
Last Updated: April 20, 2026
Protein Folding: Co- and Post-translational Processing
The process of protein synthesis, of and by itself, does not directly result in the generation of functionally and structurally complete macromolecules. Many proteins must undergo one or more forms of modification that can occur either co-translationally and/or post-translationally as described in detail in the following sections. However, an equally critical process that must be undertaken to ensure protein function is the folding of the protein into a defined three-dimensional structure. Within the environment of the cell, newly synthesized proteins are at great risk of aberrant folding and aggregation. Improper folding and protein aggregation can lead to the formation of potentially toxic species. To reduce and prevent these negative outcomes, cells harbor a complex network of molecular chaperones whose functions are to promote efficient folding and to prevent protein aggregation. The structure of proteins within the cell is in a highly dynamic state and, therefore, constant molecular chaperone surveillance is required to ensure protein homeostasis.
Protein Folding Chaperones
Human cells express several families of molecular chaperones with the most abundant families being those that were originally designated as responding to the stresses induced by heat in the fruit fly Drosophila melanogaster. These protein families are, therefore, classically termed heat shock proteins (Hsp) and also referred to as stress proteins. Although defined by the initial observation of heat-induced expression, Hsp genes can be induced by inflammation, ischemia, infections, irradiation, and exposure to heavy metals, oxidants, and organics. Many Hsp proteins possess intrinsic ATPase activity and it is the hydrolysis of ATP that powers the protein folding processes.
The Hsp superfamily is composed of multiple protein subfamilies that are loosely designated by molecular weights. These Hsp families include the Hsp40, Hsp60, Hsp70, Hsp90, and Hsp100 families. In addition, there is the small heat shock protein family often referred to as the Hsp25 family or the HSPB family. Protein folding chaperones in the ER belong to the Hsp40, Hsp70, Hsp90, and Hsp100 families.
Two additional folding chaperones are calnexin and calreticulin that function in the endoplasmic reticulum (ER) in the processes of glycoprotein maturation.
Additional folding chaperones possess distinct (non-ATPase-mediated) enzymatic activity. Protein disulfide isomerases (PDI) catalyze the formation of disulfide bonds between cysteine residues in a substrate protein. Peptidylprolyl cis-trans isomerases (PPI) catalyze isomerization of peptide bonds involving proline residues.
Hsp40 Family
The proteins of the Hsp40 family are also referred to as DnaJ proteins. The DnaJ nomenclature is derived from the E. coli Hsp70 co-chaperone DnaJ. Hsp40 proteins are correctly referred to as obligate co-chaperones since they function in conjunction with proteins of the Hsp70 family whose ATPase activities require the interactions with Hsp40 proteins. The J-domain in Hsp40/DnaJ family of chaperones is an approximately 70 amino acid domain composed of four α-helices found at the N-terminus of most proteins in the family. The J-domain was originally identified as being involved in the regulation of the ATPase activity of Hsp70 family member proteins. Within the J-domain is a HPD (His-Pro-Asp) tripeptide motif which is essential for stimulation of the ATPase activity of Hsp70 proteins. In addition to the J-domain, Hsp40 proteins possess a zinc-finger domain that is critical for the sequestration of a denatured substrate as well as in assisting Hsp70 proteins in the folding reaction.
The Hsp40/DnaJ proteins are involved in processes that regulate gene expression and translational initiation as well as those controlling the folding and unfolding, translocation, and degradation of proteins. Hsp40/DnaJ proteins also bind to unfolded or non-native polypeptides in order to prevent their aggregation.
Based primarily upon structural characteristics the Hsp40/DnaJ protein family can be divided into four subtypes: type I, type II, type III, and type IV, where only the type I, II, and III Hsp40 families exist in humans.
The type I proteins contain all of the domains originally identified in the E. coli DnaJ protein. These domains include the J-domain, the G/F-rich (Gly/Phe) region, and cysteine-rich zinc finger (ZF) domain.
Type II proteins possess the J-domain and the G/F-rich region but lack the zinc-finger domain.
Type III proteins possess only the J-domain which is in the C-terminus.
Humans express a total of 49 genes that encode proteins of the Hsp40/DnaJ family. These genes include the DNAJA1-DNAJA4 genes, the DNAJB1-DNAJB9 and DNAJB11-DNAJB14 genes. The DNAJC1-DNAJC19, DNAJC21, DNAJC22, DNAJC24, DNAJC25, DNAJC27, DNAJC28, DNAJC30, DNAJC5B, and DNAJC5G genes. The remainder of the Hsp40 family genes are the HSCB, SEC63, GAK, and SACS genes.
Hsp60 Family
Humans express a single gene of the Hsp60 family identified as the HSPD1 gene. Hsp60 is also referred to as a chaperonin, specifically a group I chaperonin. Proteins that are classified as chaperonins are large (800-900 kDa) double-ring complexes that function by globally enclosing substrate proteins for folding. Proteins up to a size of around 60 kDa can be acted upon by the chaperonins.
There are 15 genes in the human genome that encode proteins termed chaperonins with the HSPE1 encoded protein (chaperonin 10/Hsp10) being required as a co-chaperone for Hsp60 function. Human Hsp60 was originally characterized as a mitochondrial chaperone involved in correct folding of mitochondrial proteins during their import into this organelle. The activity of Hsp60 is catalyzed, in part, by its intrinsic ATPase activity.
In addition to its role in mitochondrial protein folding, Hsp60 is also involved in the recognition and ATPase-mediated unfolding of non-native and aggregated proteins so that they can efficiently refold into their native conformations. Non-protein folding functions of Hsp60 include a role in mitochondrial DNA replication. The group II chaperonins are members of the CCT [chaperonins containing TCP1 (T-complex 1)] subfamily and these proteins do not require the Hsp10 co-chaperone for activity.
Hsp70 Family
The Hsp70 family represents the most ubiquitous class of chaperones and is highly conserved in all organisms. Proteins of the Hsp70 family possess intrinsic ATPase activity. The Hsp70 family of chaperones control all aspects of intracellular protein homeostasis, which includes nascent protein folding, protein import into organelles, unfolding of non-native and aggregated proteins, and the assembly of multi-protein complexes. Several proteins of the Hsp70 family of chaperones also function extracellularly mediating immune modulation and cytoprotection.
All Hsp70 proteins possess intrinsic ATPase activity which regulates their ability to interact with exposed hydrophobic surfaces of substrate proteins. Regulation of Hsp70 functions are also effected by other proteins that interact with subdomains such as is the case for the obligate co-chaperone, Hsp40/DnaJ.
Humans express 17 genes that encode Hsp70 family member proteins. The major stress-inducible Hsp70 proteins are identified as Hsp70-1 (encoded by the HSPA1A gene) and Hsp70-2 (encoded by the HSPA1B gene). These two proteins differ by only two amino acids and are often collectively referred to as Hsp70 or Hsp70-1. The HSPA8 encoded protein (Hsp70-8) is an essential housekeeping chaperone responsible for the bulk of Hsp70-mediated protein folding and protein transport across membranes. The HSPA9 encoded protein (Hsp70-9) is predominantly found in the mitochondria and the protein contains 46 amino acid mitochondrial targeting sequence.
Proteins of the Hsp70 family are highly conserved at the amino acid level and possess a set of common domains. The N-terminal nucleotide binding domain (NBD) binds and hydrolyzes ATP. The NBD is composed of four subdomains, termed IA, IB, IIA, and IIB, that surround the ATP-binding pocket. The C-terminal substrate binding domain (SBD) is the domain that binds extended polypeptide substrates. There is also a central domain defined by the presence of several protease sensitive sites. The NBD is conserved in all of the Hsp70 family members except for the proteins encoded by the HSPA12A and HSP12B genes which contain a more divergent NBD.
The Hsp70 family member proteins that are predominantly cytosolic also contain a glycine and proline-rich (G/P-rich) C-terminal region that contains the tetrapeptide, EEVD (GluGluValAsp). This tetrapeptide motif is involved in binding of co-chaperones and other Hsp proteins. The EEVD motif is not present in the mitochondrial- nor the ER-localized Hsp70 proteins, Hsp70-9 (HSPA9) and Hsp70-5 (HSPA5), respectively. Additionally, Hsp70-5 contains the C-terminal ER retention signal, KDEL (LysAspGluLeu).
One critically important member of the Hsp70 family of molecular chaperones is the protein commonly identified as glucose-regulated protein 78-kDa, GRP78. GRP78 is also known as BiP (Binding immunoglobulin heavy chain Protein). The human GRP78 protein is encoded by the HSPA5 [heat shock protein family A (Hsp70) member 5] gene. GRP78 is referred to as the master regulator of the ER. The function of GRP78 is not only to participate in protein folding but to also maintain the permeability barrier of the ER by sealing the luminal side of inactive translocons (protein complexes through which nascent polypeptides are extruded), to facilitate the translocation of growing polypeptide chains into the ER lumen, to regulate the aggregation of nonnative polypeptides, and to contribute to calcium homeostasis in the ER. Of significance to the role of ER function in apoptosis, GRP78 plays an important role in the regulation of the unfolded protein response, UPR.
Proteins of the Hsp70 family also function as potent inhibitors of apoptosis. Hsp70-1 blocks the mitochondrial translocation and activation of mitochondrial outer membrane-associated BAX complex. As described in detail in the Protein, Organelle, and Cell Turnover page, activation of BAX (as well as the related pore complex BAK) results in the release of cytochrome c which leads to activation of the intrinsic apoptosis pathway. Hsp70-1 also inhibits assembly of the death-inducing signaling complex (DISC) typical of the extrinsic apoptosis pathway.
Within the context of the intrinsic apoptosis pathway, Hsp70-1 binds directly to apoptotic protease activating factor 1 (APAF1) and blocks the recruitment of procaspase-9 to the mitochondrial apoptosome which includes cytochrome c and ATP. Hsp70-1 also interacts with the pro-apoptotic mitochondrial protein known as apoptosis-inducing factor (AIF; encoded by the AIFM1 gene) which results in inhibition of caspase-independent apoptosis. Hsp70-1 also regulates ER-stress-mediated apoptosis by interfering with the activities of c-Jun N-terminal kinase (JNK), p38 mitogen-activated protein kinase (p38 MAPK; encoded by the MAPK14 gene), and extracellular signal-regulated kinase (ERK1/ERK2; encoded by the MAPK1 and MAPK3 genes) in the apoptotic pathway. Hsp70-1 also stimulates proteasomal degradation of apoptosis signal-regulated kinase 1 (ASK1; encoded by the MAP3K5 gene) which normally phosphorylates and activates JNK and p38 MAPK. This latter function of Hsp70-1 occurs in conjunction with the E3 ubiquitin ligase known as C-terminal Hsp70-interacting protein (CHIP).
Hsp90 Family
The Hsp90 family member proteins function as “holdases” (similar to the activity of the small Hsp family members) to keep substrate proteins in the non-aggregated state before transferring the substrate to an Hsp70 protein for refolding. Humans express four genes in the Hsp90 family, HSP90AA1, HSP90AB1, HSP90B1, and TRAP1 (TNF receptor-associated protein 1). The HSP90AA1 and HSP90AB1 encoded proteins are localized to the cytosol, the HSP90B1 encoded protein is localized to the ER, and the TRAP1 encoded protein is localized to the mitochondria. The domain structure of the Hsp90 family proteins is very similar to that of the Hsp70 family proteins which includes an N-terminal nucleotide binding domain (NBD), a central (middle) domain, and a C-terminal substrate/co-factor binding domain. Like the C-terminal domain in most Hsp70 proteins, the C-terminal domain of the Hsp90 family proteins contains an EEVD motif. However, the Hsp90 proteins contain a pentapeptide motif, MEEVD.
Hsp100 Family
Humans express a single Hsp100 family member identified as Hsp110 which is encoded by the HSPH1 gene. Hsp110 functions in a heterodimeric complex with Hsp70 family member proteins. This complex functions as a nucleotide exchange factor (NEF) whereby one protein serves to exchange ATP for ADP in the other protein of the complex and vice versa. Both proteins function cooperatively in the process of disassembling stable protein aggregates.
Calnexin and Calreticulin
Calnexin is encoded by the CNX gene and is a transmembrane protein chaperone associated with the ER. The function of calnexin is to assist in the folding of N-glycosylated proteins within the ER. The function of calnexin ensures that only glycoproteins that are properly folded and assembled continue further along the secretory pathway. Calnexin only binds to N-glycoproteins that have a Glc1Man9GlcNAc2 oligosaccharide attached to an Asp residue. This structure results from glucosidase actions on the en bloc oligosaccharide (Glc3Man9GlcNAc2) catalyzed first by glucosidase I (GluI) and then by glucosidase II (GluII). GluI is encoded by the mannosyl-oligosaccharide glucosidase (MOGS) gene. Functional GluII is composed of an α-subunit and a β-subunit. The GluII α-subunit gene is GANAB (glucosidase II alpha subunit) and the β-subunit gene is PRKCSH (protein kinase C substrate 80K-H). Calreticulin is encoded by the CALR gene. Calreticulin is a multifunctional protein whose primary function is to bind and sequester Ca2+ ions in the lumen of the ER. Calreticulin also binds to misfolded proteins preventing them from being exported from the ER to the Golgi apparatus.
Protein Disulfide Isomerases
The protein disulfide isomerase (PDI) family of enzymes are thiol oxidoreductase chaperones that are members of the thioredoxin superfamily. The PDI family of enzymes consists of 21 members. The original member of this family (initially identified as protein disulfide isomerase) is identified as PDIA1.
The PDIA1 protein is encoded by the prolyl 4-hydroxylase subunit beta (P4HB) gene. The PDIA1 protein is an endoplasmic reticulum (ER) localized enzyme that catalyzes the formation and breakage of disulfide bonds between cysteine residues within proteins as they fold. The general function of this large family of oxidoreductases is to catalyze exchange reactions between thiols and disulfides. Specifically, within the ER, these enzymes introduce and isomerize disulfide bonds in newly synthesized proteins, that since they are in the ER are typically destined for extracellular secretion or insertion into membranes. Thus, unlike other chaperones that utilize the energy of ATP hydrolysis to catalyze protein folding, PDI enzymes contribute to folding through oxidative folding.
PDI enzymes contain multiple domains initially characterized in thioredoxin and are, therefore, referred to as thioredoxin domains. The N- and C-terminal thioredoxin domains possess the catalytic disulfide/dithiol centers and contain the canonical CXXC motif. The internal thioredoxin domains impart structure to the PDI enzymes as well as providing sites for additional interactions with the protein substrates.
The primary function of PDI enzymes is in the process of redox protein folding in the within the lumen of the ER lumen. As such most PDI family enzymes contain the ER localization motif, Lys-Asp-Glu-Leu (KDEL), that promotes retrograde transport back to the ER. Some PDI family member enzymes do not contain the KDEL sequence and function instead as transmembrane localized enzymes. Two PDI family member proteins (encoded by the ERP27 and ERP29 genes) do not possess the dithiol redox active domains.
During the course of PDI-mediated introduction and isomerization of disulfide bonds the enzymes themselves are oxidized. The major mechanism for the reduction of oxidized PDI is via the coupled oxidation of glutathione (GSH) generating a reduced, and functional PDI, and oxidized glutathione (GSSG). In the cytosol GSSG is normally reduced via the activity of glutathione reductases. However, there is no glutathione reductase in the lumen of the ER so the oxidized glutathione (GSSG) needs to be transported out of the ER to the cytosol where it can be reduced. The transport of GSSG out of the ER is catalyzed by the SLC family transporter encoded by the SLC33A1 gene.
Clinical significance of the SLC33A1 encoded transporter is evident from the fact that mutation in the gene are associated with a lethal disorder identified as Huppke-Brendel syndrome (HPBDS). HPBDS is associated with severe psychomotor impairment, congenital hearing loss, bilateral cataracts, and low serum ceruloplasmin and copper.
A PDI related to the Hsp40 protein family is encoded by the DNAJC10 gene and the encoded protein was originally called PDIA19 and also identified as ERdj5. The DNAJC10 encoded protein is a major ER reductase that also participates in the reduction of oxidized PDI enzymes.
In addition to ensuring the proper folding of newly synthesized proteins, PDI enzymes function as effectors of the translocation of misfolded and terminally unfolded proteins from the ER to the cytosol where they are ultimately degraded by the proteasomal machinery. The process of translocation of misfolded proteins from the ER to the cytosol is referred to as ER-assisted protein degradation (ERAD). The process of ERAD increases significantly under conditions of increased cellular oxidative stress and ultimately triggers the unfolded protein response, UPR.
Peptidylprolyl Isomerases
Peptidylprolyl cis-trans isomerases (peptidylprolyl isomerases, PPIases) represent a family of enzymes that catalyze the interconversion of cis and trans isomers of peptide bonds made with the amino acid proline. Humans express PPIase genes that are divided into two large families, the parvulins and the immunophilins.
The immunophilin family is further divided into the FKBP prolyl isomerase and the cyclophilin peptidylprolyl isomerase subfamilies. The immunophilins are so-called because they were first identified as modifying the activity of immunosuppressive drugs such as cyclosporine and tacrolimus. The drug FK506 is also known as tacrolimus. The FKBP prolyl isomerase subfamily is composed of 18 proteins. The term FKBP refers to the fact that the founding member was identified by its binding to the immunosuppressant FK506, thus it was called FK506-binding protein (FKBP). The cyclophilin peptidylprolyl isomerase subfamily is composed of 19 proteins where cyclophilin A is the founding member (encoded by the PPIA gene).
There are two genes in the parvulin family of PPIases. Unlike both the FKBP and cyclophilin family PPIases, the parvulins do not bind to immunosuppressants. The two parvulin family members are peptidylprolyl cis/trans isomerase, NIMA-interacting 1 (encoded by the PIN1 gene) and peptidylprolyl cis/trans isomerase, NIMA-interacting 4 (encoded by the PIN4 gene)
The activity of human PPIases is dependent, in part, on the presence of various different functional domains. One of the most common domains found in human PPIases is the TPR (tetratricopeptide repeats) motif. A TPR motif consists of multiple repeats of 34 amino acids defined by a specific pattern of hydrophobic amino acids. Clustered TPR motifs in a protein result in the arrangement of parallel helix-turn-helix domains. In most proteins with TPR motifs it is common to find three consecutive motifs in the organization of the repeat. When present in multiple repeats the TPR motifs form a right-handed superhelix. This superhelix forms a groove with a large surface area to which the appropriate ligand can bind. TPR motifs are found in PPIases of both the cyclophilin and FKBP families. Another domain found in several human PPIases is the Ca2+-binding EF hand domain. The human PPIases that have an EF-hand domain are all FKBP family members and all of the EF-hand-containing FKBP proteins are localized to the endoplasmic reticulum (ER).
Within eukaryotic proteins the trans isomer of proline peptide bonds is the more common form. However, several important proteins, including several ribonucleases and interleukins, possess cis-prolines in the native state. Given that the energy for cis-trans isomerization is quite high, the transformation will not occur spontaneously and represents the rate-limiting step in protein folding. Several PPIases possess autocatalytic activity (e.g. Pin1 and FKBP) that catalyzes intrachain cis-trans isomerizations, thus participating in their own folding.
Secreted and Membrane-Associated Proteins
Proteins that are membrane bound or are destined for excretion, as well as glycoproteins, are synthesized by ribosomes associated with the membranes of the endoplasmic reticulum (ER). The ER associated with ribosomes is termed rough ER (RER). These classes of protein all contain an N-terminus termed a signal sequence or signal peptide. The signal peptide is usually 13-36 predominantly hydrophobic amino acid residues in length. Proteins that contain a signal peptide are called preproteins to distinguish them from proproteins (proteins that undergo proteolysis to become active). However, some proteins that are destined for secretion are also further proteolyzed following secretion and are, therefore, termed preproproteins. The insulin precursor protein is a prime example of a preproprotein.
Of significance is that at least one-third of the human genome codes for proteins that will be synthesized by membrane-bound ribosomes and translocated into the ER where they will undergo post-translational modifications, oxidative folding, and maturation to their functional tertiary or quaternary state. These error-susceptible steps in protein biosynthesis are both aided and monitored by resident ER chaperones and their cofactors in a process termed ER quality control.
Two major chaperone families exist in the ER that interact with a wide variety of clients: the lectin chaperones, which generally recognize incompletely folded glycosylated proteins, and the Hsp70 family member, GRP78 (glucose-regulated protein 78; also known as immunoglobulin heavy chain binding protein, BiP), which can interact with both non-glycosylated and glycosylated proteins.
In addition to playing a major role in chaperoning newly synthesized proteins, GRP78/BiP is also responsible for maintaining the permeability barrier of the ER during protein translocation, targeting misfolded proteins for retrograde translocation so they can be degraded by the proteasome, contributing to ER calcium stores, and sensing conditions of stress in this organelle to activate the mammalian unfolded protein response (UPR). The binding of adenosine nucleotides by GPR78/BiP plays a vital role in all of these activities except calcium binding, and its interaction with specific ER-localized DnaJ (ERdj) family members allows it to contribute to diverse cellular functions.
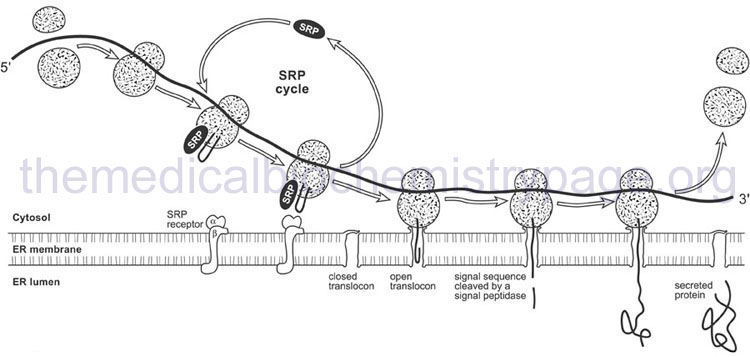
The signal peptide is recognized by a ribonucleoprotein complex termed the signal recognition particle (SRP). Recognition occurs as the signal peptide emerges from the exit side of the ribosome. The eukaryotic SRP is composed of six proteins and an RNA termed the 7SL RNA. The six proteins of the SRP are SRP9, SRP14, SRP19, SRP54, SRP68, and SRP72. Humans express at least three genes encoding the 7SL RNA identified as RN7SL1, RN7SL2, and RN7SL3.
When SRP binds to the emerging signal peptide it induces a translational elongation arrest until the entire translational complex and SRP binds to the SRP receptor on the ER. The SRP receptor (termed SR) is a heterodimeric complex composed of an α-subunit (SR-α; encoded by the SRPRA gene) and a β-subunit (SR-β; encoded by the SRPRB gene). Associated with the SRP receptor is a translocation channel through which the emerging polypeptide is extruded into the lumen of the ER. The translocation channel is referred to as the translocon. Although the translocon is composed of multiple protein subunits the critical channel is formed from the heterotrimeric Sec61 complex which contains the Sec61α, Sec61β, and Sec61γ proteins. The Sec61α subunit is encoded by the SEC61A1 gene, the Sec61β subunit is encoded by the SEC61B gene, and the Sec61γ subunit is encoded by the SEC61G gene.
The signal peptide is removed from the elongating protein following passage through the ER membrane. The removal of the signal peptide is catalyzed by enzymes of the serine protease family known as signal peptidases. The human ER signal peptidase is a multiprotein complex identified as the signal peptidase complex, SPC. Human SPC is composed of five subunits encoded by the SPCS1, SPCS2, SPCS3, SEC11A, and SEC11C genes. The protein encoded by the SEC11A gene constitutes the catalytic portion of the SPC.
Targeting Proteins to the Endoplasmic Reticulum
Many proteins, both soluble and membrane associated, remain integral components of the endoplasmic reticulum, ER. This includes all of the proteins whose functions are critical for the correct processes carried out by the ER. Proteins that are localized to the ER contain specific recognition motifs that are composed of amino acids at either the N-terminus or the C-terminus (predominant site) of the protein. The first, and considered the classical, ER retention signal identified was the tetrapeptide composed of Lys-Asp-Glu-Leu and designated the KDEL sequence or the KDEL motif. The KDEL motif is found at the C-terminus of most soluble proteins retained in the ER.
The recognition of the KDEL motif is carried out by specific receptors. Humans express three KDEL receptor genes identified as KDELR1, KDELR2, and KDELR3. The function of KDEL receptors is to bind to ER chaperones that contain the KDEL sequence (such as the Hsp70 proteins as discussed above). These chaperones are recognized by the KDEL receptor in compartments of the ER that are in association with the Golgi apparatus. When the KDEL receptors bind a ligand the complexes are packaged into coat protein complex I (COPI) vesicles which undergo retrograde transport back to the ER.
The other major ER retention signal is the KKXX motif (where K is Lys and X is any amino acid) that is also most often associated with the C-terminus of retained proteins. The KKXX motif is found in most ER membrane retained proteins. Proteins harboring the KKXX motif also interact with COPI, and do so in the cis Golgi compartment, thereby, stimulating retrograde transport back to the ER.
Tail-Anchoring of Membrane Proteins
Most membrane anchored proteins become embedded in the membrane via the mechanisms just outlined that involve the N-terminal signal peptide targeting the protein to the membrane. However, there is another important class of membrane anchored proteins whose membrane anchoring signal resides in the C-terminus. These membrane proteins are members of the tail-anchored membrane protein family. As described, proteins that are membrane targeted via the presence of an N-terminal signal peptide become embedded co-translationally.
Tail-anchoring of membrane proteins, on the other hand, does not occur co-translationally but is, in fact, uncoupled from ongoing protein synthesis. All tail-anchored proteins contain a single transmembrane (TM) sequence at their extreme C-terminus. The presence of this TM sequence not only targets the protein to the appropriate subcellular organelle membrane but it also serves to ensure the protein remains anchored to the membrane. The amino acids adjacent to the TM anchoring segment participate in the correct subcellular localization of a given tail-anchored protein.
All tail-anchored proteins expose the larger N-terminal portion of the protein to the cytosol, regardless of subcellular localization. Tail-anchored proteins are found in the mitochondrial membranes, peroxisomal membranes, and the intracellular membrane compartments that are connected to the exocytotic and endocytotic pathways. These latter membranes include endoplasmic reticulum (ER), Golgi, endosome, and lysosome membranes. With respect to the processes of exocytosis and endocytosis, the large family of proteins required for vesicle trafficking and membrane fusion, the SNARE proteins (see Pathways for Protein Exocytosis and Endocytosis section below), are all tail-anchored.
The processes by which tail-anchored proteins become embedded in a membrane are of three main types. In one pathway, first identified in the membrane targeting of SNARE family member protein identified as synaptobrevin 2 (encoded by the VAMP2 gene), the protein becomes transiently associated with the signal peptide-binding domain of the signal recognition particle (SRP) which is involved in the classic co-translational membrane targeting pathway outlined above. The interaction of the tail-anchored protein with SRP is followed by membrane insertion, a process involving the SRP receptor. The SRP-dependent tail-anchoring process is associated with a subset of tail-anchored proteins whose TM segments are highly hydrophobic. In the case of SRP-mediated tail-anchoring, the energy for the process is supplied via GTP hydrolysis. However, the vast majority of tail-anchored proteins are inserted into their target membrane via an ATP-dependent process.
In the second pathway for tail-anchoring, ATPases of the heat shock protein 70 (HSP70) family of molecular chaperones catalyze the targeting and insertion. Humans express 17 different genes that encode proteins of the HSP70 family with the cytosolic ATPase commonly identified as HSC70 (encoded by the HSPA8 gene) being involved in the tail-anchoring process. HSC70 and its co-chaperone identified as HSP40 (encoded by the DNAJB1 gene) promote the ATP-dependent insertion of a subfamily of tail-anchored proteins. A characteristic of HSC70-HSP40 catalyzed tail-anchoring is that their substrate proteins possess a TM segment that has a low hydrophobic character.
The third major tail-anchoring pathway involves what is called the GET pathway. Proteins of the GET pathway were initially identified in experiments aimed at dissecting the secretory pathways in yeast and their names derive from Golgi-ER Trafficking. Subsequent to these initial characterizations it was found that human homologs of the yeast GET genes encode proteins involved in the process of tail-anchoring membrane proteins. The designation GET is now used to refer to Guided Entry of Tail-anchored proteins. In the GET pathway the membrane proteins identified as Get1 and Get2 form the receptor for a complex referred to as the TM-recognition complex, TRC. In humans a potential Get1 homolog is encoded by the WRB (tryptophan rich basic protein) gene and the Get2 homolog is likely to be encoded by the CAMLG (calcium modulating ligand) gene. Within the cytosol Get3, Get4, and Get5 interact to form the TRC. The targeted tail-anchored protein is bound by the Get3 protein of the TRC. In humans the Get3 protein is encoded by the ASNA1 (arsA arsenite transporter, ATP-binding, homolog 1) gene. Humans express a GET4 gene and the likely gene encoding the human Get5 homolog is the UBL4A (ubiquitin-like 4A) gene. Following binding of the appropriate tail-anchored protein to the TRC, the complex interacts with the Get1/Get2 receptor and protein anchoring ensues.
Targeting Proteins to Specific Subcellular Organelles
Numerous proteins are unique to the organelle in which they serve their functions whether they be structural or enzymatic. Because various proteins are destined for specific subcellular organelles there must be mechanisms in place to accurately and efficiently target them to their functional destination. The process of organelle targeting involves both direct amino acid sequences in the proteins and proteins involved in the recognition and transport of organellar-specific proteins. For example, proteins that are members of the lysosomal hydrolase family are targeted to the lysosomes via a specific carbohydrate modification which is then recognized by a specific receptor in the lysosomal membrane. Proteins destined for the nucleus, the mitochondria, and the peroxisomes are all targeted to these organelles by the presence of specific peptide sequences and protein structures.
Lysosomal Targeting of Degradative Hydrolases
The lysosomes represent the major intracellular membrane-bound organelles that are responsible for the degradation and turnover of numerous cellular constituents such as proteins, carbohydrates, lipids, nucleic acids, and membranes as well as contributing to the degradation of invading viruses and bacteria. The lysosomes are also required in the process of autophagy and are involved in the process of apoptosis. The interior of the lysosome is maintained at an acidic pH through the action of transmembrane V-type ATPase complex.
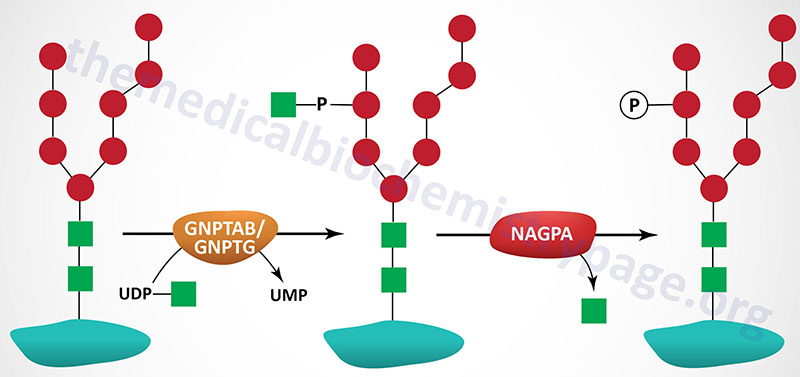
The majority of enzymes that are destined for the lysosomes (lysosomal enzymes) are directed there by a specific carbohydrate modification. During transit through the Golgi apparatus a residue of GlcNAc-1-phosphate (GlcNAc-1-P) is added to the carbon-6 hydroxyl group of one or more specific mannose residues that have been added to these enzymes.
The GlcNAc is activated by coupling to UDP and is transferred by UDP-GlcNAc:lysosomal enzyme GlcNAc-1-phosphotransferase complex (commonly called GlcNAc-phosphotransferase), yielding a phosphodiester intermediate: GlcNAc-1-P-6-Man-protein. The GlcNAc-phosphotransferase complex is a heterohexameric complex composed of two α-, two β-, and two γ-subunits.
The α- and β-subunits are both derived from the same gene following proteolysis of the encoded precursor protein. This gene is identified as N-acetylglucosamine-1-phosphate transferase alpha and beta subunits (GNPTAB). The GNPTAB gene is located on chromosome 12q23.2 and is composed of 23 exons that encode a 1256 amino acid precursor protein. Processing of the precursor protein results in the generation of the α- and β-subunits.
Mutations in the GNPTAB gene result in the lethal lysosomal storage disease identified as I-cell disease (mucolipidosis II; also known as mucolipidosis II alpha/beta) and the less severe related disorder, pseudo-Hurler polydystrophy (mucolipidosis IIIA; also known as mucolipidosis III alpha/beta). More than 70 mutations in the GNPTAB gene have been found in I-cell disease patients with the most common (40%) being a nonsense mutation: R1189X.
The γ-subunits are encoded by the N-acetylglucosamine-1-phosphate transferase gamma subunit (GNPTG) gene. The GNPTG gene is located on chromosome 16p13.3 and is composed of 11 exons that encode a 305 amino acid precursor protein. Mutations in the GNPTG gene are associated with a disease referred to as mucolipidosis IIIC (also called mucolipidosis III gamma).
A second reaction, catalyzed by N-acetylglucosamine-1-phosphodiester α-N-acetylglucosaminidase (encoded by the NAGPA gene), removes the GlcNAc leaving mannose residues phosphorylated in the 6 position, identified as Man-6-P-protein. The NAGPA gene is also located on chromosome 16p13.3 and is composed of 11 exons that encode a 515 amino acid precursor protein.
The protein encoded by the NAGPA gene is often referred to as uncovering enzyme (UCE). The preproprotein encoded by the NAGPA gene is processed into a functional homotetrameric complex composed of two disulfide bonded homodimers.
A specific mannose-6-phosphate receptor (MPR) is present in the membranes of the Golgi apparatus and binding of Man-6-P to this receptor targets proteins to the lysosomes. Two distinct MPRs have been identified and both are members of the P-type lectin family. Both are type I integral membrane glycoproteins that contain an N-terminal extracellular domain, a single transmembrane domain and a C-terminal cytoplasmic domain. One receptor is large with a molecular weight of approximately 300kDa, the other receptor is smaller with a molecular weight of approximately 46kDa. Structural similarities between these two receptors indicates they are derived from a single ancestral gene with the larger receptor arising through multiple gene duplications. The extracellular portion of the larger receptor contains 15 repeating elements, each of which is highly similar to the extracellular domain of the smaller receptor. Both receptors exist as dimers embedded in the membrane.
The large receptor binds two moles of Man-6-P and the smaller binds one mole of Man-6-P per subunit, thus 4 and 2 moles of Man-6-P per dimer, respectively. The bovine and murine versions of the smaller receptors require divalent cations for ligand binding and thus the receptor has been termed the cation-dependent Man-6-P receptor (CD-MPR). However, the human counterpart may not require cations for its activity. The CD-MPR protein is encoded by the M6PR (mannose-6-phosphate receptor, cation dependent) gene.
The larger receptor does not require divalent cations for ligand binding and is therefore, commonly referred to as the cation-independent Man-6-P receptor (CI-MPR). However, the CI-MPR has been shown to bind the non-glycosylated polypeptide hormone, insulin-like growth factor 2 (IGF-2) and as such the larger MPR is more frequently identified as the IGF-2 receptor (encoded by the IGF2R gene). The IGF-2 receptor is available at the cell surface and its role in binding IGF-2 is to target this hormone for degradation in the lysosomes. In addition to IGF-2, the IGF-2 receptor has been shown to bind a diverse array of Man-6-P-containing proteins as well as several non-glycosylated proteins. Although the IGF-2 receptor and CD-MPR exhibit distinct activities, both receptors function to target newly synthesized lysosomal enzymes to the lysosomes.
Nuclear Protein Targeting and Import
The nucleus is the organelle in which the genome is isolated from the rest of the cellular processes. The nucleus is enclosed by a membrane system composed of two lipid bilayers. The nuclear membrane is most often referred to as the nucleolemma and is composed of closely associated inner and outer lipid bilayers. The space between the inner and outer nuclear membranes is referred to as the perinuclear space. This space is in contact with the lumen of the endoplasmic reticulum (ER) via connections between the outer nuclear membrane and the ER membranes. The inner and outer nuclear membranes are also connected at thousands of locations via multiprotein complexes that generate pores in the nuclear membrane called nuclear pore complexes, NPC.
The nuclear pores are through which RNA and proteins are transported. The genome is transcribed into the various RNA forms within the interior of the nucleus in what is referred to as the nucleoplasm and then the RNAs that are destined for the cytosol are transported through the NPCs. Nuclear proteins such as histones, RNA polymerases, transcription factors, etc., are all translated in the cytosol and then transported into the nucleus via the actions of the proteins of the NPC. The import and export of nuclear macromolecules through the NPC involves proteins identified as importins and exportins and the monomeric G-protein Ran (with bound GTP; RanGTP).
The NPC is a large protein complex that consists of a central channel surrounded by three large ring-like structures, the cytoplasmic ring, the central spoke ring, and the nuclear ring. In addition to the proteins forming the NPC channel and the ring-like structures there are proteins associated with both the cytoplasmic and nuclear sides of the core structure. Each of the more than 30 proteins of the NPC are referred to as nucleoporins. Molecules that are less than 40 kDa in mass can move through the NPC channel passively. Larger molecules require active transport and this process involves a family of nuclear chaperones called importins and exportins which are members of a family of proteins called the karyopherin-β family. Humans express numerous importin (at least 16) and exportin (at least 7) proteins that are defined, in part, by the type of protein cargo they are involved in actively transporting through the NPC.
Importins bind to cargo proteins on the cytoplasmic side of the NPC and do so through the recognition of a specific nuclear localization sequence, NLS. The NLS is composed of a Lys (K) and Arg (R) rich peptides in nuclear proteins. The K/R-rich peptides can be composed of a monopartite or a bipartite motif. The prototypical monopartite NLS is K-K/R-X-K/R, where X represents any amino acid. The prototypical bipartite NSL is two clusters of K/R-rich peptides separated by a spacer of approximately 10 amino acids. A more complex NLS, that is recognized by the importin-β2 protein, is called the PY-NLS. At least 100 human proteins have been identified that contain this PY-NLS. Following transport through the NPC the cargo protein is released from the importin via interaction with RanGTP. Following cargo release the importin-RanGTP complex is transported back to the cytosol. In the cytosol the GTP bound to Ran is hydrolyzed to GDP by the GTPase activating protein (GAP) RanGAP. The hydrolysis of GTP causes the release of the importin so that it can be made available for nuclear import.
Exportins bind their cargo proteins inside the nucleus, along with RanGTP, for transport out of the nucleus. The recognition of cargo proteins by exportins involves a nuclear export signal (NES) in the target protein. The NES is a short peptide of hydrophobic amino acids with a common consensus of LXXXLXXLXL, where X represents any amino acid. Like the import of cytoplasmic proteins into the nucleus, the export process is coupled to GTP hydrolysis in the RanGTP complex through the action of RanGAP. Following GTP hydrolysis the exported protein is released to the cytosol. The exportin protein is then transported back into the nucleoplasm. Since RNA molecules do not contain amino acid sequences they do not, directly, possess nuclear export signals. Therefore, cytoplasmic RNAs (e.g. mRNA, rRNA, and tRNA) form ribonucleoprotein complexes in the nucleoplasm and the protein components of these complexes are recognized by exportins. For example the exportin identified as exportin-t is responsible for the nuclear export of tRNAs.
In addition to directionally specific importins and exportins, all of which are members of the karyopherin-β family of proteins, there are several bidirectional karyopherin-β transporters. Human importin 13, exportin 4, and exportin 5 all have bidirectional nuclear transport capabilities.
Mitochondrial Protein Targeting and Import
The mitochondria are critically vital organelles, second only to the nucleus, whose functions are required for cell viability. The major function of the mitochondria is to generate the high energy molecule ATP, through the utilization of the energy contained in the reduced electron carriers, NADH and FADH2. This vital process is explained in detail in the Oxidative Phosphorylation and Related Mitochondrial Functions page. Through the process of oxidative phosphorylation the mitochondria interconnects the metabolic processes of carbohydrate, lipid, and amino acid catabolism.
In addition, the mitochondria serve as important conduits in the processes of urea synthesis, heme synthesis, and steroidogenesis. Mitochondria are able to carry out these highly diverse, but interrelated, biochemical processes via the catalytic activity of proteins encoded by both the mitochondrial (mtDNA) and the nuclear genomes. Proteomic analysis has determined that the mitochondrial proteome consists of nearly 1,000 different proteins, the vast majority of which are derived from nuclear genes. The mitochondrial genome consists of a total of 16,569 bp that encoded 13 proteins, 22 tRNAs, and 2 rRNAs.
The mitochondrial proteins that are encoded by the nuclear genome are translated in the cytoplasm and then these proteins are imported into the mitochondria by specialized recognition and transport proteins and complexes. Mitochondrial protein import and localization is controlled by specific amino acid sequences in the precursor proteins. These sequences not only target the proteins to the mitochondria but also ensure that individual proteins are properly distributed into the four mitochondrial compartments: outer mitochondrial membrane (OMM), intermembrane space (IMS), inner mitochondrial membrane (IMM), and matrix.
Like all but the mitochondrial genome encoded proteins, mitochondrial proteins are synthesized in the cytosol. Proteins destined for the mitochondria posses a presequence akin to the leader peptide of ER targeted proteins. Transfer of these proteins to their appropriate location in the mitochondria requires specific chaperones and transport complexes. The import of proteins into the mitochondria occurs via a co-translational process. Unlike the co-translational synthesis of proteins in the endoplasmic reticulum (ER) where the leader sequence is captured by the signal recognition particle (SRP) after emergence from the ribosome, mitochondria targeted proteins do not interact with the outer mitochondrial membrane until 200–400 additional amino acids of the nascent protein have been synthesized.
At the level of the outer mitochondrial membrane the transport process involves a complex called the translocase of outer mitochondrial membrane, TOM. The TOM is composed of several proteins that are receptors for mitochondrially targeted proteins and that compose the channel of the TOM itself. The TOM complexes are localized to specialized domains of the outer mitochondrial membrane that are closely associated with openings in the inner mitochondrial membrane that are referred to as cristae junctions. The channel of the TOM complex is formed from the Tom40, Tom22, and Tom7 proteins while the preprotein receptors are Tom20 and Tom70. Accessory subunits of the TOM that are required for assembly and stabilization of the TOM are Tom5 and Tom6. The human Tom proteins are encoded by genes with the designation TOMM. As an example the Tom40 protein is encoded by the TOMM40 gene that is located on chromosome 19q13.32 and is composed of 10 exons that generate three alternatively spliced mRNAs all of which encode the same 361 amino acid protein.
Mitochondrial precursor proteins that contain an appropriate presequence are initially recognized by Tom20 and then they interact with Tom22 prior to import. Other mitochondrial proteins that are hydrophobic precursors contain an integral targeting sequences that is recognized by Tom70. Whether recognized by Tom20 or Tom70, both classes of mitochondrial protein are imported through the Tom40 channel. Many proteins that are embedded in the outer mitochondrial membrane such as Tom20 and Tom70 are referred to as signal-anchored proteins. These proteins are embedded in the outer membrane via sequences in the N-terminus and have their C-terminal domains extended into the cytosol. Other classes of outer membrane anchored proteins have a C-terminal transmembrane domain and are referred to as tail-anchored proteins. The proteins of the Bcl-2 family of apoptosis regulating proteins are members of the mitochondrial tail-anchored family of proteins.
Proteins of the intermembrane space (IMS) are generally small soluble proteins that are imported via the mitochondrial IMS import and assembly (MIA) pathway. The MIA pathway is unique in that it couples the process of protein import to the folding and oxidation of the imported protein leading to the formation of internal disulfide bonds in the process. The introduction of the disulfide bonds in IMS proteins is catalyzed by a protein identified as Mia40 which acts as the chaperone and the sulfhydryl oxidase identified as growth factor, augmenter of liver regeneration (encoded by the GFER gene; is the human homolog of the yeast Erv1 enzyme). The human Mia40 protein is encoded by the coiled-coil-helix-coiled-coil-helix domain containing 4 (CHCHD4) gene.
Transport of mitochondrial proteins to the inner membrane and the mitochondrial matrix involves complexes identified as translocase of inner mitochondrial membrane, TIM. Like the TOM the TIM complexes are composed of central channel forming proteins. The TIM that is responsible for matrix targeted proteins contains the Tim23 (encoded by the TIMM23 gene) protein which is the primary channel protein, along with Tim17. Proteins that are transported via the Tim23 mediated TIM contain an amphipathic helix at their N-termini that, that like TOM transport, is referred to as a presequence. The TIM23 complex contains several additional proteins such as Tim50 which regulates channel opening and Tim 21 which regulates docking functions. Matrix proteins that contain the appropriate presequence have it proteolytically removed during the transport process.
Mitochondrial inner membrane proteins also contain a TIM recognized presequence. However, multipass integral inner membrane proteins are inserted into the membrane via the TIM complex identified as TIM22. The central channel of the TIM22 complex is generated from the Tim22 protein encoded by the TIMM22 gene. The Tim22 protein is referred to as the carrier translocase. Like the TIM23 complex, the TIM22 complex is composed of several additional subunits such as Tim12, Tim18, and Tim54. The functionally most significant proteins inserted into the inner mitochondrial membrane via the TIM22 complex are the metabolite transporters such as the dicarboxylate transporter encoded by the SLC25A10 gene.
Mitochondrial matrix proteins have their targeting sequence removed upon import into the matrix via the action of a heterodimeric complex termed the mitochondrial processing peptidase, MPP. The MPP is composed of subunits encoded by the PMPCA (α-subunit) and PMPCB (β-subunit) genes. The removal of the targeting sequence is required for the function of matrix-associated proteins.
The PMPCA gene is located on chromosome 9q34.3 and is composed of 13 exons that generate three alternatively spliced mRNAs, each of which encode a distinct protein isoform.
The PMPCB gene is located on chromosome 7q22.1 and is composed of 14 exons that encode a 489 amino acid precursor protein.
Peroxisomal Protein Targeting and Import
The peroxisomes are a single membrane organelle, similar to lysosomes, present in virtually all eukaryotic cells. The peroxisome is a specialized enzyme “factory” that contains in excess of 50 different enzymes involved in a variety of metabolic processes of lipid metabolism that includes β-oxidation of very long chain fatty acids, α-oxidation of fatty acids and synthesis of cholesterol, bile acids, and ether-lipids. Proteins that are involved in, and necessary for correct peroxisome biogenesis, are called peroxins (PEX). At least 16 PEX genes have been identified in humans with three (PEX11A, PEX11B, and PEX11G) comprising subunits of the PEX11 complex.
The biogenesis of peroxisomes is directly tied to the endoplasmic reticulum (ER) with the lipids of the membranes of the peroxisomes being derived from the ER and most peroxisomal membrane proteins (PMP) being synthesized in, and trafficking through, the ER. Two PEX proteins, PEX3 and PEX19 are critical for the correct targeting of PMPs to the peroxisomal membrane. PMP that are dependent on PEX19 (most all PMP) are referred to as type I PMP while type II PMP utilize PEX3 (as well as PEX22) for correct targeting. PEX3 and PEX19 also are involved in peroxisomal targeting of peroxisomal proteins that are synthesized in the cytosol. Defects in peroxisomal biogenesis genes result in a family of disorders referred to as peroxisomal biogenesis disorders, PBD. The most severe PBD is Zellweger syndrome which represents a cluster of disorders that results from mutations in at least eight different PEX genes.
Enzymes that are targeted to the peroxisomes contain either of two amino acid consensus elements called peroxisome targeting sequences (PTS). The PTS1 is a C-terminal consensus sequence of –(S/A/C)(K/R/H)(L/M) referred to as the SKL motif. This sequence element is recognized by a cytosolic PTS1 receptor encoded by the PEX5 gene. There are two primary isoforms of PEX5 encoded proteins in humans identified as Pex5pS and Pex5pL (for short and long forms, respectively). The Pex5pL protein has an internal 37 amino acid insertion, hence the “long” designation. The PTS2 is an N-terminal consensus sequence of –(R/K)(L/V/I/Q)XX(L/V/I/H/Q)(L/S/G/A/K)X(H/Q)(L/A/F)–, where X represents any amino acid. The PTS2 receptor is encoded by the PEX7 gene and the encoded protein is referred to as Pex7p.
Pex5pS, Pex5L, and Pex7p interact with newly synthesized target proteins in the cytosol and direct them to the peroxisome. On the membrane of the peroxisome is a component of the protein import machinery encoded by the PEX14 gene called Pex14p. Following interaction of Pex5pS or Pex5pL, to which a PTS1-containing protein is bound, with Pex14p, the PTS1 containing protein is transferred into the peroxisome. The activity of Pex7p in peroxisome protein import actually requires Pex5pL as well. PTS2 containing proteins interact with Pex7p and then, in conjunction with Pex5pL, the complex interacts with Pex14p and the PTS2 containing protein is transferred into the peroxisome. Very few proteins contain a PTS2 sequence but one enzyme of note is phytanoyl-CoA hydroxylase (PHYH) which is defective in classic Refsum disease.
Pathways for Protein Exocytosis and Endocytosis
The release of cellular substances, particularly proteins, hormones, and neurotransmitters, involves controlled and regulated processes collectively referred to as exocytosis. The reverse process, exemplified by ligand-bound receptor internalization, also involves controlled and regulated processes and these processes are referred to as endocytosis. The membrane vesicles of exocytosis originate from the trans-Golgi network or from the recycling of endosomes. Their migration to the plasma membrane involves the cytoskeletal machinery of the cell. Exocytosis serves numerous biologically important functions in the cell. Exocytosis is the cellular means by which lipids and proteins are delivered to the plasma membrane facilitating cellular growth. Exocytosis allows a cell to signal to the external environment by the release of vesicle contents. The process of exocytosis allows proteins that are embedded in the vesicle membrane to be delivered to the plasma membrane such as is the case for transport proteins, protein channels, and signaling receptors.
The processes of exocytosis and endocytosis are connected, in most instances, since the exocytosed vesicles are retrieved via the processes of endocytosis. The processes of exocytosis and endocytosis can be collectively categorized into three main modes. In one mode (classic) the exocytotic vesicles fuse into the plasma membrane followed by an endocytotic process that involves membrane invagination and vesicle reformation. In the second mode (kiss-and-run) a fusion pore opens to release vesicle contents and then recloses. In the third mode (bulk) giant vesicles, that have formed via vesicle-vesicle fusion, are exocytosed and then the giant vesicles are retrieved by bulk endocytosis. Exocytosis is dependent upon endocytosis in order to prevent vesicle membrane exhaustion. Indeed, endocytosis of neurotransmitter vesicle membranes is required to maintain the size of nerve terminals.
Although the process of exocytosis is coupled to endocytosis as a means to maintain and replenish membranes, there are several highly specific endocytotic processes that are not directly coupled to exocytosis. The controlled uptake of small molecules and fluids by cells within small vesicles is a process of endocytosis termed pinocytosis. One particular type of pinocytotic uptake is receptor-mediated endocytosis where ligand binding to a receptor results in the complex being internalized in the target cell. This process involves receptors that are embedded in membrane domains formed by the protein clathrin, referred to as clathrin-coated pits. The process of invading pathogen uptake by cells of the immune system is also a highly specialized form of endocytosis term phagocytosis.
The overall processes of exocytosis and endocytosis are, in all modes, controlled via calcium influx through voltage dependent calcium channels and via intracellular calcium sensor proteins such as calmodulin (in endocytosis) and the synaptotagmins, SYT (in exocytosis). The role of calcium fluxes and calcium sensors in the regulation of exocytosis and endocytosis can be clearly demonstrated with the use of drugs that inhibit the activity of calmodulin or those that inhibit calcium influx. In either case endocytosis is inhibited leading to a rapid loss in membrane replenishment at synaptic terminals in nerve cells.
One of the major downstream targets of calmodulin in the process of endocytosis is the phosphatase, calcineurin (CaN; also known as protein phosphatase 3). Calcineurin functions as a heterodimer composed of a catalytic subunit (calcineurin A) and a Ca2+-binding regulatory subunit (calcineurin B). Humans express three catalytic subunit genes (PPP3CA, PPP3CB, PPP3CC) and two regulatory subunit genes (PPP3R1 and PPP3R2). Humans express 17 synaptotagmin genes identified as SYT1–SYT17.
Equally important to the processes of exocytosis and endocytosis are members of the large RAB family of monomeric G-proteins. There are 65 genes in the human genome that encode RAB family proteins and each protein is known to serve distinct roles in membrane identity, exocytotic and endocytotic vesicle budding, and membrane fusion through the recruitment of various effector proteins. For example the RAB5 protein is associated with clathrin-coated pits and as such is involved in the control of receptor-mediated endocytosis. Another important example is the intracellular vesicles in skeletal muscle and adipose tissue cells that contain the GLUT4 glucose transporter. These vesicles are stimulated to fuse with the plasma membrane in response to insulin signaling and the migration and fusion of these vesicles involves the RAB8, RAB10, and RAB14 proteins.
A large family of proteins, the SNARE family, is required for the processes of membrane fusion that are necessary intermediate steps in the exocytotic and endocytotic pathways. The term SNARE is derived from SNAP REceptor, where SNAP is Soluble NSF (N-ethylmaleimide-sensitive factor) Attachment Protein. The NSF protein is a member of the AAA subfamily of ATPases. The SNARE superfamily contains 15 genes in humans and two additional gene subfamilies identified as the vesicle-associated membrane proteins (VAMP) subfamily and the syntaxins (STX) subfamily. Humans express 8 genes of the VAMP family with the best known member being more commonly called synaptobrevin. Humans express two forms of synaptobrevin identified as isoform 1 (encoded by the VAMP1 gene) and isoform 2 (encoded by the VAMP2 gene). There are 16 genes of the STX family with syntaxin 1A being the most well characterized via its interactions with the calcium sensors of the synaptotagmin family.
Many aspects of membrane fusion involve SNARE proteins, not just the processes of exocytosis and endocytosis. For example, intracellular vesicle fusion with target membrane compartments results in the formation of peroxisomes and lysosomes. In addition to specific gene families, the SNARE proteins can be defined as being a v-SNARE (vesicle) or a t-SNARE (target). The v-SNARE proteins are incorporated into the membranes of the transport vesicles, while t-SNARE proteins are found in the membranes of the target membrane. For example, neurotransmitter vesicles contain v-SNARE proteins while the nerve terminal membrane contains t-SNARE proteins.
The overall process of exocytosis requires multiple distinct steps each of which can be regulated by many different factors. The transport of exocytotic cargo (be it a secreted hormone or a membrane channel protein) begins when the macromolecules are packaged into vesicles that bud from the trans-Golgi network. These exocytotic vesicles are carried from point of origin to final destination by motor proteins of the myosin and kinesin families. The motor proteins utilize actin filaments and microtubule tracks to guide their movement and the hydrolysis of ATP serve as the source of the energy required for the movement. The specificity of the overall migration is controlled by the RAB proteins.
Once a vesicle reaches the plasma membrane exocytosis involves a the formation of a multisubunit tethering complex called the exocyst. The exocyst facilitates SNARE family member protein-mediated membrane fusion. The fusion process involves interactions between the particular vesicle SNARE (v-SNARE) and the particular target SNARE (t-SNARE) which results in SNARE proteins “zippering” together. Following fusion and completion of the exocytosis process the SNARE protein interactions need to be disassembled in order to allow membrane recycling carried out via endocytosis. The two proteins that carry out the disassembly process are NSF and α-SNAP (soluble NSF attachment protein).