
Last Updated: October 28, 2025
Introduction to Protein C
Protein C (PC) is a trypsin-like serine protease that serves as a major regulator of the blood coagulation process. Protein S (PS) serves as a co-factor for the functions of activated protein C (abbreviated aPC, and also APC).
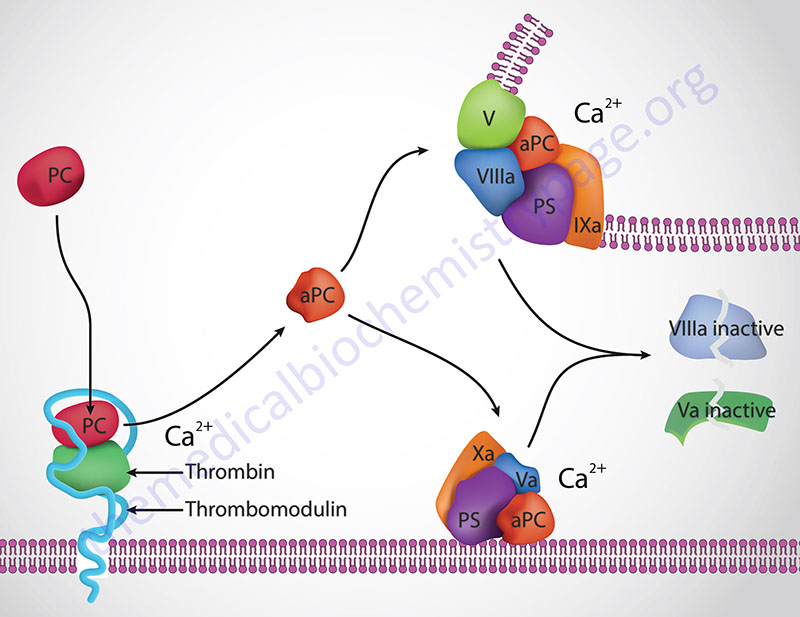
Thrombin cleavage of PC removes the activation peptide generating activate protein C, aPC. When activated through cleavage by thrombin, aPC cleaves both factor activated factor V (Va) and activated factor VIII (VIIIa) into inactive enzymes. This results in the termination of the role of VIIIa as the scaffold for the formation of the tenase complex and Va as a co-factor in the conversion of prothrombin to thrombin in the prothrombinase complex. The net effect, at the level of coagulation, of the activation of PC is termination of further increases in thrombin production and a halt to further fibrin clot formation.
The activation of PC by thrombin occurs on the surface of the endothelium when thrombin binds to thrombomodulin and “captures” circulating PC. Following activation, aPC interacts with PS and cleaves VIIIa and Va.
Molecular Biology of Protein C
Protein C is encoded by the PROC gene. The PROC gene is located on chromosome 2q14.3 and is composed of 8 exons that generate 12 alternatively spliced mRNAs that collectively encode 10 distinct protein isoforms.
Protein C undergoes a series of post-translational modifications that include several N-linked glycosylation sites and γ-carboxylation of nine glutamine residues (gla residues) in the amino terminus. These gla residues in the amino terminus of PC constitute the “gla domain” of the protein. In addition to the gla domain, PC contains two epidermal growth factor-like (EGF) domains), the serine protease domain, and an activation peptide domain.
Protein C in Blood Coagulation
The importance of aPC in controlling Va activity can be seen in the hypercoagulopathy (thrombophilia) referred to as factor V Leiden thrombophilia. This thrombophilia is caused by a mutation in the factor V gene resulting in a protein that is not effectively degraded by aPC. Factor V Leiden is the most common inherited thrombophilia in Caucasian populations of European descent. Overall, 5% of the world population harbors the factor V Leiden mutation. The symptoms of factor V Leiden are deep vein thrombosis (DVT) and pulmonary embolism, both of which can be fatal. In fact it is estimated that in as many as 30% of patients who experience DVT and/or pulmonary embolisms are carriers of the factor V Leiden mutation. Loss of protein C results in massive and usually lethal thrombotic complications in infants with homozygous PC deficiency. In individuals who are heterozygous for PC deficiencies there is an increased risk for venous thrombosis.
Although the role of aPC in the termination of coagulation is extremely important it also serves many additional functions that alter the inflammatory processes occurring in the vasculature. Activated PC binds to the endothelial protein C receptor (EPCR: encoded by the PROCR gene) and leads to the activation of protease-activated receptor-1 (PAR-1) which elicits cytoprotective and anti-inflammatory responses within endothelial cells. The EPCR functions as a co-receptor for aPC-mediated cleavage and activation of PAR-1. The EPCR is also found on monocytes, neutrophils, fibroblasts, and keratinocytes. The cytoprotective effects elicited via aPC activation of PAR-1 include protection of the endothelial cell barrier and induction of anti-apoptotic signaling pathways as well as expression of eNOS. Additional endothelial responses to aPC activation of PAR-1 occur via inhibition of the expression and actions of the potent pro-inflammatory transcription factor NFκB. The suppression of NFκB action results in downregulation of the synthesis of endothelial pro-inflammatory cytokines such as IL-6 and IL-8, the chemokine MCP-1 (monocyte chemoattractant protein-1), and the cell adhesion molecule ICAM-1 (intercellular adhesion molecule-1).
The binding of aPC to the EPCR on monocytes leads to inhibition of the synthesis and release of pro-inflammatory cytokines (e.g. IL-1, IL-6 and TNFα) from these cells which results from inhibition of the actions of NFκB. Additional monocyte effects of aPC include decreased tissue factor (factor III) expression and inhibition of the release of the chemokines MIP-1α (macrophage inflammatory protein-1α) and MCP-1.
The anti-inflammatory and cytoprotective effects of aPC are so potent that the drug Xigris® (dotrecogin alpha) was an FDA approved recombinant aPC used in the treatment of sepsis. The drug was voluntarily removed from the market by Eli Lilly and Co. Sepsis is initiated by infection whereby microbes and/or microbial toxins released into the blood trigger a systemic and uncontrolled activation of both the coagulation cascade and inflammatory pathways. Sepsis is the leading cause of death in intensive care patients who are not coronary patients. Severe sepsis afflicts more than 700,000 people in the United States each year with a mortality rate of 30% to 50%.
In addition to its demonstrated efficacy in the treatment of sepsis, aPC is currently being investigated for the treatment of numerous conditions. These include the treatment of stoke since in mouse models the anti-inflammatory and anticoagulant actions of aPC have the additional benefit of exerting neuroprotective effects. Ischemia-reperfusion injury (I/R) results when tissues are temporarily deprived of oxygenated blood (ischemia) and the return of blood flow (reperfusion) results in additional tissue damage. The secondary damage of I/R is a consequence of the intense inflammatory reactions that are initiated in response to anoxia. The damage from reperfusion can occur in tissues or organs that were not affected by the initial ischemic episode. In mouse models it has been shown that infusion of aPC attenuates the oxidative tissue damage of ischemia and this effect may be due to direct aPC-mediated inhibition of neutrophil activation. Additional conditions that may be treated with aPC infusion include acute lung injury, asthma, and acute pancreatitis. Studies have also demonstrated that aPC may promote wound healing and angiogenesis.
Clinical Features of Protein C Deficiency
Because protein C is a vitamin K-dependent hemostasis factor, biosynthesis of the active protein is inhibited by oral anticoagulants that function via inhibition of vitamin K-dependent carboxylation reactions (e.g. coumadin). Inheritance of deficiencies in protein C occur in an autosomal dominant manner. There are both heterozygous and homozygous deficiencies in protein C. Defects in protein C are the most commonly occurring causes of the hereditary predisposition to thrombosis.
Homozygous deficient infants develop purpura fulminans (hemorrhagic condition usually associated with infection or sepsis) shortly after birth. These complications can be treated by protein C supplementation.
Heterozygous individuals are quite frequent in the general population with estimates of 0.3%. Many remain asymptomatic but up to 5% of familial thrombophilias are the result of protein C (or protein S) deficiency.
It should be noted that because APC requires interaction with protein S for full activity, deficiencies in protein S are phenotypically similar to protein C deficiencies. In addition, some factor V mutants result in the inability of APC to bind to activated factor V (Va) resulting in similar phenotypes in factor V mutants. The clinical manifestations of heterozygous protein C, protein S and factor V mutants overlap and include venous thrombosis and pulmonary embolism.