exert a portion of their effects at the level of enhanced translation do so by affecting the state of eIF-4E phosphorylation. However, it should be noted that not all signals that lead to increased eIF-4E phosphorylation lead to increased rates of translation. Changes in eIF-4E phosphorylation correlate well with progression through the cell cycle. In resting (G0) cells eIF-4E phosphorylation is low, it increases during G1 and S phase and then declines again in M phase. Phosphorylation of eIF-4E occurs at one major site which is Ser209 (in the human and mouse proteins).
The primary signal transduction pathway leading to eIF-4E phosphorylation is that involving the RAS gene. Many growth factors stimulate activation of RAS in response to binding their cognate receptors. Subsequently, RAS activation leads to the phosphorylation and activation of MAPK-interacting serine/threonine kinase-1 (Mnk1: encoded by the MKNK1 gene) which in turn phosphorylates eIF-4E. Although the exact effect of eIF-4E phosphorylation is not clearly defined, it may be necessary to increase affinity of eIF-4E for the mRNA cap structure and affinity for eIF-4G.
The principal mechanism utilized in the regulation of eIF-4E activity is through its interaction with a family of binding/repressor proteins termed 4EBPs (4E binding proteins) which are widely distributed in numerous vertebrate and invertebrate organisms. Humans express nine genes in the 4EBP family (EIF4BP1–EIF4BP9) where the EIF4EBP1 and EIF4EBP2 encoded proteins are also identified as PHAS-1 and PHAS-2 (PHAS refers to phosphorylated heat- and acid-stable protein).
Binding of 4E-BP to eIF-4E does not alter the affinity of eIF-4E for the cap structure but prevents the interaction of eIF-4E with eIF-4G which in turn suppresses the formation of the eIF-4F complex (see Table of Initiation Factors above). The ability of 4EBP to interact with eIF-4E is controlled via the phosphorylation of specific Ser and Thr residues in 4EBP. When hypophosphorylated, 4EBP bind with high efficiency to eIF-4E but lose their binding capacity when phosphorylated. Numerous growth and signal transduction stimulating effectors lead to phosphorylation of 4EBP just as these same responses can lead to phosphorylation of eIF-4E.
There are several signal transduction pathways whose activations lead to phosphorylation of 4EBP. These include pathways that lead to activation of phosphatidylinositol 3-kinase (PI3K), the AKT (AK strain transforming) Ser/Thr kinase (which is also called protein kinase B, PKB) and the mechanistic target of rapamycin (mTOR) family of proteins. AKT/PKB was originally identified as the tumor inducing gene in the AKT8 retrovirus found in the AKR strain of mice. Humans express three genes in the AKT family identified as AKT1 (PKBα), AKT2 (PKBβ), and AKT3 (PKBγ). The mTOR proteins are homologs of the yeast TOR proteins that were identified in a screen for yeast mutants resistant to rapamycin. The mTOR kinases possess catalytic domains that share significant homology with lipid kinases of the PI3K family.
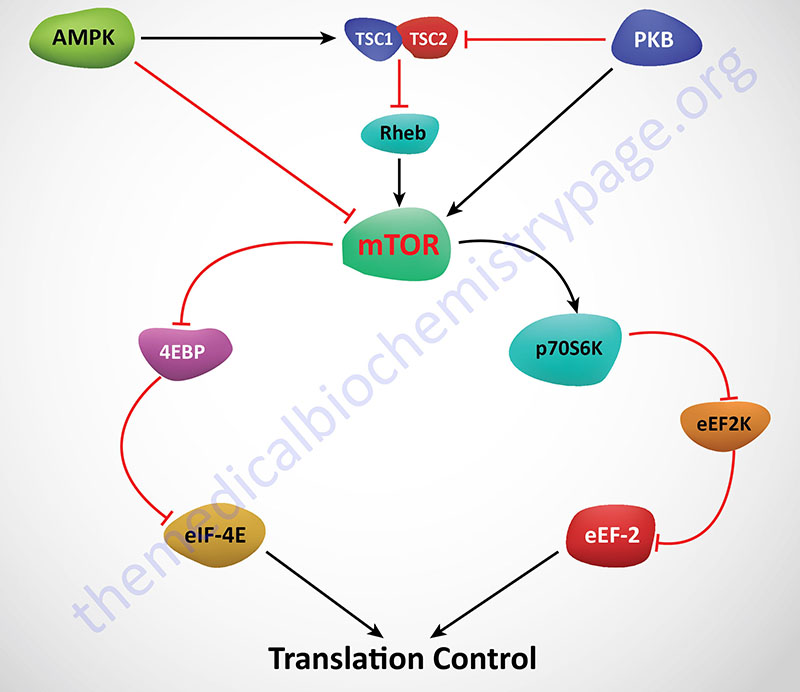
mTOR is actually a component of two distinct multiprotein complexes termed mTORC1 and mTORC2 (mTOR complex 1 and mTOR complex 2). The activity of mTORC1 is sensitive to inhibition by by rapamycin whereas mTORC2 is not. Within the context of mTOR activity, mTORC1 is the central complex as it is responsible for integrating a diverse series of signal transduction cascades initiated by changes in both intra- and extracellular events. Activation and/or regulation of mTORC1 is involved in the control of cell proliferation, survival, metabolism and stress responses. These events can be triggered by nutrient availability, glucose, oxygen, and numerous different types of cell surface receptor activation, each of which eventually impinge on the activity of mTORC1.
One of the major effects of insulin is increased protein synthesis and this effect is elicited, in part, via activation of mTOR function. For more information on the regulation of protein synthesis by insulin see the Insulin Function, Insulin Resistance, and Food Intake Control of Secretion page.
Regulation of mTOR activity is effected via several mechanisms. Activation of AMPK results in phosphorylation and activation of the TSC1/TSC2 complex which results in inhibition of mTOR. AMPK can also phosphorylate and inhibit mTOR. Conversely, activation of AKT/PKB (as in the case of insulin receptor activation) leads to activation of mTOR either by inhibition of the TSC1/TSC2 complex or by phosphorylation and activation of mTOR directly. Activation of mTOR leads to phosphorylation of p70S6K and 4EBP1. The net effect of phosphorylation of 4EBP1 is that it is released from eIF-4E allowing eIF-4E to actively bind eIF-4G and recognize the cap structure of mRNAs. Activated p70S6K phosphorylates and inhibits eEF-2K. If eEF-2K does not phosphorylate eEF-2 then translation elongation proceeds uninhibited.
Heme Control of Translation
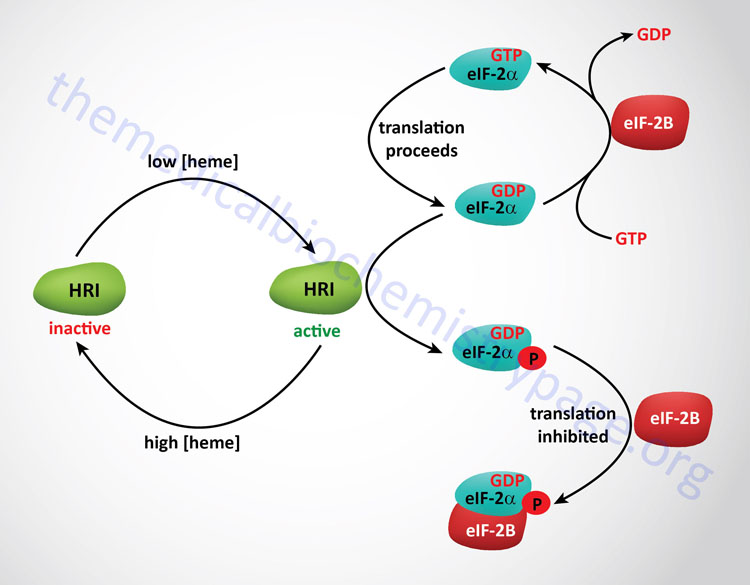
As discussed above, the regulation of initiation in eukaryotes is effected by phosphorylation of the α-subunit of eIF-2. In red blood cells there is a need to restrict the translation of the globin mRNAs if there is insufficient levels of heme to generate functional hemoglobin. Thus, when heme levels are low an eIF-2α kinase is active to limit translational initiation until there is adequate heme to generate functional hemoglobin. The regulating eIF-2α kinase in this system is called the heme regulated inhibitor (HRI). HRI is also known as the heme controlled inhibitor (HCI) or heme controlled repressor (HCR). The gene encoding HRI is identified as EIF2AK1. When heme levels rise in the red blood cell eIF-2α is protected from phosphorylation by a specific 67kDa protein that associates with the γ-subunit of eIF-2. Removal of the phosphate from eIF-2α is catalyzed by a specific eIF-2 phosphatase which is unaffected by heme. The presence of HRI was first seen in in vitro translation system derived from lysates of reticulocytes. Other stressors that include heat shock and oxidative stress result in activation of HRI in the red blood cell.
Expression of the EIF2AK1 gene is also important in hepatic tissues. In acute heme-deficient states in the liver HRI is activated leading to reductions in global hepatic translational. Additional activators of hepatic HRI include inducers of cytochrome P450 enzymes such as xenobiotics. One important xenobiotic metabolizing enzyme whose translation is regulated by HRI is the CYP2B6 gene which is a member of the large family of CYP2 family (16 members) genes. The CYP2B6 gene is also one of the phenobarbitol-inducible cytochrome P450 enzymes. Activation of HRI in hepatocytes would normally protect cells by ensuring adequate cellular energy and nutrients during acute heme deficiency. However, in genetically predisposed individuals, such as those with any of several porphyrias, the activation of HRI could lead to global translational arrest of physiologically important enzymes and proteins. This could play a significant role in the severe and often fatal clinical symptoms of the acute hepatic porphyrias.
Role of HRI in Responses to Mitochondrial Dysfunction
A major contributor to the integrated responses that cells undertake to stress and mitochondrial dysfunction is the phosphorylation of the alpha subunit of the heterotrimeric translation initiation factor, eIF-2. Human cells express four genes that encode kinases that phosphorylate eIF-2α as described in detail the Regulation of Translation Initiation: The eIF-2α Kinases section above. A role for HRI, as well as other eIF-2α kinases, in cellular stress responses and mitochondrial dysfunction, referred to as the integrated stress response (ISR), has been shown to be associated with their ability to phosphorylate eIF-2α.
When mitochondria are stressed or when they sense nuclear DNA damage, a mitochondria-localized protease known as OMA1 zinc metallopeptidase, or simply as OMA1, is activated. The term OMA1 refers to Overlapping activity with the M-AAA protease A1. M-AAA protease A1 is a member of the Mitochondrial ATPases Associated with Any diverse cellular activities (AAA) protease family.
One of the substrates of OMA1 is the protein DAP3 binding cell death enhancer 1 which is encoded by the DELE1 gene. DELE is derived from DAP3-binding, Enhancer of Ligand-mediated Endopeptidase. The DELE1 encoded protein is a signaling peptide that is critical to the activation of the ISR. Another target for OMA1 the OPA1mitochondrial dynamin-like GTPase encoded by the OPA1 gene. The OPA1 encoded protein can induce mitochondrial outer membrane permeabilization.
When OMA1 cleaves DELE1 a short form results identified as DELE1s. DELE1s is transported out of the mitochondria to the cytosol where it interacts with HRI. The interaction of DELE1s with HRI activates the kinase which in turn phosphorylates eIF-2α. The phosphorylation of eIF-2α results in the inhibition of global protein synthesis and the activation of the translation of specific proteins required to initiate the ISR. As a result of eIF-2α phosphorylation translation of the transcription factor, activating transcription factor 4 (ATF4) is enhanced. ATF4 is the master transcriptional regulator of the ISR.
Interferon Control of Translation
Regulation of translation can also be induced in virally infected cells. It would benefit a virally infected cell to turn off protein synthesis to prevent propagation of the viruses. This is accomplished by the induced synthesis of interferons (IFs). There are 3 classes of interferons: the leukocyte or α-IFs, the fibroblast or β-IFs and the lymphocyte or γ-IFs. Interferons are induced by dsRNAs and themselves induce a specific kinase termed RNA-dependent protein kinase (PKR) that phosphorylates the α-subunit of eIF-2 thereby shutting off translation in a similar manner to that of stress and heme control of translation. The PKR gene is identified as EIF2AK2.
Additionally, interferons induce the synthesis of 2′-5′-oligoadenylate, pppA(2’p5’A)n, that activates a pre-existing ribonuclease, RNase L. RNase L degrades all classes of mRNAs thereby shutting off host cell translation.
Iron Control of Translation
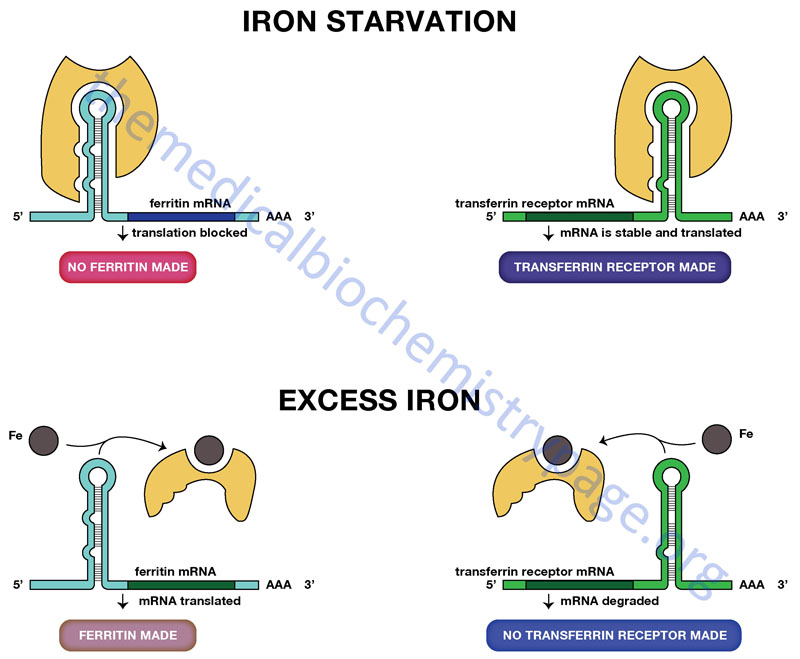
Regulation of the translation of certain mRNAs occurs through the action of specific RNA-binding proteins. Proteins of this class have been identified that bind to sequences in either the 5′-untranslated region (5′-UTR) or 3′-UTR of target mRNAs. Two particularly interesting and important regulatory schemes related to iron homeostasis encompass an RNA-binding protein that binds to either the 5′-UTR of the ferritin mRNA or to the 3′-UTR of the transferrin receptor mRNA.
The ferritin multimeric complex is composed of heavy (H-ferritin) and light (L-ferritin) subunits encoded by distinct genes (FTH1 and FTL, respectively). Both the FTH1 and FTL encoded mRNAs are translationally regulated by RNA-binding protein interaction. This RNA-binding protein is called iron response element binding protein (IRBP) and it binds to a specific iron response element (IRE) formed by the overall structure of its target mRNAs. This IRE forms a hair-pin loop structure that is recognized by IRBP.
This IRBP is an iron-deficient form of aconitase which is structurally similar to the iron-requiring aconitase of the TCA cycle. Humans express two distinct aconitase genes, ACO1 and ACO2. The protein encoded by the ACO1 gene is a cytoplasmic protein that is the IRBP involved in the translational control of the transferrin receptor and ferritin mRNAs. The ACO1 gene located on chromosome 9q21.1 which encodes a protein of 889 amino acids. The protein encoded by the ACO2 gene is mitochondrially localized and is the TCA cycle enzyme. In addition to the ferritin and transferrin receptor mRNAs, additional mRNAs encoding proteins involved in overall iron homeostasis also contain an IRE. These additional mRNAs include those encoding the membrane bound iron transporters, divalent metal transporter 1 (DMT1; encoded by the SLC11A2 gene) and ferroportin, and the heme biosynthetic enzymes, ALAS2 and ACO2.
The transferrin receptor is a protein located in the plasma membrane that binds the protein transferrin. Transferrin is the major iron transport protein in the plasma. When iron levels are low the rate of synthesis of the transferrin receptor mRNA increases so that cells can take up more iron. When iron levels are low, IRBP is free of iron and can therefore, interact with the IRE in the 3′-UTR of the transferrin receptor mRNA. Transferrin receptor mRNA with IRBP bound is stabilized from degradation. Conversely, when iron levels are high, IRBP binds iron then cannot interact with the IRE in the transferrin receptor mRNA. The effect is an increase in degradation of the transferrin receptor mRNA.
A related, but opposite, phenomenon controls the translation of the ferritin mRNA. Ferritin is an iron-binding protein that prevents toxic levels of ferrous iron (Fe2+) from building up in cells. The ferritin mRNA has an IRE in its 5′-UTR. As with the transferrin receptor mRNA, when iron levels are high, IRBP cannot bind to the IRE in the 5′-UTR of the ferritin mRNA. This allows the ferritin mRNA to be translated. Conversely, when iron levels are low, the IRBP binds to the IRE in the ferritin mRNA preventing its translation.
Prokaryotic Protein Synthesis Inhibitors
Antibiotics are broadly divided into two categories, the bactericidal (bactericidal) compounds and the bacteriostatic compounds. Bacteriostatic compounds do not kill bacteria but instead prevent them from reproducing. The bactericidal agents do kill bacteria. Many of the antibiotics reviewed in this discussion are bacteriostatic. The majority of antibiotics that have been developed to interfere with prokaryotic protein synthesis, exert their effects at the level of the functions associated with primarily the large ribosomal subunit (50S) or the small ribosomal subunit (30S). The enzymes that activate the amino acids used in protein synthesis, the aminoacyl-tRNA synthetases, are not antibiotic targets. By interfering with ribosomal functions, the vast majority of antibiotics interfere with the formation of several distinct components of the translational machinery. These include the 30S pre-initiation complex which comprises the mRNA, the small 30S ribosomal subunit, and the formylated methionine initiator tRNA (fmet-tRNAifmet); the 70S initiation complex which is composed of the 30S pre-initiation complex and the large 50S ribosome; or the processes of peptide elongation. A brief list of several antibiotics is presented in the Table below the following detailed discussion of the major types of large and small ribosomal subunit antibiotics.
Inhibitors of Prokaryotic Large (50S) Ribosomal Subunit Functions
MLS Group: Macrolides
The MLS group of antibiotics includes the macrolides, the lincosamides and the streptogramins. These three families of compounds are structurally distinct, yet they share a common mode of action and show similar antibacterial profiles. The MLS group of compounds interfere with the growth of staphylococci (any of the various spherical Gram-positive parasitic bacteria of the genus Staphylococcus), streptococci (any of the various spherical Gram-positive bacteria of the genus Streptococcus), Mycoplasma (a large genus of bacteria lacking a cell wall), and Campylobacter (the genus of spiral Gram-negative bacteria that are motile due to the presence of flagella). The various MLS class antibiotics are produced by the more than 500 strains of Streptomyces by means of various isoforms of polyketide synthase.
The macrolides belong to the polyketide class of naturally occurring compounds. All of the macrolides are derived from a precursor macrocyclic lactone to which one or more deoxy sugar residues may be attached. The macrolides also include a derivative class of compound called the ketolides. The specific classifications of the macrolides are determined on the basis of the number of atoms in the ring of the macrocyclic lactone which can range from 12 to 17. However, the macrolides used as antibiotics in humans are all derived from the 14-, 15-, or 16-membered macrocyclic lactones. The deoxy sugars attached to the macrocyclic ring include either cladinose or desosamine. All of the macrolides, and semi-synthetic macrolide derivatives, that are formed from the 14- and 15-membered rings contain the suffix -thromycin in the drug name.
The macrolides that contain a 14-membered ring include clarithromycin, erythromycin, roxithromycin, and the ketolide semi-synthetic derivatives such as telithromycin. The ketolide derivatives are used primarily to treat respiratory infections and macrolide-resistant bacteria.
The macrolides that contain a 15-membered ring include azithromycin which is a semi-synthetic derivative of erythromycin with a methyl-substituted nitrogen atom incorporated into the lactone ring. Azithromycin exhibits a similar spectrum to that of erythromycin, with increased activity against Gram-negative bacteria. The macrolides that contain a 16-membered ring include josamycin, midecamycin, spiramycin and tylosin. However, it should be noted that none of these 16-membered ring macrolides are approved for use in the United States.
Another important macrolide that is not used as an antibiotic is tacrolimus. This drug is a 23-membered ring macrolide isolated from Streptomyces tsukubaensis that is used as an immunosuppressant, principally following allogenic organ transplantation. Tacrolimus is also known as FK-506 or fujimycin.
MLS Group: Lincosamides
The lincosamides have some structural similarities to the macrolides but do not contain the lactone ring. This class of antibiotic consists of lincomycin and its more robust semi-synthetic derivative, clindamycin. Lincomycin was originally isolated from Streptomyces lincolnensis. This bacterium was discovered in a soil sample from Lincoln Nebraska, hence the derivation of its name. The lincosamides are effective against Gram-positive bacteria including Streptococci, Staphylococci, and Mycoplasma. In addition, the lincosamides are effective against Actinomyces, some species of Plasmodium, and against the protozoan Toxoplama gondii. Due to the toxicity spectrum associated with lincomycin use, clindamycin is currently the only lincosamide routinely prescribed. Clindamycin is effective in patients with infections caused by anaerobic bacteria, in some malaria patients, and in the treatment of methicillin-resistant Staphylococcus aureus (MRSA) infections. Lincomycin is only utilized to treat patients that are allergic to penicillin or who have contracted an infection by an antibiotic-resistant strain of Gram-positive bacteria.
MLS Group: Streptogramins
The streptogramin family of antibiotics are natural antibiotics produced by various strains of Streptomyces. The streptogramin antibiotics are classified into two distinct groups: streptogramin A and streptogramin B. The first isolated mixture of streptogramins came from Streptomyces graminofaciens found in a soil sample in Texas, hence the derived name of these antibiotics. Although most streptogramins are produced by Streptomyces, other bacterial strains such as Actinoplanes, Actinomadura, and Micromonospora are known to produce streptogramins. Very few streptogramins are utilized as human antibiotics, whereas the majority of these compounds are utilized as growth promoters in animal husbandry. The first streptogramin, virginiamycin, was isolated from Streptomyces virginiae in 1952 from a soil sample of Roanoke, Virginia. The streptogramins function as antibiotics by binding to the 23S rRNA close to the peptidyltransferase center of the 50S subunit. In so doing, these antibiotics block extension of the peptide chain and lead to dissociation of peptidyl-tRNA.
The group A streptogramins are members of the polyketide family synthesized by the bacteria Streptomyces virginiae. Each of the group A streptogramins are composed of a 23-membered polyunsaturated macrolactone ring with peptide bonds. The group B streptogramins are cyclic hexadepsipeptides of the non-ribosomal peptide antibiotic family. The major streptogramins of the group A class are virginiamycin M, pristinamycin IIA, and dalfopristin, and those of the group B class include pristinamycin IA, quinupristin, and virginiamycin S. Although the two classes of streptogramin are structurally different, they act synergistically, allowing them to exert greater antibiotic activity than the either class do separately, for example pristinamycin IA + IIA, quinupristin + dalfopristin, and virginiamycin M + S. The pristinamycins are close structural relatives of the virginiamycins and they are highly effective human antibiotics. Pristinamycins are highly active against a wide range of Gram-positive bacteria, including methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant Staphylococcus aureus (VRSA), vancomycin-resistant strains of Enterococcus faecium (VREF), and drug-resistant Streptococcus pneumoniae. In addition to these Gram-positive bacteria, pristinamycin is effective against a few Gram-negative bacteria, such as Haemophilus influenzae and Haemophilus parainfluenzae as well as other pathogens that cause atypical pneumonia such as Mycoplasma pneumoniae, Chlamydophila pneumoniae, and Legionella pneumophila.
The Amphenicols
The amphenicols are a class of antibiotics with a phenylpropanoid structure that function by blocking prokaryotic peptidyltransferase in the 50S ribosome. Amphenicols are effective against a wide variety of Gram-positive and Gram-negative bacteria, including most anaerobic organisms. Their activity profile classifies the amphenicols as broad spectrum antibiotics. Chloramphenicol (structure shown in the Table below) was the first isolated amphenicol and was obtained from Streptomyces venezuelae. Chloramphenicol, and its derivatives thiamphenicol and florfenicol, are now chemically synthesized. Due to bone marrow toxicity, chloramphenicol is now very rarely used except for the treatment of brain abscesses and eye infections, or in developing countries because it is inexpensive. The advantage of thiamphenicol is that, despite its inhibitory effect on the red cell count, it has never been associated with aplastic anemia, whereas, florfenicol does carry a risk for development of aplastic anemia. Bacterial resistance to chloramphenicol (and thiamphenicol) is the result of enzymatic modification by chloramphenicol acetyltransferases (CAT). These enzymes covalently link an acetyl group from acetyl-CoA to chloramphenicol, preventing it from binding to the ribosomes. The CAT enzymes do not confer resistance to florfenicol.
Linezolid (Zyvox®)
Linezolid is a member of a class of drug referred to as the oxazolidinone class. Linezolid is active against most Gram-positive bacteria and is used to treat infections such as pneumonia (especially in a hospital setting), and infections of the skin and blood. Linezolid is used principally to treat vancomycin-resistant strains of Enterococcus (VRE) and methicillin-resistant Staphylococcus aureas (MRSA). Enterococci are Gram-positive lactic acid bacteria that belong to the Phylum Firmicutes. The most significant negative side-effect associated with the use of Linezolid is myelosuppression, which includes anemia, leukopenia, pancytopenia, and thrombocytopenia. The discontinuance of Linezolid results in a return to normal hematological parameters. Therefore, it is highly recommended that patients taking this drug for more than two weeks have their blood counts monitored on a weekly basis.
Inhibitors of Prokaryotic Small (30S) Ribosomal Subunit Functions
The small ribosomal subunit (30S) of prokaryotes is composed of the 16S RNA and several ribonucleoproteins (designated with the letter S followed by their size). The functions of the 30S subunit are inhibited by drugs of the aminoglycoside and tetracycline families.
Aminoglycosides
The aminoglycosides are antibiotics that have been isolated from various strains of Streptomyces or Micromonospora. The names of the drugs isolated from Streptomyces all contain the suffix –mycin, whereas those isolated from Micromonospora all contain the suffix –micin. The aminoglycosides are composed of a 6-carbon aminocyclitol ring (2-deoxystreptamin in many instances) linked by glycosidic bonds to one or more sugar derivatives. The initially characterized aminoglycoside was streptomycin isolated from the actinobacterium Streptomyces griseus. Streptomycin was the first drug found to be effective against tuberculosis.
The aminoglycosides exerts bactericidal activity against Gram-negative bacteria and some facultative anaerobic bacilli. These drugs generally are not effective against Gram-positive bacteria nor anaerobic Gram-negative bacteria. In order to gain access to the ribosome, the aminoglycosides must first cross the lipopolysaccharide (LPS) covering (Gram-negative organisms), the bacterial cell wall, and finally the cell membrane. Because of their polarity a specialized active transport process is required for their entry into the bacterium.
Amikacin (synthesized from kanamycin), gentamicin and tobramycin are the major aminoglycosides used in human clinical settings. In addition to these three aminoglycosides, sisomicin, and netilmicin all exhibit an extended spectra that includes Pseudomonas aeruginosa. The additional aminoglycosides, neomycin, framycetin (neomycin B), paromomycin (aminosidine), and kanamycin have broader bactericidal spectra than streptomycin that includes several Gram-positive as well as many Gram-negative aerobic bacteria.
The major toxicities reported with the use of the aminoglycosides are nephrotoxicity and ototoxicity. A number of therapeutic drugs, such as the aminoglycosides, are toxic to functions of the kidney proximal tubule resulting in renal failure due to acute tubular necrosis with secondary interstitial damage (referred to as renal Fanconi syndrome). In addition to the aminoglycosides, the anti-cancer drugs cisplatin and ifosfamide, the anti-retroviral drug tenofovir, and the anti-convulsant sodium valproate can cause renal Fanconi syndrome. The average frequencies of nephrotoxicity for gentamicin and tobramycin are higher than that for amikacin. The average frequency of ototoxicity is highest for amikacin.
Due to the associated nephrotoxicity, renal function should be monitored during therapeutic use of any aminoglycoside. Polyuria, decreased urine osmolality, enzymuria, proteinuria, cylindruria (urinary casts), and increased fractional sodium excretion are indicative of aminoglycoside nephrotoxicity. Aminoglycoside-mediated ototoxicity, usually manifests as either auditory or vestibular dysfunction. Vestibular injury leads to nystagmus, incoordination, and loss of the righting reflex. These effects of aminoglycosides are most often irreversible.
In addition to nephro- and ototoxicity, all aminoglycosides, when administered in doses that result in high plasma concentrations, have been associated with muscle weakness and respiratory arrest attributable to neuromuscular blockade. This blockade is due to the chelation of calcium and competitive inhibition of the release of acetylcholine from pre-synaptic neurons.
Tetracyclines
The tetracyclines are a family of structurally related compounds derived from the polyketide synthesis pathway functioning in various strains of Streptomyces. The name tetracycline is derived from the four-ring carbocyclic skeleton of their structure (see the Table below). The tetracyclines exhibit a broad spectrum of activity, including Gram-positive and Gram-negative bacteria. Tetracycline was an important antibiotic used historically in the treatment of cholera. Today tetracycline is used in humans primarily to treat acne and rosacea. Tetracyclines function as antibiotics by inhibiting the stable binding of aminoacyl-transfer (t)RNA to the bacterial ribosomal A-site resulting in termination of protein synthesis.
Eukaryotic Protein Synthesis Inhibitors
Shiga and Shiga-Like Toxins
The Shiga toxins are a family of structurally and functionally related exotoxins whose principal effects are exerted at the level of protein synthesis but are not exclusive to this biochemical pathway. Due to historical perspectives there are numerous related terms that describe these toxin family members. This toxin family includes Shiga toxin from Shigella dysenteriae serotype 1 and the Shiga toxins (Stx) that are produced by enterohemorrhagic strains of Escherichia coli (EHEC). The Shiga toxins in S. dysenteriae and Shiga-like toxin-producing E. coli (STEC) are encoded by a diverse group of lambda bacteriophages. There are seven identified toxins produced in the various strains of STEC.
In 1897 the Japanese microbiologist Kiyoshi Shiga characterized that the bacterium, S. dysenteriae was a major cause of the potentially lethal disorder referred to as dysentery. Dysentery is a form of inflammatory gastroenteritis that can result from bacterial, viral, protozoal, or parasitic worm infection. In 1977, a group of E. coli were identified that produced a toxin whose activity was tested on Vero cells in culture. The toxin killed the Vero cells and so it was termed verotoxin, and the bacteria were termed verotoxin-producing E. coli (VTEC). Several strains of E. coli were identified in the 1980’s that produced toxins that were related to Shiga toxin and therefore, these bacteria were named Shiga-like toxin-producing E. coli (STEC). The toxins produced by VTEC and STEC strains of E. coli are now identified with the abbreviation: Stx. Shiga toxin from S. dysenteriae and the E. coli-produced Shiga toxin 1 (Stx1) differ only by a single amino acid. There are two distinct families of E. coli Stx toxins identified as the Stx1 and Stx2 families. The Stx1 family is composed of Stx1 and Stx1c, while the Stx2 family is composed of Stx2, Stx2c, Stx2d, Stx2e, and Stx2f.
The most common strain of STEC causing hemorrhagic colitis in humans is the serotype O157:H7. The O and H designations for strains of E. coli define the O-antigens and the H-antigens, immunologic determinants on the surface of the bacteria. The O-antigens are determined by portions of the lipopolysaccharide (LPS) component of the outer membrane. There are several hundred characterized O-antigens. The H-antigens are determined by the bacterial flagella. Up to 15% of patients with hemolytic colitis, due to ingestion of the O157:H7 strain, go on to develop hemolytic-uremic syndrome (HUS). HUS is characterized by hemolytic anemia, acute renal failure (uremia), and a low platelet count (thrombocytopenia).
Shiga toxin and all of the Stx toxins gain entry into host cells via attachment to the cell surface glycosphingolipid, globotriaosylceramide (Gb3; also known as CD77 or the Pk blood group antigen). Following attachment the toxin is subsequently internalized. The toxin, Stx2e, can also bind to the cell surface glycosphingolipid, globotetraosylceramide (Gb4). Following the binding to Gb3 (or Gb4) receptor molecules, Shiga toxin and the Stx family toxins are internalized by endocytosis from clathrin-coated pits. Although the primary uptake mechanism involves clathrin-coated pits, when clathrin-dependent endocytosis is inhibited, Shiga toxin is still efficiently internalized.
Following uptake, Shiga toxins localize to the endosomes and are then transferred to the trans-Golgi network, then to the ER. During membrane transfer host furin endoproteases cleave the toxins into their active forms similar to the process described below for the ADP-ribosylating toxins. Finally the host cell machinery translocates the catalytic A subunit to the cytosol. Shiga toxin and the Stx toxins possess a highly specific RNA N-glycosidase activity that cleaves an adenine base in the 28S rRNA in the large (60S) ribosomal subunit. The 3′ end of the 28S rRNA functions in aminoacyl-tRNA binding, peptidyltransferase activity and ribosomal translation. The result of toxin modification of the 28S rRNA is inhibition of elongation factor-dependent aminoacyl-tRNA binding and subsequent peptide elongation resulting in cell death.
Although Shiga toxins are extremely potent ribosome-modifying enzymes, their effects within the host cell are not limited to the inhibition of protein synthesis. These toxins exert several different cellular effects, including the induction of cytokine expression by macrophages and the activation of what is referred to as the ribotoxic stress response due to the modification of ribosomes. Following Shiga toxin modification of the 28S rRNA, JUN N-terminal kinase (JNK) proteins and p38 family mitogen-activated protein kinases (MAPK) are activated and the signaling exerted by extracellular signal-regulated kinase 1 (ERK1; also known as MAPK3) and ERK2 (also known as MAPK1) are disrupted.
ADP-Ribosylating Pathogens
Bacterial ADP-ribosyltransferase toxins (bARTT: also called bacterial ADP-ribosylating exotoxins, bARE) are encoded by a range of bacteria, including the human pathogens, Corynebacterium diphtheriae, Vibrio cholerae, Bordetella pertussis, Pseudomonas aeruginosa, Clostridium botulinum, and Streptococcus pyogenes, as well as certain strains of Escherichia coli. The bARTT enzymes are encoded by genes that belong to the family of ADP-ribosyltransferase (ADPRT) encoding genes found in both prokaryotes and eukaryotes. Like all members of the ADPRT family of enzymes the bARTT enzymes use a nicotinamide adenine dinucleotide (NAD+) donor molecule to catalyze the transfer of the ADP-ribose moiety to a given substrate. The acceptor for these ADP-ribosylation reactions can be an Arg, Asn, Thr, Cys or Gln residue, depending on the individual toxin. Members of the bARTT family catalyze the addition of ADP-ribose on various eukaryotic substrates, including monomeric G-proteins of the RHO family (e.g. Clostridium botulinum toxin), heterotrimeric G-proteins (Vibrio cholerae and Bordetella pertussis toxins) and actin (Clostridium botulinum toxin). The toxins produced by Corynebacterium diphtheriae and Pseudomonas aeruginosa induce the ADP-ribosylation of the critical protein synthesis elongation factor, eEF-2.
Cholera Toxin
The well-characterized toxin produced by Vibrio cholerae (identified as CT or CTx) is known to induce the ADP-ribosylation of a specific Arg residue in the α-subunit of a heterotrimeric Gs-type G-protein in intestinal enterocytes resulting in the classic “rice water” diarrhea that is associated with cholera.
The unregulated activation of the Gs-type G-protein leads to continuous activation of adenylate cyclase and consequent cAMP production. The increased cAMP activates PKA which leads to unregulated phosphorylation of numerous proteins, one of which is the cystic fibrosis transmembrane conductance regulator (CFTR) Cl– channel. The phosphorylation of CFTR leads to increased Cl– transport out of intestinal enterocytes. The enterocytes attempt to compensate for the loss of the loss of Cl– by activating a Cl–/HCO3– exchanger resulting in increased HCO3– excretion into the intestinal lumen. The enterocyte Na+/H+ (NHE1) cation exchanger is also activated and the resultant increase in H+ and HCO3– efflux to the lumen is the cause of the increased H2O [H+ + HCO3– ↔ H2CO3 ↔ H2O + CO2] in the lumen and the consequence watery diarrhea.
The major strains of V. cholerae that are the causes of cholera are the O1 and O139 strains. However, an additional toxin produced by several clinical strains of V. cholerae, identified as ChxA-I (encoded by the CHX gene), has been characterized that also induces ADP-ribosylation of eEF-2. The ChxA-I toxin is not produced by the O1 nor the O139 strains of V. cholerae.
Pertussis Toxins
Bordetella pertussis exerts its pathological effects in several functionally distinct ways. The bacterium binds to sulfatides on the surface of airway epithelial cells which stimulates the production of a toxin called tracheal cytotoxin (TCT). TCT functions by interfering with the beating of cilia preventing the clearing of debris from the lungs. This effect of TCT is what causes the coughing associated with B. pertussis infection. Another toxin produced by B. pertussis, identified as PTx, is a member of the bARTT family of enzymes. PTx is known to induce the ADP-ribosylation of the α-subunit of a heterotrimeric Gi-type G-protein in phagocytes that infiltrate the site of B. pertussis infection. The effect of PTx is inhibition of the normal immune response to B. pertussis infection. The PTx-mediated inhibition of the inhibitory action of Gi-type G-proteins on adenylate cyclase activity causes the phagocytes to produce too much cAMP. The elevated cAMP levels then disturb the normal signal transduction processes of the phagocytes that is required for normal responses to infection. B. pertussis produces another toxin identified as adenylate cyclase toxin that possesses intrinsic adenylate cyclase activity. The combined effects of adenylate cyclase toxin and PTx are dramatic increases in cAMP within the phagocytes furthering the degradation of cellular signaling needed to mount a proper immune response.
Diphtheria Toxin
Diphtheria toxin (DT or DTx) is encoded on a lysogenic bacteriophage in C. diphtheriae. C. diphtheriae colonize the upper respiratory tract epithelium. Following bacteriophage lysogeny, C. diphtheriae release DT, which then disseminates throughout the body. DT targets many different tissues through its ability to bind to the ubiquitous heparin-binding epidermal growth factor-like growth factor (HB-EGF) receptor. After receptor binding, the DT-receptor complex undergoes clathrin-mediated endocytosis and traffics to an early endosome. Once internalized, DT is processed by host proteases, which results in the formation of a disulfide bonded dipeptide consisting of an A subunit and a B subunit (A-B toxin). Endosomal acidification promotes the insertion of the B subunit into the membrane to deliver the A domain across the endosomal membrane, where reduction of the disulfide bond releases the A domain into the cytosol. The A subunit then catalyzes the ADP-ribosylation of eEF-2 resulting in the inhibition of the function of eEF-2 and consequent inhibition of protein synthesis.
The site of toxin-induced ADP-ribosylation of eEF-2 was originally discovered from work with C. diphtheriae and shown to be a His residue (H715) in eEF-2 that contains a modification that was subsequently termed a diphthamide residue. The precise purpose for the diphthamide residue, in the function of eEF-2, remains elusive. In eukaryotes, there are seven genes that have been identified in the diphthamide synthesis pathway. These genes are identified as DPH1 through DPH7. The first step in diphthamide residue synthesis requires the DPH1 through DPH4 genes, while the second step requires the DPH5 gene. The terminal steps in diphthamide synthesis involves the DPH6 and DPH7 genes. The DPH5 encoded enzyme is a histidine protein methyltransferase (HPMT) that trimethylates the H715 residue in eEF-2.
The DPH1 gene has been found to be frequently deleted in ovarian and breast cancer, therefore, this gene is also identified as the OVCA1 (ovarian cancer gene 1) gene. Deletion of the DPH3 gene leads to embryonic death and DPH4 mutants are associated with retarded growth and development. Thus, there is clear evidence for the biological significance of this particular form of modification of eEF-2. The diphthamide residue has also been shown to be important in maintaining translational fidelity since a loss of the modification is associated with elevated frameshifting during protein synthesis.
Strains of Escherichia coli also produce an ADP-ribosylating toxin, termed the heat-labile enterotoxin, that catalyzes the ADP-ribosylation of the same Arg residue in the Gs-type G-protein in the gut as does cholera toxin. The consequences of the E. coli toxin are often referred to as “traveler’s diarrhea”.
Pseudomonas Toxin
The toxin produced by Pseudomonas aeruginosa is an A-B toxin of 613 amino acids that is closely related to DT. This toxin is referred to as Pseudomonas aeruginosa exotoxin (PE, encoded by the TOXA gene). Despite limited primary amino acid homology, the A domains of PE and DT are structurally related. PE uses low density lipoprotein receptor-related protein 1 (LRP1) as a host receptor. Like DT, PE is cleaved by a cellular protease and is internalized via clathrin-mediated endocytosis. However, unlike the mechanism for delivery of the A subunit of CT to the cytosol, PE undergoes retrograde trafficking through the Golgi to the ER. Within the ER, reduction of the disulfide bond and use of the cellular ER-associated degradation (ERAD) system enables the A domain to traverse the ER membrane into the cytosol. Once in the cytosol, the PE A subunit catalyzes the same mechanism as DT, ADP-ribosylation of the diphthamide residue of eEF-2.
Table of Antibiotic and Toxin inhibitors of Translation (non-inclusive)
| Chloramphenicol |  |
| Streptomycin | inhibits prokaryotic peptide chain initiation, also induces mRNA misreading |
| Tetracycline |  |
| Neomycin | similar in activity to streptomycin |
| Erythromycin | inhibits prokaryotic translocation through the ribosome large subunit |
| Fusidic acid | similar to erythromycin only by preventing EFG from dissociating from the large subunit |
| Puromycin | resembles an aminoacyl-tRNA, interferes with peptide transfer resulting in premature termination in both prokaryotes and eukaryotes |
| Diphtheria toxin | protein from Corynebacterium diphtheriae which causes diphtheria; catalyzes ADP-ribosylation and inactivation of eEF-2; eEF-2 contains a modified His residue known as diphthamide, it is this residue that is the target of the toxin |
| Ricin | found in castor beans; is a heterodimer of A chain (267 amino acids) and B chain (262 amino acids) proteins; functions similar to shiga toxin of Shigella dysenteriae and the shiga-like toxins of enterohemorrhagic E. coli; the A chain is an N-glycoside hydrolase that catalyzes cleavage of the eukaryotic large subunit 28S rRNA, the B chain is a lectin that binds to galactose residues on cell-surface glycoproteins |
| Cycloheximide |  |