
Last Updated: October 30, 2025
Introduction to Erythrocyte PK Deficiency
Deficiency of erythrocyte pyruvate kinase is the most common enzyme deficiency resulting in inherited nonspherocytic hemolytic anemia. In contrast, spherocytic anemia is characterized as an auto-hemolytic disorder that results from molecular defects in erythrocyte (red blood cell, RBC) membrane proteins leading to spherical erythrocytes instead of the normal bi-concave disc shaped cells.
Overall, erythrocyte pyruvate kinase deficiency is the second leading cause of inherited hemolytic anemia. Mutations in the glucose-6-phosphate dehydrogenase gene represent the leading causes of inherited forms of hemolytic anemia.
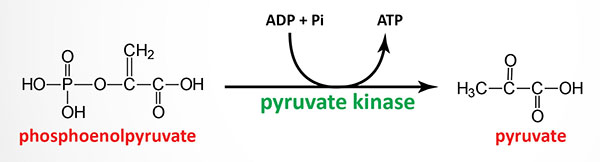
Erythrocyte pyruvate kinase deficiency is inherited as an autosomal recessive disorder and mutations in the erythrocyte pyruvate kinase gene occur with a frequency of 1 in 10,000. The disorder is characterized by lifelong chronic hemolysis of variable severity. Red blood cells from heterozygous individuals possess only 40%–60% of the pyruvate kinase activity of normal individuals yet these individuals are almost always clinically normal.
Molecular Biology of Erythrocyte PK Deficiency
The liver (L-PK or PKL) and red blood cell forms (R-PK or PKR) of pyruvate kinase are encoded by the same gene (PKLR) with differences resulting from differential transcriptional initiation. The PKLR gene is located on chromosome 1q22 spanning 9.5 kb and is composed of 13 exons that generate two mRNAs as a result of alternative promoter utilization.
The R-PK promoter is active exclusively in erythrocytes due to a strong erythroid cell-specific enhancer element. The L-PK promoter is active in hepatocytes and pancreatic β-cells. Of the 13 exons in the PKLR gene, exons 3-12 encode identical portions of both the liver and erythrocyte mRNAs. The erythrocyte PK mRNA also includes exon 1, whereas exon 2 is included in the liver PK mRNA. The R-PK protein is 574 amino acids in length and the L-PK protein is 543 amino acids.
The pyruvate kinase isoform, originally thought to be muscle specific, is encoded by a separate gene (symbol PKM) located on chromosome 15q23 and is composed of 17 exons spanning 32 kb. The PKM gene generates nine alternatively spliced mRNAs that encode multiple forms of two PKM family enzymes identified as PKM1 and PKM2.
Numerous mutations (over 260 to date) have been identified in the PKLR gene resulting in erythrocyte PK deficiency with the vast majority of those mutations being single nucleotide changes. In persons of northern European descent over 50% of individuals with PK deficiency harbor a nucleotide change at position 1529 (encoding amino acid 510) that changes a G to an A residue. This mutation results in an Arg to Gln change in the protein and is identified as the R510Q mutation and also as the 1529A mutation. A common mutation found in individuals of southern European descent is a substitution of Trp for Arg at amino acid position 486, the R486W mutation.
Although many of the identified mutations in the PKLR gene, that are associated with erythrocyte PK deficiency, also inflict loss of the liver enzyme from the PKLR gene, the necessary function of hepatic PK is accommodated through expression of the PKM gene.
Clinical Features of PKLR Deficiency
The clinical severity of the anemia resulting from PKLR deficiencies varies from mild and fully compensated hemolysis to severe cases that require transfusions for survival. In the most severe cases an affected fetus will die in utero. Most PKLR deficient patients are diagnosed in infancy or early childhood. Severely affected patients may require multiple transfusions or splenectomy. The requirement for transfusion in many infants will diminish during childhood or following splenectomy. There are many patients with mild PKLR deficiency who do not manifest symptoms until adulthood. In these individuals, the symptoms, that include mild to moderate splenomegaly, mild chronic hemolysis, and jaundice, often manifest incidental to an acute viral infection or during an evaluation in pregnancy.
Biochemically, the loss of fully functional pyruvate kinase in erythrocytes results in impaired production of ATP given that the activity of pyruvate kinase in glycolysis is the only means of net ATP production in erythrocytes. The loss of ATP leads to cessation of all active processes in the cell. As a result of the loss of ATP, plasma membrane Na+,K+-ATPases are some of the first activities to cease functioning. Na+,K+-ATPases normally transport three Na+ ions out of the cell while transporting two K+ ions into the cell. The erythrocyte plasma membrane is more permeable to K+ than it is to Na+ which results in loss of intracellular K+. The erythrocyte becomes hypotonic leading to loss of water and shrinkage of the cell contributing to hemolysis.
In addition to loss of ATP production impaired pyruvate kinase activity leads to the accumulation of upstream glycolytic intermediates upstream. In particular the measurement of plasma 2,3-biphosphoglycerate (2,3-BPG) can show levels up to threefold above normal. Accumulation of 2,3-BPG further impairs glycolytic flux as a result of the inhibition of the hexokinase reaction. Increased levels of 2,3-BPG will reduce the affinity of hemoglobin for O2 resulting in increased delivery of O2 per unit hemoglobin molecule. This latter phenomenon tends to result in a better patient tolerance of anemia than would otherwise be expected.
Clinical evaluation of blood from patients with PKLR deficiency will show normocytic anemia (sometimes macrocytic) which is reflective of the reticulocytosis that is invariably present. Blood smears from erythrocyte PK deficient patients often show the presence of abnormal erythrocytes. These abnormal cells include anisocytes (erythrocytes of unequal size), poikilocytes (erythrocytes of variable shapes including tear-drop, ovate, or sickle), and echinocytes (typically referred to as burr cells) which are characterized by the presence of small projections (spiculations) in the plasma membrane that are of a uniform shape and distribution. Echinocytes are distinct from acanthocytes in that acanthocytes have larger irregularly dispersed spiculations in the plasma membrane.
When peripheral blood is stained by new methylene blue or brilliant cresyl blue, reticulocytes will be seen as cells containing a fine basophilic network. Reticulocytes are immature red blood cells that have lost their nuclei just prior to entering the circulation. In normal individuals reticulocytes comprise 0.5% to 1.7% of the total erythrocytes. An increase in the percentage of reticulocytes (reticulocytosis) is an important sign in any patient suffering anemia. In PKLR deficiency the level of reticulocytosis results in a reticulocyte level of 4% to 15%. In association with the reticulocytosis there is a reduction in hematocrit (packed cell volume) to around 17% to 37% where it ranges from 41% to 50% in normal individuals. Hemoglobin measurement also shows a decrease to 6–12g/dL where normal ranges from 12–16.5g/dL. Examination of bone marrow from PKLR deficient patients will show normoblastic erythroid hyperplasia.