

Last Updated: July 6, 2026
Structure and Function of Hormones
The integration of body functions in humans and other higher organisms is carried out by the nervous system, the immune system, and the endocrine system. The endocrine system is composed of a number of tissues that secrete their products, endocrine hormones (primarily peptide hormones), into the circulatory system; from there they are disseminated throughout the body, regulating the function of distant tissues and maintaining homeostasis.
In a separate but related system, exocrine tissues secrete their products into ducts and then to the outside of the body or to the intestinal tract. Classically, endocrine hormones are considered to be derived from amino acids, peptides, or sterols and to act at sites distant from their tissue of origin. However, the latter definition has begun to blur as it is found that some secreted substances act at a distance (classical endocrines), close to the cells that secrete them (paracrine), or directly on the cell that secreted them (autocrine). Insulin-like growth factor-I (IGF-I), which behaves as an endocrine, paracrine, and autocrine, provides a prime example of this difficulty.
Hormones are normally present in the plasma and interstitial tissue at concentrations in the range of 10-7M to 10-10M. Because of these very low physiological concentrations, sensitive protein receptors have evolved in target tissues to sense the presence of very weak signals. In addition, systemic feedback mechanisms have evolved to regulate the production of endocrine hormones.
Once a hormone is secreted by an endocrine tissue, it generally binds to a specific plasma protein carrier, with the complex being disseminated to distant tissues. Plasma carrier proteins exist for all classes of endocrine hormones. Carrier proteins for peptide hormones prevent hormone destruction by plasma proteases. Carriers for steroid and thyroid hormones allow these very hydrophobic substances to be present in the plasma at concentrations several hundred-fold greater than their solubility in water would permit. Carriers for small, hydrophilic amino acid-derived hormones prevent their filtration through the renal glomerulus, greatly prolonging their circulating half-life.
Tissues capable of responding to endocrines have two properties in common: they posses a receptor having very high affinity for hormone, and the receptor is coupled to a process that regulates metabolism of the target cells. Receptors for most amino acid-derived hormones and all peptide hormones are located on the plasma membrane. Activation of these receptors by hormones (the first messenger) leads to the intracellular production of a second messenger, such as cAMP, which is responsible for initiating the intracellular biological response.
Steroid and thyroid hormones are hydrophobic and diffuse from their binding proteins in the plasma, across the plasma membrane to intracellularly localized receptors termed nuclear receptors. The resultant complex of steroid and receptor bind to response elements of nuclear DNA, regulating the production of mRNA for specific proteins.
Receptors for Peptide Hormones
With the exception of the thyroid hormone receptor, the receptors for amino acid-derived and peptide hormones are located in the plasma membrane. Receptor structure is varied: some receptors consist of a single polypeptide chain with a domain on either side of the membrane, connected by a membrane-spanning domain. Some receptors are comprised of a single polypeptide chain that is passed back and forth in serpentine fashion across the membrane, giving multiple intracellular, transmembrane, and extracellular domains. Other receptors are composed of multiple polypeptides. For example, the insulin receptor is a disulfide-linked tetramer with the β-subunits spanning the membrane and the α-subunits located on the exterior surface.
Subsequent to hormone binding, a signal is transduced to the interior of the cell, where second messengers and phosphorylated proteins generate appropriate metabolic responses. The main second messengers are cAMP, Ca2+, inositol-1,4,5-triphosphate (IP3; also designated Ins-1,4,5-P3), and diacylglycerol (DAG). The generation of cAMP occurs via activation of Gs-type G-protein coupled receptors (GPCR) whose associated G-proteins activated adenylate cyclase. For more information on GPCR and G-proteins visit the Signal Transduction Pathways: G-Proteins and GPCR page. Adenylate cyclase then converts ATP to cAMP and the subsequent increases in cAMP lead to activation of cAMP-dependent protein kinase (PKA) as shown in the Figure below.
GPCR also couple to Gq-type G-proteins that result in the activation of phospholipase C-β (PLCβ). Activated PLCβ hydrolyzes membrane phospholipids (as described below) resulting in increased levels of IP3 and DAG. Downstream signaling proteins are phosphorylated on serine and threonine by PKA and DAG-activated protein kinase C (PKC) leading to alterations in their activities. Additionally, a series of membrane-associated and intracellular tyrosine kinases phosphorylate specific tyrosine residues on target enzymes and other regulatory proteins.
The hormone-binding signal of most, but not all, plasma membrane receptors is transduced to the interior of cells by the binding of receptor-ligand complexes to a series of membrane-localized GDP/GTP binding proteins known as G-proteins. The classic interactions between receptors, G-protein transducer, and membrane-localized adenylate cyclase are illustrated below using the pancreatic hormone glucagon as an example receptor-mediated activation of a Gs-type G-protein. When Gs-type G-proteins bind to receptors, GTP exchanges with GDP bound to the α subunit of the G-protein. The Gαs-GTP complex binds adenylate cyclase, activating the enzyme. The activation of adenylate cyclase leads to cAMP production in the cytosol and to the activation of PKA, followed by regulatory phosphorylation of numerous enzymes. For more information on G-proteins and GPCR go to the Signal Transduction Pathways: G-Proteins and GPCR page.
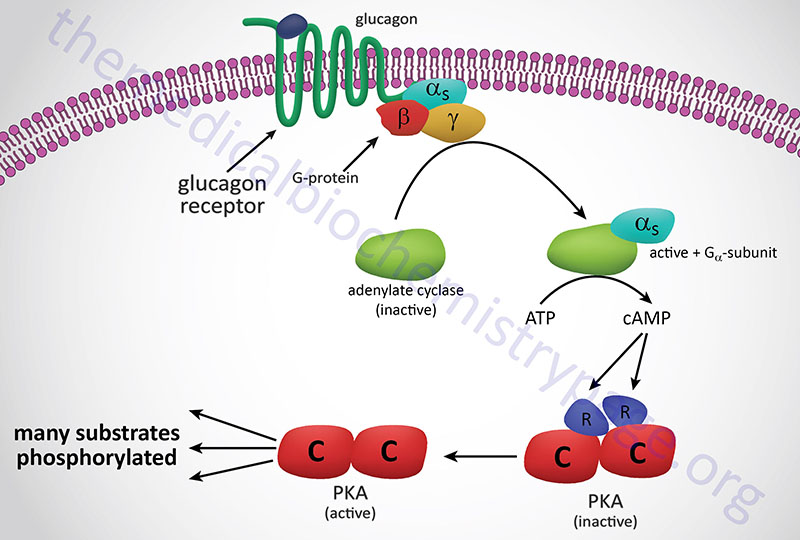
A second class of peptide hormones induces the transduction of two second messengers, DAG and IP3 (diagrammed below for α1-adrenergic stimulation by epinephrine). Hormone binding to receptor is followed by interaction with a stimulatory G-protein which is followed in turn by G-protein activation of membrane-localized PLCβ. G-proteins that are coupled to receptor-activation of PLCβ are termed Gq-proteins. PLCβ hydrolyzes membrane-localized phosphatidylinositol-4,5-bisphosphate (PIP2; PtdIns-4,5-P2) to produce two messengers: IP3, which is soluble in the cytosol, and DAG, which remains in the membrane phase. Cytosolic IP3 binds to specific receptors in the endoplasmic reticulum (ER) membranes [sarcoplasmic reticulum (SR) in muscle cells], which are ligand-gated Ca2+ channels allowing stored Ca2+ to be released to the cytosol.
Humans express three distinct IP3 receptors encoded by the ITPR1, ITPR2, and ITPR3 genes. The ITPR1 gene is located on chromosome 3p26.1 and is composed of 63 exons that generate three alternatively spliced mRNAs encoding three distinct isoforms of the receptor. ITPR1 isoform 1 is a 2710 amino acid protein, isoform 2 is a 2695 amino acid protein, and isoform 3 is a 2743 amino acid protein. The ITPR2 gene is located on chromosome 12p11.23 and is composed of 62 exons that encode a 2701 amino acid protein. The ITPR3 gene is located on chromosome 6p21.31 and is composed of 60 exons that encode a 2671 amino acid protein.
Each of the IP3 receptors possesses a cytoplasmic N-terminal ligand-binding domain and is comprised of six membrane-spanning helices that forms the core of the ion pore. The released ER/SR Ca2+ activates numerous enzymes, many by activating their calmodulin or calmodulin-like subunits. DAG has two roles: it binds and activates protein kinase C (PKC), and it opens Ca2+ channels in the plasma membrane, reinforcing the effect of IP3. Like PKA, PKC phosphorylates serine and threonine residues of many proteins, thus modulating their catalytic activity.
Only one receptor class, that for the natriuretic peptides (e.g. atrial natriuretic peptide, ANP: also sometimes called atrial natriuretic factor, ANF), has been shown to be coupled to the production of intracellular cGMP. ANP, a peptide secreted by cardiac atrial tissue, is much like other peptide hormones in that it is secreted into the circulatory system and has effects on distant tissues. The principal sites of ANP action are within vascular smooth muscle cells leading to vasodilation and in the kidney glomerulus, where it modulates the rate of filtration, increasing Na+ excretion in the urine.
The receptors for the natriuretic factors are integral plasm membrane proteins, whose intracellular domains possess intrinsic guanylate cyclase activity that catalyzes the formation of cGMP from GTP following natriuretic factor-binding. Intracellular cGMP itself exerts effects in vascular smooth muscle and in addition it activates cGMP-dependent protein kinase (PKG), which phosphorylates and modulates enzyme activity, leading to additional biological effects of the natriuretic factors.
Basics of Peptide Hormones
Many amino acid and peptide hormones are elaborated by neural tissue, with ultimate impact on the entire system. When their composition was still unknown, hypothalamic secretory products were known as releasing factors, since their effect was to release endocrine hormones from the pituitary. More recently the releasing factors have been renamed releasing hormones. Currently, both names are in common use.
Releasing hormones are synthesized in neural cell bodies of the hypothalamus and secreted at the axon terminals into the portal hypophyseal circulation, which directly bathes the anterior pituitary. These peptides initiate a cascade of biochemical reactions that culminate in hormone-regulated, whole-body biological end points. Cells of the anterior pituitary, with specific receptors for individual releasing hormones, generally respond through a Ca2+, IP3, PKC-linked pathway that stimulates exocytosis of preexisting vesicles containing the various anterior pituitary hormones. The pituitary hormones are carried via the systemic circulation to target tissues throughout the body. At the target tissues they generate unique biological activities.
The secretion of hypothalamic, pituitary, and target tissue hormones is under tight regulatory control by a series of feedback and feedforward loops. This complexity can be demonstrated using the growth hormone (GH) regulatory system as an example. The stimulatory substance, growth hormone releasing hormone (GHRH), and the inhibitory substance, somatostatin (SS), both products of the hypothalamus, control pituitary GH secretion. Somatostatin is also called growth hormone-inhibiting hormone (GHIH). Under the influence of GHRH, growth hormone is released into the systemic circulation, causing the target tissue to secrete insulin-like growth factor-1, IGF-1. Growth hormone also has other more direct metabolic effects; it is both hyperglycemic and lipolytic. The principal source of systemic IGF-1 is the liver, although most other tissues secrete and contribute to systemic IGF-1. Liver IGF-1 is considered to be the principal regulator of tissue growth. In particular, the IGF-1 secreted by the liver is believed to synchronize growth throughout the body, resulting in a homeostatic balance of tissue size and mass. IGF-1 secreted by peripheral tissues is generally considered to be autocrine or paracrine in its biological action.
Systemic IGF-1 also has hypothalamic and pituitary regulatory targets. The negative feedback loops cause down-regulation of GH secretion directly at the pituitary. The longer positive feedback loop, involving IGF-1 regulation at the hypothalamus, stimulates the secretion of GHIH, which in turn inhibits the secretion of growth hormone by the pituitary. The latter is a relatively unusual negative feed-forward regulatory process. In addition, a shorter negative feedback loop is shown that involves direct IGF-1 action on the pituitary, leading to down-regulation of GH secretion. Similar feedback loops exist for all the major endocrine hormones, and many subtle nuances modulate each regulatory loop.
Pancreatic Hormones
The primary function of the pancreatic hormones is the regulation of whole-body energy metabolism, principally by regulating the concentration and activity of numerous enzymes involved in catabolism and anabolism of the major cell energy supplies.
Insulin
The earliest of these hormones recognized was insulin, whose major function is to counter the concerted action of a number of hyperglycemia-generating hormones and to maintain low blood glucose levels. Because there are numerous hyperglycemic hormones, untreated disorders associated with insulin generally lead to severe hyperglycemia and shortened life span. Insulin is a member of a family of structurally and functionally similar molecules that include the insulin-like growth factors (IGF-1 and IGF-2), and relaxin. The tertiary structure of all 4 molecules is similar, and all have growth-promoting activities, but the dominant role of insulin is metabolic while the dominant roles of the IGFs and relaxin are in the regulation of cell growth and differentiation. For an extended discussion of the production and processing of insulin, the insulin receptor, and the actions of insulin go to the Insulin Function, Insulin Resistance, and Food Intake Control of Secretion page.
Insulin is synthesized as a preprohormone in the β-cells of the islets of Langerhans of the endocrine pancreas. The signal sequence of the preproinsulin protein is removed in the lumen of the endoplasmic reticulum and the proinsulin protein is packaged into secretory vesicles in the Golgi. Within these secretory vesicles to proprotein undergoes proteolysis to release the carboxy terminal A peptide, the amino terminal B peptide, and the C peptide which represents the central third of the proprotein. The A and B peptides are then folded into the native structure of functional insulin by the formation of two disulfide bonds between these two peptides.
Insulin secretion from β-cells is principally regulated by plasma glucose levels and is referred to as glucose-stimulated insulin secretion, GSIS. The increased uptake of glucose by pancreatic β-cells leads to a concomitant increase in metabolism. The increase in metabolism leads to an elevation in the ATP/ADP ratio. This in turn leads to an inhibition of an ATP-sensitive K+ channel. The net result is a depolarization of the cell leading to Ca2+ influx and insulin secretion.
Chronic increases in numerous other hormones (including GH, hPL, estrogens, and progestins), up-regulate insulin secretion, probably by increasing the preproinsulin mRNA and enzymes involved in processing the increased preprohormone. The adrenergic hormone, norepinephrine, diminishes insulin secretion through its binding to α2-adrenergic receptors of pancreatic β cells.
Conversely, the adrenergic hormone epinephrine, by binding to β2-adrenergic receptors on pancreatic β cells, inhibits insulin secretion. Epinephrine counters the effect of insulin in liver where it binds to both α1– and β2-adrenergic receptors. Activation of α1-adrenergic receptors increases release of stored intracellular Ca2+ which binds to the calmodulin subunit of phosphorylase kinase resulting in increased glycogenolysis and glucose release to the blood. The activation of hepatic β2-adrenergic receptors induces adenylate cyclase activity, increases cAMP, and activates PKA. These latter events induce both glycogenolysis and gluconeogenesis, both of which lead to increased serum glucose.
Insulin secreted by the pancreas is directly infused via the portal vein to the liver, where it exerts profound metabolic effects. In most other tissues insulin increases the number of plasma membrane glucose transporters, but in liver, glucose uptake is dramatically increased because of increased activity of the glycolytic enzymes glucokinase, phosphofructokinase-1 (PFK-1), and pyruvate kinase (PK), the key regulatory enzymes of glycolysis. The latter effects are induced by insulin-dependent activation of phosphodiesterase, with decreased PKA activity and diminished phosphorylation of the regulatory glycolytic enzymes. In addition, phosphatases specific for the phosphorylated forms of the glycolytic enzymes increase in activity under the influence of insulin. All these events lead to conversion of the glycolytic enzymes to their active forms and consequently a significant increase in glycolysis. In addition, glucose-6-phosphatase activity is down-regulated. The net effect is an increase in the content of hepatocyte glucose and its phosphorylated derivatives, with diminished blood glucose.
In addition to the latter events, diminished cAMP and elevated phosphatase activity combine to convert glycogen phosphorylase to its inactive form and glycogen synthase to its active form, with the result that not only is glucose funneled to glycolytic products, but glycogen content is increased as well.
Insulin generates its intracellular effects by binding to a plasma membrane receptor, which is the same in all cells. The receptor is a disulfide-bonded glycoprotein. One function of insulin (aside from its role in signal transduction) is to increase glucose transport into extrahepatic tissue, primarily skeletal muscle and adipose tissue, by increasing the number of glucose transporters (GLUT4) in the plasma membrane. Glucose transporters are in a continuous state of turnover. Increases in the plasma membrane content of transporters stem from an increase in the rate of recruitment of new transporters into the plasma membrane, deriving from a special pool of preformed transporters localized in the cytoplasm.
In addition to its role in regulating glucose metabolism, insulin stimulates lipogenesis, diminishes lipolysis, and increases amino acid transport into cells. Insulin also modulates transcription, altering the cell content of numerous mRNAs. It stimulates growth, DNA synthesis, and cell replication, effects that it holds in common with the IGF family of growth factors and relaxin.
Glucagon
Glucagon is a 29-amino acid hormone synthesized by the α-cells of the islets of Langerhans of the endocrine pancreas. Glucagon was originally identified as a contaminant of insulin that induced rapid elevation in serum glucose levels following injection. This effect served as the derivation of the name glucagon as an amalgam of glucose and agonist.
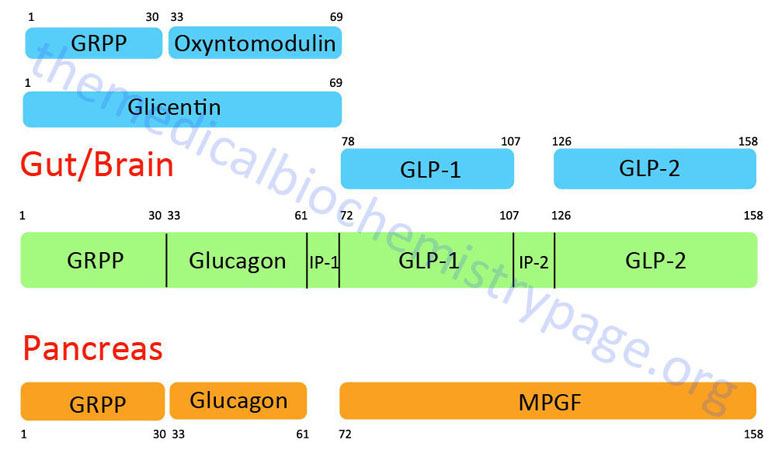
The glucagon peptide is derived by proteolytic processing from the very much larger proglucagon protein. The proglucagon protein is encoded by the GCG gene. The GCG gene is located on chromosome 2q24.2 and is composed of 6 exons that encode a 180 amino acid preproprotein.
The organization and differential processing of the proglucagon protein is described in greater detail in the Gut-Brain Interrelationships and Control of Feeding Behavior page. Like insulin, glucagon lacks a plasma carrier protein, and like insulin its circulating half life is also about five minutes. As a consequence of the latter trait, the principal effect of glucagon is on the liver, which is the first tissue perfused by blood containing pancreatic secretions.
Glucagon Secretion
The control of the secretion of glucagon from pancreatic α-cells is complex and involves metabolic, paracrine, hormonal, and neuronal signals. The primary metabolic signal that initiates the release of glucagon is falling serum glucose levels. Normal fasting plasma glucose (FPG) concentration is around 90mg/dL (~5mM). When blood glucose falls to the range of 80–85mg/dL secretion of glucagon in triggered.
Although it is well established that a drop in plasma glucose drives an elevation in glucagon secretion, the precise mechanisms that control this response of α-cells to hypoglycemia remain incompletely understood. It is most likely that a combination of three broad mechanisms are at play in the regulation of hypoglycemia-induced glucagon secretion. The mechanisms include direct α-cell sensing of serum glucose changes, indirect modulation of α-cell function through paracrine actions in the pancreatic islets, and autonomic nervous system-mediated stimulation of α-cell secretion. Paradoxically, individuals with type 1 and type 2 diabetes exhibit increased glucagon secretion with increasing serum glucose concentration.
Multiple signals also negatively regulate glucagon secretion. Insulin, amylin, and zinc, released from pancreatic β-cells, exert paracrine effects on α-cells resulting in restricted glucagon release, Additional factors, including somatostatin (released from pancreatic δ-cells), serotonin, GABA and urocortin-3 (both secreted from pancreatic β-cells) also exert paracrine inhibition of glucagon secretion. Serotonin exerts its effects on the α-cell by binding to the 5-HT1F receptor which is a Gi-coupled GPCR. Insulin exerts direct effects on pancreatic α-cells due to the presence of its receptor in these cells. Zinc is associated with insulin in the secretory vesicles of pancreatic β-cells such that it is released in concert with insulin release. Zinc effects on α-cells include opening of the α-cell KATP channel resulting in inhibition of the normal electrical activity of the α-cell that is associated with glucagon release.
Urocortin-3 (encoded by the UCN3 gene) is a member of the corticotropin-releasing factor (CRF) family of peptides. Urocortin-3 is synthesized in pancreatic β-cells and stimulates insulin secretion. When released from β-cells urocortin-3 binds to the type 2 CRF receptor (CRFR2) present on pancreatic δ-cells. Activation of CRFR2 stimulates release of somatostatin which then inhibits glucagon secretion from β-cells. Secretion of urocortin-3 is triggered by high blood glucose as well as by GLP-1, signals that also lead to enhanced insulin secretion.
As opposed to inhibition of glucagon secretion, there are several signals that are associated with stimulation of glucagon secretion. These signals include epinephrine, GIP, and glucagon itself. Epinephrine directly stimulates increased blood glucose levels by activating hepatic gluconeogenesis. By stimulating glucagon release from pancreatic α-cells, epinephrine also indirectly enhances blood glucose levels. Epinephrine binds to α1-adrenergic receptors on α-cells triggering activation of PLCβ. The activation of PLCβ results in the generation of diacylglycerol (DAG) and inositol-1,4,5-trisphosphate (IP3; Ins-1,4,5-P3) from membrane PIP2 (PtdIns-4,5-P2). IP3 binds to receptors in the endoplasmic reticulum (ER) membrane which results in release of stored Ca2+ leading to enhanced glucagon exocytosis. The Ca2+ and DAG also activate PKC which potentiates the release of stored Ca2+. Both GIP and glucagon enhance α-cell release of glucagon by binding to their Gs-coupled receptors leading to increased levels of cAMP. The increased levels of cAMP activate PKA which then phosphorylates numerous substrates that promote the interaction of glucagon secretory vesicles with the plasma membrane enhancing secretion.
Pancreatic α-cells are capable of sensing the level of blood glucose in a manner similar to that of the β-cells. Pancreatic α-cells express the GLUT1 glucose transporter for glucose uptake. GLUT1 has a very high affinity for glucose which ensure rapid uptake and modulation of α-cell glucose concentration in response to changes in serum glucose levels. In addition, α-cells express the ATP-sensitive potassium channel (KATP) that is found in β-cell membranes. The KATP in α-cells is sensitive to much lower levels of ATP than the KATP in β-cells meaning that α-cells are depolarized at low serum glucose concentration contributing to glucagon secretion. As glucose levels rise, the further increase in ATP production results in full closure of the KATP. This results in closure of sodium (Na+) channels that, in turn, prevents the depolarization that is normally responsible for the opening of calcium channels.
The sodium channels that are involved in the regulation of sodium currents in the pancreatic α-cells are composed of proteins encoded by the SCN3A (sodium voltage-gated channel alpha subunit 3) and SCN3B (sodium voltage-gated channel beta subunit 3) genes. The SCN3A encoded protein is commonly identified as Nav1.3.
The calcium channels that are involved in glucagon secretion are the P/Q type (Cav2.1 family channels) calcium channels. The inability of these calcium channels to open leads to inhibition of exocytosis so that at high glucose concentration secretion of glucagon by α-cells in inhibited. However, the glucose-mediated inhibition of glucagon secretion still involves paracrine effects from the β-cells.
The various calcium channels involved in α-cell functions, calcium currents and glucagon secretion, include members of the L-type (Cav1.x) family and the P/Q (Cav2.x) family channels. These calcium channels are composed of proteins that are encoded by the CACNA1A (Cav2.1), CACNA1C (Cav1.2), CACNA1D (Cav1.3), CACNA1H, CACNA2D1 (Cav3.2), CACNA2D2, and CACNG4 genes.
Of significance to the pathology of type 1 diabetes (T1D) is the observation that α-cell responses to hypoglycemia and hyperglycemia are impaired, yet the responses of α-cells to serum amino acids are normal. In T1D hypoglycemia does not lead to the same level of glucagon secretion as in a normal individual, often times type 1 diabetics exhibit no glucagon secretion during hypoglycemia. In most T1D patients the administration of insulin can lead to severe hypoglycemia as a result of these altered responses of α-cells to hypoglycemia. On the other end of the spectrum is that in T1D the inhibition of glucagon release by hyperglycemia is also defective leading to episodes of inappropriately high serum glucagon. One of the pathological consequences of this elevated glucagon is its contribution to the potential for ketoacidosis, a frequent problem in type 1 diabetics.
The incretin hormones, GLP-1 and GIP, inhibit glucagon secretion in a glucose-dependent. The effects of GLP-1 and GIP are most likely to be indirect through their effects on insulin secretion although the GLP-1 receptor has been shown to be expressed on pancreatic α-cells. That GLP-1 binding to its receptor on α-cells exerts a direct effect on glucagon secretion has been demonstrated by the fact that a GLP-1 receptor antagonist increases glucagon secretion.
Glucose-sensing neurons in the nucleus of the solitary tract (NTS, for the Latin term nucleus tractus solitarii) of the brain stem contribute to hypoglycemia-induced increases in glucagon secretion from the pancreas. A specific subset of GLUT2 expressing neurons in the NTS. These GLUT2 expressing neurons represent of subset of glucose inhibited neurons whose responses are determined by intracellular glucose metabolism. This process couples hypoglycemia to changes in membrane excitability. The response of the GLUT2-expressing NTS localized neurons to low glucose is mediated by reduced intracellular
glucose metabolism resulting in an increase in the AMP to ATP ratio that results in activation of AMPK activity which in turn leads to closure of leak K+ channels. These NTS neurons are GABAergic neurons and they send projections to the dorsal motor nucleus of the vagus (DMNX). The increase in vagal activity, resulting from hypoglycemia-induced activation of GLUT2-expressing neurons in the NTS, triggers increased glucagon secretion.
Glucagon Effects Through Receptor Activation
The role of glucagon is well established. It binds to a plasma membrane G-protein coupled receptor (GPCR). The glucagon receptor is derived from the GCGR gene. The GCGR gene is located on chromosome 17q25.3 and is composed of 15 exons that encode a protein of 477 amino acids.
The major site of expression of the GCGR gene is in hepatocytes of the liver with the second highest level of expression seen in the heart and the kidney. Within the kidney the expression of the GCGR gene is found in the proximal tubule, the descending and thin ascending limbs of the loop of Henle, the thick ascending limb of the loop of Henle, in podocytes and endothelial cells of the glomerulus, the distal convoluted tubule, the connecting tubule, and the collecting duct. Lower, yet physiologically relevant expression of the glucagon receptor has been found in adipocytes. However, there is evidence that at physiological concentrations, glucagon does not exert effects in human adipose tissue.
Glucagon binding to its receptor results in activation of an associated Gs-type G-protein, which in turn activates adenylate cyclase causing increased production of cAMP. The resultant increases in cAMP lead to activation of the kinase, PKA, which in turn phosphorylates numerous substrates resulting in a reversal of most of the effects that insulin exerts upon the liver as described above. The increases in PKA activity also lead to a marked elevation of circulating glucose, with the glucose being derived from liver gluconeogenesis and liver glycogenolysis.
Glucagon-mediated activation of gluconeogenesis occurs through direct modulation of enzyme activity as well as by alteration in the level of expression of several genes encoding gluconeogenic enzymes. These mechanisms of glucagon mediated increases in hepatic glucose production via gluconeogenesis are discussed in detail in the Gluconeogenesis page.
Within the liver, glucagon activation of its receptor also results in increased amino acid transport into hepatocytes and increased amino acid metabolism as a means to provide the carbon skeletons necessary for glucose synthesis via gluconeogenesis. The increase in amino acid metabolism also results in an increase in urea production.
The uptake and oxidation of free fatty acids from the blood provides some of the energy required by hepatocytes to carry out gluconeogenesis. However, glucagon effects are also exerted on hepatic lipid metabolism such that fatty acid synthesis is inhibited and fatty acid oxidation is enhanced. One primary site of action of glucagon on fatty acid metabolism is via PKA-mediated phosphorylation and inhibition of the rate-limiting enzyme of fatty acid synthesis, acetyl-CoA carboxylase 1 (ACC1). The inhibition of ACC1 results in direct inhibition of fatty acid synthesis but also results in decreased production of its product, malonyl-CoA, which is an inhibitor of the outer mitochondrial membrane-localized fatty acid transporter, carnitine palmitoyltransferase 1 (CPT1). The de-repression of CPT1 allows higher rates of fatty acyl-CoA import into the mitochondria allowing for increased β-oxidation.
Activation of the glucagon receptor, in hepatocytes, has also been shown to be coupled to activation of a Gq-type G-protein and activation of phospholipase C-β (PLCβ) resulting in increased production of diacylglycerol (DAG) and inositol trisphosphate (IP3; Ins-1,4,5-P3) from membrane phosphatidylinositol-4.5-bisphosphate (PIP2; PtdIns-4,5-P2). The released IP3 binds to specific receptors on the ER membrane which, when activated, leads to the mobilization of stored Ca2+ into the cytosol. Of significance to hepatic glucose homeostasis, the release of stored intracellular Ca2+ results in binding to the calmodulin subunits of several kinases such as Ca2+/calmodulin-dependent protein kinase II (CaMKII).
Increased CaMKII activity leads to phosphorylation of glycogen synthase and consequent inhibition of the activity of the enzyme. This contributes to the ability of hepatocytes to release glucose stored in glycogen. CaMKII also phosphorylates the lipolytic enzyme, ATGL leading to increased triglyceride breakdown in hepatocytes. The release of fatty acids from triglycerides leads to increased fatty acid oxidation which increases the concentration of acetyl-CoA. Acetyl-CoA is the activator of pyruvate carboxylase which ultimately leads to increased gluconeogenesis.
In addition to direct changes in metabolic pathway activity, exerted primarily via PKA-mediated phosphorylation events, the actions of PKA and CaMKII lead to changes in gene expression in these same cells. PKA phosphorylates numerous substrates including the transcription factor, cAMP response element-binding protein, CREB. Active PKA migrates to the nucleus where it phosphorylates CREB bound to cAMP-response elements (CRE) resulting in altered transcription. Important CREB target genes are those encoding the gluconeogenic enzymes, phosphoenolpyruvate carboxykinase, (PEPCK) and glucose-6-phosphatase.
As indicated, glucagon binds to its receptor in the liver and kidneys and to a lesser extent, in heart, endocrine pancreas, adrenal glands, spleen, adipose tissue, and cerebral cortex. Glucagon actions have been shown to result in increased activation of hormone-sensitive lipase, HSL in adipose tissue but there is controversy as to whether or not the effects in adipose tissue are direct or indirect. Nonetheless, the activation of HSL leads to increased release of fatty acids stored in the triglycerides in adipose tissue. The released fatty acids enter the circulation, are bound by albumin and transported to various tissues for oxidation. In the liver the oxidation of fatty acids is necessary to provide the energy needed for gluconeogenesis which is activated in liver in response to glucagon. Within the endocrine pancreas, the glucagon receptor is found on the β-cells that secrete insulin. The effect of glucagon on these cells is to stimulate insulin release so that there results a fine regulatory control over the overall level of circulating glucose.
Amylin
Amylin is a 37 amino acid peptide that is secreted from β-cells of the pancreas simultaneously with insulin. Amylin was originally identified as a major component of diabetes-associated islet amyloid deposits, hence its original name of islet amyloid polypeptide preprotein, IAPP.
The amylin protein is encoded by the IAPP gene. The IAPP gene is located on chromosome 12p12.1 and is composed of 4 exons that generate two alternatively spliced mRNAs, both of which encode the same 89 amino acid preproprotein.
The secretion of amylin, like that of insulin, occurs in response to nutrient intake and hormonal signals such as those exerted by GLP-1. Like insulin, the primary regulator of amylin synthesis and secretion is glucose. Indeed, both the amylin gene and the insulin gene possess multiple binding sites for the homeodomain-containing transcription factor, pancreatic/duodenal homeobox 1 (PDX1).
The structurally active form of amylin exists with an intrachain disulfide bond and an amidated C-terminus. When assayed by immunohistochemical means, approximately 60% of amylin peptide present in the plasma is glycosylated. The functional significance of the glycosylation is currently unknown and when assayed in vitro the glycosylated peptide is biologically inactive.
The primary actions attributable to amylin secretion are reduction in the rate of gastric emptying, suppression of food intake, and suppression of post-meal glucagon secretion. Collectively these three actions compliment the plasma glucose concentration regulating actions of insulin. The anorexigenic actions of amylin are most likely mediated within the CNS via neurons in the area postrema (AP) as evidenced by the fact that peripheral administration of amylin to animals results in neuronal activation in this region of the brain. The AP is a sub-structure of the region of the brain identified as the dorsal vagal complex (DVC). In addition to the AP, the DVC encompasses the nucleus of the solitary tract (NTS) and the dorsal motor nucleus of the vagus.
The plasma half-life of amylin is quite short being less than 15 minutes. The clearance of amylin from the plasma occurs via the kidneys both through renal excretion and renal peptidases associated with the vascular supply.
A stable analog of amylin called pramlintide (Symlin ®) is used as an adjunct to insulin treatment for type 1 and type 2 diabetes. Patients who use pramlintide show a modest degree of weight loss. Current trials are being undertaken to establish the efficacy of pramlintide in the treatment of obesity in patients without diabetes.
Amylin exerts its effect via interaction with GPCR complexes of the secretin-like receptor family (GPCR class B receptors). There are three distinct receptor complexes that bind amylin. These complexes all contain the calcitonin receptor (CALCR) as a core protein and either one of three receptor activity-modifying proteins (RAMPs), RAMP1, RAMP2 or RAMP3.
The specific amylin receptors result from the dimerization of various splice variants of the calcitonin receptor (CALCRa or CALCRb) with either RAMP1, RAMP2 and RAMP3, respectively. These receptors are commonly referred to as AMY1 (also designated AMY1R: CALCR and RAMP1), AMY2 (also designated AMY2R: CALCR and RAMP2) and AMY3 (also designated AMY3R: CALCR and RAMP3) with either an “a” or “b” in the subscript designating which CALCR splice variant of the calcitonin receptor is in the complex.
Amylin receptors are expressed in the nucleus accumbens, the dorsal raphe and the area postrema in the hind brain. Studies in rats have demonstrated that AMY2a and AMY3a are the amylin receptor subtypes localized to the area postrema which indicates that the satiety inducing effects of amylin are the result of activation of these two receptor subtypes. Within the area postrema, the key second messenger system associated with the amylin receptors appears to be cGMP.
The calcitonin receptor-like receptor (CRLR) and both RAMP1 and RAMP2 are expressed in the subfornical organ and are likely responsible for the involvement of amylin in drinking behaviors.
RAMP1 and RAMP2 but not RAMP3 have been shown to be expressed in the rat nucleus accumbens suggesting that the amylin receptor in the nucleus accumbens is either AMY1 or AMY2. The precise role of these amylin receptors in the nucleus accumbens haven not been well-established but it has been proposed that they may link food intake behavior and motor activity to amylin function. Peripheral injection of amylin demonstrates that the peptide crosses the blood-brain barrier resulting in access to a number of brain regions such as the cerebellum, midbrain, frontal cortex, parietal cortex, and occipital cortex.
Somatostatin
Somatostatin is produced and secreted by enteroendocrine D cells of the stomach and duodenum, δ-cells of pancreatic islets, and is also secreted by the hypothalamus.
In the pancreas, somatostatin acts as a paracrine inhibitor of other pancreatic hormones and, thus, also has systemic effects. It has been speculated that somatostatin secretion responds principally to blood glucose levels, increasing as blood glucose levels rise leading to down-regulation of glucagon secretion. In the gut, somatostatin is involved in the inhibition of gastric acid secretion.
Somatostatin is a cyclic peptide hormone that is derived from the SST gene. The SST gene is located on chromosome 3q27.3 and is composed of two exons that encode a 116 amino acid preproprotein. There are two forms of somatostatin generated from the preproprotein and they are identified as SS-28 and SS-14. Both forms have identical C-terminal sequences. The SS-28 form is the predominant form within the gut and the SS-14 form predominates in the central nervous system. In neural tissue somatostatin inhibits GH secretion and, thus, has systemic effects.
Somatostatin has been shown to bind to six receptors encoded by five distinct genes. The somatostatin receptor genes are identified as SSTR1–SSTR5, each of which encodes a GPCR-type receptor protein with the SSTR2 gene encoding two distinct receptor subtypes as a result of alternative mRNA splicing. The two SSTR2 encoded receptors are identified as SSTR2A and SSTR2B, although the level of SSTR2B mRNA in humans is extremely low and not likely to be of any physiologic significance. All five SSTR genes are expressed throughout the central nervous system as well in several peripheral tissues such as the gut, pancreas, liver, kidney, lung, pituitary, thyroid, and cells of the immune system.
The primary function of the somatostatin receptors is the suppression of secretory activities of numerous cell types. Somatostatin suppresses the secretion of growth hormone, prolactin, ACTH, cholecystokinin (CCK), gastrin, secretin, glucose-dependent insulinotropic peptide (GIP: also known as gastric inhibitory peptide), vasoactive intestinal peptide (VIP), glucagon, insulin, renin and aldosterone. Within the CNS, somatostatin functions as a neurotransmitter and neuromodulator.
The somatostatin receptors form complex signaling networks as a result of homodimerization and heterodimerization. All five receptor types are coupled to Gi-type G-proteins, the activation of which results in inhibition of adenylate cyclase, decreased levels of cAMP, and reduced activation of PKA. However, there is also coupling to other signal transduction processes. Somatostatin receptor activation is also coupled to activation of MAPK and protein tyrosine phosphatases. The SSTR2, SSTR3, SSTR4, and SSTR5 receptors couple to regulation of inwardly rectifying K+ channel function with the SSTR2 receptor also coupled to the inhibition of voltage-dependent Ca2+ channel activity. The SSTR1 receptor also couples to voltage-dependent Ca2+ channel activity. The SSTR2 and SSTR5 receptors also couple to activation of PLCβ while the SSTR4 receptor also couples to activation of PLA2. Heterodimerization between the SSTR2 and SSTR3 proteins leads to inactivation of SSTR3-mediated signaling. Activation of SSTR2 and SSTR4 on parietal cells of the stomach result in the inhibition of gastric acid production.
The Hypothalamic-Pituitary Axis
The hypothalamus is located below the thalamus and just above the brain stem and is composed of several domains (nuclei) that perform a variety of functions. The hypothalamus forms the ventral portion of the region of the brain called the diencephalon. Anatomically the hypothalamus is divided into three broad domains termed the posterior, tuberal, and anterior regions. Each of these three regions is further subdivided into medial and lateral areas.
The various nuclei of the hypothalamus constitute the functional domains of the various hypothalamic areas. A few of the specific nuclei of the hypothalamus include the paraventricular nucleus (PVN) which is located in the anterior medial area and is involved in oxytocin and vasopressin release from the pituitary and the arcuate nucleus of the hypothalamus (ARC, also abbreviated ARH), the dorsomedial hypothalamic nucleus (DMH), and the ventromedial nucleus (VMN) all of which are located in the tuberal medial area.
The ARC is involved in control of feeding behavior as well as secretion of various pituitary releasing hormones, the DMH is involved in stimulating gastrointestinal activity, and the VMN is involved in satiety (sensation of being full). The most important overall function of the hypothalamus is to link the central nervous system to the endocrine system via the pituitary gland (also termed the hypophysis).
The hypothalamus is involved in the control of certain metabolic processes as well as other functions of the autonomic nervous system. With respect to this discussion the hypothalamus synthesizes and secretes a variety of neurohormones, referred to as hypothalamic-releasing factors, that act upon the pituitary to direct the release of the various pituitary hormones.
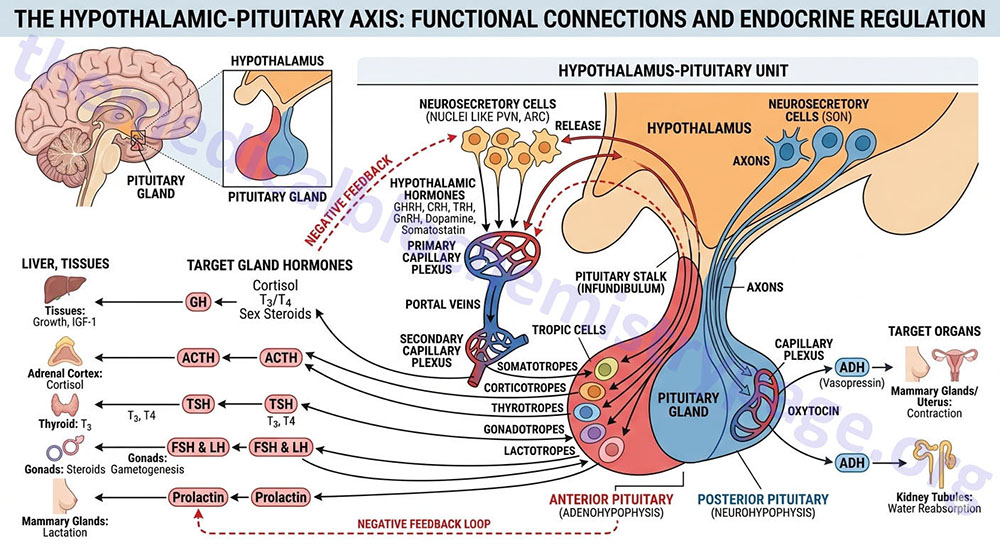
The pituitary gland has two lobes called the posterior and anterior lobes. Each lobe secretes peptide hormones that exert numerous effects on the body. Each of the pituitary hormones is described in detail in the following sections. It is the aim of this discussion to provide the background for understanding what pituitary hormones are released and what are the triggers for their release. The posterior pituitary excretes the two hormones, oxytocin and vasopressin. The anterior pituitary secretes six hormones: adrenocorticotropic hormone (ACTH, also called corticotropin), thyroid-stimulating hormone (TSH), follicle-stimulating hormone (FSH), luteinizing hormone (LH), growth hormone (GH), and prolactin (PRL). The hormone ACTH is derived from a large precursor protein identified as pro-opiomelanocortin (POMC). The secretion of the anterior pituitary hormones is under control of the hypothalamus, hence the description of the system as the hypothalamic-pituitary axis. The secretion of the hormones ACTH, TSH, FSH , LH, and GH are stimulated by signals from the hypothalamus, whereas, PRL secretion is inhibited by hypothalamic signals.
Hypothalamic Releasing Hormones
The secretion of anterior pituitary hormones results in response to hypophysiotropic hormones that are carried in the portal hypophysial vessels from the hypothalamus to the pituitary. These hypothalamic hormones are commonly referred to as releasing or inhibiting hormones. There are six primary hypothalamic releasing and inhibiting hormones: corticotropin-releasing hormone (CRH; also corticotropin-releasing factor, CRF), thyrotropin-releasing hormone (TRH), gonadotropin-releasing hormone (GnRH), luteinizing hormone-releasing hormone (LHRH), growth hormone-releasing hormone (GHRH), growth hormone release-inhibiting hormone (GHIH, more commonly called somatostatin), and prolactin release-inhibiting hormone (PIH or PIF).
Several peptides have been identified as prolactin releasing factors (PRFs) but none of these peptides are major contributors to direct release of prolactin. The major regulator of prolactin secretion is dopamine. One of the proteins identified as a prolactin releasing peptide (PrRP; encoded by the PRLH gene) exerts its effect in the brain to regulate food intake and overall energy expenditure. PrRP exerts its effects by binding to the prolactin releasing hormone receptor (encoded by the PRLHR gene; was also known as GPR10). PrRP has also been shown to have some affinity for the neuropeptide FF (NPFF) receptor type 2 (NPFF-R2; encoded by the NPRFFR2 gene.)
GnRH has been shown to stimulate the release of both FSH and LH and as a consequence the term GnRH is more appropriately used than LHRH.
The hypothalamic releasing and inhibiting hormones are secreted from the median eminence of the hypothalamus. The GnRH-secreting neurons are primarily in the medial preoptic area of the hypothalamus. The somatostatin-secreting neurons reside in the periventricular nuclei. The TRH-secreting and CRH-secreting neurons are found in the medial parts of the periventricular nuclei. The GHRH-secreting neurons reside in the arcuate nuclei which is the same region that contains dopamine-secreting neurons. Most of the receptors for the hypophysiotropic hormones are GPCR.
The Gonadotropins
The glycoprotein hormones are the most chemically complex family of the peptide hormones. All members of the family are highly glycosylated. Each of the glycoprotein hormones is an (α:β) heterodimer, with the α-subunit being identical in all members of the family. The biological activity of the hormone is determined by the β-subunit, which is not active in the absence of the α-subunit. The α-subunit gene (identified as chorionic gonadotropin, alpha: CGA) is located on chromosome 6q14.3 and is composed of 5 exons that generate two alternatively spliced mRNAs which encode isoform 1 precursor (147 amino acids) and isoform 2 precursor (116 amino acids) proteins.
The molecular weight of the gonadotropins (follicle stimulating hormone, FSH; luteinizing hormone, LH, and human chorionic gonadotropin, hCG) is about 25,000 Daltons, whereas that of the thyroid tropic hormone, thyroid stimulating hormone (TSH) is about 30,000. Synthesis of FSH and LH occurs in the same cells of the anterior pituitary and secretion of both is controlled by the hypothalamic decapeptide hormone GnRH. All members of the glycoprotein family transduce their intracellular effects via their respective receptors and the associated G-protein, adenylate cyclase, second-messenger systems. The gonadotropins (LH, FSH and hCG) bind to cells in the ovaries and testes, stimulating the production of the steroid sex hormones estrogen, testosterone (T), and dihydrotestosterone (DHT). The synthesis of the sex hormones is reviewed in the steroid hormones page.
FSH and LH
The FSH β-chain gene (gene symbol: FSHB) is located on chromosome 11p14.1 and is composed of 3 exons that generate two alternatively spliced mRNAs, both of which encode the same 129 amino acid precursor protein. The LH β-chain gene (gene symbol: LHB) is located on chromosome 19q13.33 and is composed of 3 exons that encode a 141 amino acid precursor protein. The synthesis and release of FSH and LH is controlled by the action of the hypothalamic releasing factor GnRH. The function of GnRH is to induce an episodic release of both FSH and LH that determines the onset of puberty and ovulation in females. GnRH binds to its receptor on gonadotrophs and initiates a signaling cascade that results in release of FSH and LH. The GnRH receptor gene (symbol: GNRHR) is located on chromosome 4q13.2 and is composed of 3 exons that generate two alternatively spliced mRNAs, both of which encode distinct protein isoforms. The GNRHR encoded proteins are members of the GPCR and Ca2+-dependent receptor family. The control of the hypothalamic-pituitary axis at the level of FSH and LH is controlled by several additional proteins including follistatin, activin, and leptin. Follistatin is a protein that binds to and inhibits proteins of the transforming growth factor-β family (TGFβ) of which activin is a member. Therefore, follistatin inhibits the activity of activin on promoting FSH synthesis and release.
In females FSH stimulates follicular development and estrogen synthesis by granulosa cells of the ovary. In males FSH promotes testicular growth and within the Sertoli cells of the seminiferous tubules of the testis FSH enhances the synthesis of androgen-binding proteins, ABP. The function of ABP is to bind testosterone (T) and dihydrotestosterone (DHT), as well as 17β-estradiol, resulting in the concentration of the male sex hormones within these cells. The concentration of T and DHT leads to the enhancement of spermatogenesis. In females, LH induces thecal cells of the ovary to synthesize estrogens and progesterone and promotes estradiol secretion. The surge in LH release that occurs in mid-menstrual cycle is the responsible signal for ovulation. Continuous LH secretion stimulates the corpus luteum to produce progesterone. In males, LH binds to Leydig cells of the testis resulting in the induction of the steroidogenic acute regulatory (StAR) protein. The function of StAR is to transport cholesterol from the outer mitochondrial membrane to the inner membrane where steroid hormone biosynthesis is initiated, therefore, the result of the induction of StAR synthesis is increased synthesis and secretion of T.
The FSH receptor (FSHR) is located on chromosome 2p16.3 and is composed of 14 exons that generate two alternatively spliced mRNAs that encode 695 (isoform 1) and 669 (isoform 2) precursor proteins. The predominant form of the FSH receptor is a 678 amino acid glycosylated protein that is a member of the GPCR family of receptors. Binding of FSH to its receptors results in activation of adenylate cyclase leading to increased PKA activity.
The LH receptor is referred to as the LH-choriogonadotropin receptor (LHCGR). The gene for this receptor is also found on chromosome 2p16.3 and it encodes a 699 amino acid precursor protein that is processed into a 674 amino acid glycosylated member of the GPCR family of receptors. The LHCGR contains a large extracellular domain that includes several leucine-rich repeats (LRR). There are other members of the GPCR family that contain LRR in their extracellular domains and this subfamily of receptors is referred to as the LRR-containing GPCR (LRG) family. The LHCGR is coupled a G-protein that activates adenylate cyclase resulting in increased PKA activity. The LHCGR is expressed in the ovary, thecal cells, stromal cells, luteinizing granulosa, and luteal cells of the ovary and in Leydig cells of the testes.
hCG
The β-chain of hCG is encoded by six genes (identified as CGB1, CGB2, CGB3, CGB5, CGB7, and CGB8) that are all located on chromosome 19q13.33 contiguous with the LHB gene (see above). Human chorionic gonadotropin is produced only during pregnancy. The actions of hCG are exerted by binding of the hormone to the LHCGR in the luteal cells of the ovary. Initially the developing embryo synthesizes and secretes hCG. Following implantation the cells of the syncytiotrophoblast (part of the placenta) produce and secrete hCG. The production of hCG increases markedly after implantation; its appearance in the plasma and urine is one of the earliest signals of pregnancy and the basis of many pregnancy tests. The role of hCG during pregnancy is to prevent disintegration of the corpus luteum so as to maintain the synthesis of progesterone by this tissue.
Kisspeptin
Kisspeptin is a neuropeptide (peptide hormone) that exerts critical regulatory effects, via the central nervous system, that control reproductive neural circuits. Kisspeptin exerts these effects by stimulating gonadotropin-releasing hormone (GnRH)-induced gonadotropin secretion as well as by regulating the pubertal activation of GnRH neurons.
Kisspeptin regulates the processes of female follicle development, oocyte maturation, and ovulation through its effects on the hypothalamic-pituitary-gonadal axis. Kisspeptin is also involved in the processes of male reproduction through regulation of Leydig cell functions, spermatogenesis, sperm functions, and reproductive behaviors.
In addition to its role in the regulation of reproductive processes, kisspeptin also exerts control on numerous peripheral processes of metabolism, particularly in the liver, adipose tissue, and pancreas.
The kisspeptin protein is encoded by the KISS1 (KiSS-1 metastasis suppressor) gene. The KISS1 encoded protein was first thought to be related to the processes of cancer metastasis. The name KiSS-1 is thought to have been given to the gene since it was discovered at Pennsylvania State University in Hershey, Pennsylvania, home of Hershey’s chocolate kisses.
The KISS1 gene is located on chromosome 1q32.1 and is composed of 3 exons that can generate two different preproproteins due to the presence of a polymorphism in the terminal exon. When there is an adenosine at the polymorphic site there is a translation stop codon resulting in the synthesis of a 138 amino acid preproprotein. When the adenosine is absent the protein is extended by seven amino acids resulting in a 145 amino acid preproprotein. Mutations in the KISS1 gene have been identified in humans and these mutations are associated with infertility, impaired puberty, and very low levels of gonadotropins and sex steroids.
There are actually several bioactive peptides derived from the KISS1 encoded mRNA in addition to the two potential preproproteins resulting from the terminal exon polymorphism. Following removal of the signal sequence the kisspeptin proprotein is processed into several short peptides that are designated by the number of amino acids and identified as kisspeptin-54 (Kp-54), kisspeptin-14 (Kp-14), kisspeptin-13 (Kp-13), and kisspeptin-10 (Kp-10). These peptides are collectively referred to as kisspeptins. Kisspeptin-54 is the primary bioactive protein derived from the KISS1 mRNA. When originally isolated and characterized, the KISS1 encoded protein was identified as metastatin. Kisspeptins are members of the RF amide peptide family (also termed RF peptide related family, RFRP) so-called because they contain an Arg-Phe (R-F) dipeptide at the C-terminal end of the protein with an -NH2 modification (Arg-Phe-NH2).
Kisspeptin exerts its effects by binding to the kisspeptin receptor (Kiss1R) on responsive cells. The Kiss1R is a member of the G-protein coupled receptor (GPCR) family. The Kiss1R protein is encoded by the KISS1R gene. This receptor was originally identified as GPR54. The KISS1R gene is located on chromosome 19p13.3 and is composed of 4 exons that encode a 398 amin acid protein. The highest levels of expression of the KISS1R gene are found in the brain.
Kiss1R is coupled to a Gq-type G-protein, that upon activation by kisspeptin binding, activates phospholipase C beta (PLCβ) resulting in the production of IP3 and DAG from membrane-associated PIP2.
Within the brain there are two major populations of KISS1R expressing neurons. One population is found in the arcuate nucleus (ARC) directly above the median eminence. The KISS1R expressing neurons also express the genes encoding neurokinin B and dynorphin. These neurons are referred to as KNDy neurons.
The other population resides in an area that spans the anteroventral periventricular nucleus and periventricular nucleus (AVPV/PeN). In this second domain of KISS1R expressing neurons there are more neurons in females than in males.
The kisspeptin neurons in the AVPV/PeN are positively regulated the sex hormone estradiol and also mediates the positive feedback induction of the preovulatory GnRH/LH surge in females that is exerted by estradiol. Within the ARC, kisspeptin neurons stimulate the tonic, pulsatile release of GnRH and luteinizing hormone (LH), actions that control gonadal sex steroid hormone synthesis, gametogenesis, and the menstrual cycle. Circulating sex steroids function to feed back and inhibit these effects of kisspeptin.
Thyroid Stimulating Hormone (TSH): Thyrotropin
As indicated above, TSH (also called thyrotropin) is a member of the glycoprotein hormone family and as such is composed of a common α-subunit encoded by the CGA gene and a unique β-chain. The β-chain of TSH is encoded by the TSHB gene (thyroid-stimulating hormone, β-chain) which is on chromosome 1p13.2 and contains 3 exons, the first of which is non-coding.
Secretion of TSH is stimulated by thyrotropin-releasing hormone (TRH) from the hypothalamus. TRH, a tripeptide, is synthesized by neurons in the supraoptic and supraventricular nuclei of the hypothalamus and stored in the median eminence. TRH is transported to the anterior pituitary via the pituitary portal circulation and binds to a specific receptor located on TSH- and prolactin-secreting cells. There are two TRH receptors, identified as TRH-R1 and TRH-R2, both of which are G-protein coupled receptors (GPCRs). Both TRH receptors are coupled to Gq-type G-proteins. Binding of TRH to its receptor activates a typical PLCβ-mediated signaling cascade. The TRH-induced signaling leads to TSH secretion as well as increased TSH transcription and post-translational glycosylation. Although both receptors are expressed differentially in the brain and in peripheral tissues, they exhibit indistinguishable TRH-binding affinities. However, only TRH-R1 is expressed at functional levels in the anterior pituitary. The TRH-mediated release of TSH is pulsatile with peak secretion being exerted between midnight and 4am.
The synthesis and release of TSH is controlled by two pathways. The first is exerted by the level of T3 (triiodothyronine) within thyrotropic cells which regulates TSH expression, translation and release. The second regulation is of course exerted by TRH as described above. While in the circulation TSH binds to receptors on the basal membrane of thyroid follicles. The receptors are coupled through G-protein activation of adenylate cyclase as well as PLCβ. The TSH receptor gene (symbol: TSHR) is on chromosome 14q31.1 and is composed of 12 exons that generate three alternatively spliced mRNAs. The major TSHR encoded protein is a 764 amino acid glycosylated member of the GPCR family of receptors. The TSHR and the LHCGR proteins share a significant degree of homology. TSH binding to its receptor triggers a signaling cascade that results in increased thyrocyte cAMP, PKA, IP3, and DAG leading to, in the short term, increased secretion of the thyroid hormones, thyroxin (T4) and triiodothyronine (T3). TSH-binding to its receptor also results in increased TSH synthesis and thyroid cell growth.
Chronic stimulation of the TSH receptor causes an increase in the synthesis of a major thyroid hormone precursor, thyroglobulin. Thyroglobulin produced on rough endoplasmic reticulum has a molecular weight of 660,000. It is glycosylated and contains more than 100 tyrosine residues, which become iodinated and are used to synthesize T3 and T4. Thyroglobulin is exocytosed through the apical membrane into the closed lumen of thyroid follicles, where it accumulates as the major protein of the thyroid and where maturation takes place. Mature, iodinated thyroglobulin is taken up in vesicles by thyrocytes and fuses with lysosomes. Lysosomal proteases degrade thyroglobulin releasing amino acids and T3 and T4, which are secreted into the circulation.
These compounds are very hydrophobic and require a carrier protein for delivery to target tissues. In the plasma, T3 and T4 are bound to a carrier glycoprotein known as thyroxin-binding globulin and are disseminated throughout the body in this form. The feedback loop that regulates T3 and T4 production is a single short negative loop, with the T3 and T4 being responsible for down-regulating pituitary TSH secretion. Meanwhile, continuously secreted hypothalamic TRH is responsible for up-regulating TSH production. The TSH actually secreted by thyrotrophs is the net result of the negative effects of T3 and T4 and the positive effect of TRH.
Thyroid hormones act by binding to cytosolic receptors very similar to steroid hormone receptors, and for this reason T3 and T4 are often classified along with the hydrophobic steroid hormones. The principal role of thyroid hormones is also like that of steroid hormones. In adults, the ligand receptor combination binds to thyroid hormone response elements in nuclear DNA and is responsible for up-regulating general protein synthesis and inducing a state of positive nitrogen balance.
Numerous congenital and acquired forms of hypothyroidism and hyperthyroidism are the result of alterations in the expression, processing, and function of the TSHR. The most common TSHR disorder resulting in hyperthyroidism (thyrotoxicosis) is Graves disease. Graves disease is caused by thyroid-stimulating autoantibodies (TSAb, also called thyroid-stimulating immunoglobulins, TSIs) which bind to and activate the human TSH receptor, leading to the thyrotoxicosis characteristic of this disease. TSAbs bind to the TSH receptor and mimic the TSH stimulation of the gland by increasing intracellular cAMP. The hyperactivated thyroid then secretes excessive T3 and T4. Graves disease is classified as a form of thyrotoxicosis, the name for the clinical syndrome resulting from tissues exposed to high levels of thyroid hormones. One theory proposed for the development of the TSAb is that there is a defect in suppressor T cells that allows helper T cells to stimulate B cells to produce thyroid autoantibodies. The clinical features of Graves disease are thyrotoxicosis, goiter (enlarged thyroid gland), an ophthalmopathy in the form of exophthalmos (eyes bulge out), and dermopathy in the form of pretibial myxedema (localized lesions of the skin, primarily in the lower legs, resulting from the deposition of hyaluronic acid).
At the other end of the spectrum are disorders that lead to hypothyroidism. Deficiency in iodine is the most common cause of hypothyroidism worldwide. Indeed the practice of producing iodized table salt was to stem the occurrence of hypothyroidism. When hypothyroidism is evident in conjunction with sufficient iodine intake it is either autoimmune disease (Hashimoto thyroiditis) or the consequences of treatments for hyperthyroidism that are the cause. In the embryo, thyroid hormone is necessary for normal development and hypothyroidism in the embryo is responsible for cretinism, which is characterized by multiple congenital defects and intellectual impairment. Because the neurological consequences of congenital hypothyroidism are severe neonatal screening for thyroid hormone levels at birth is routine. Most infants born with congenital hypothyroidism appear normal at birth. However, if left untreated the symptoms will include a thick protruding tongue, poor feeding, prolonged jaundice (which exacerbates the neurological impairment), hypotonia (recognized as “floppy baby syndrome”), episodes of choking, and delayed bone maturation resulting in short stature.
The Pro-Opiomelanocortin (POMC) Family
The POMC gene is located on chromosome 2p23.3. POMC is expressed in both the anterior and intermediate lobes of the pituitary gland. The primary protein product of the POMC gene is a 285 amino acid precursor that can undergo differential processing to yield at least 8 peptides, dependent upon the location of synthesis and the stimulus leading to their production. POMC is produced in the pituitary, the ARC of the hypothalamus, the nucleus of the solitary tract (NTS for Latin term nucleus tractus solitarii; specialized cells within the medulla responsible for sensations of taste and visceral sensations of stretch), as well as in several peripheral tissues such as the skin and reproductive organs. Within the brain neurons that respond to POMC-derived peptides (termed POMCm neurons) are critical in the regulation of overall energy balance via the melanocortin peptides (primarily α-MSH; this is N-terminally acetylated MSH).
The processing of POMC involves glycosylations, acetylations, and extensive proteolytic cleavage at sites shown to contain regions of basic protein sequences. The proteases that recognize these cleavage sites are tissue-specific; thus, the physiologically active product of the anterior pituitary is ACTH (discussed in detail in the section below). Aside from ACTH, the activities of the melanocortins (primarily α-MSH) produced in the anterior pituitary are the best understood peptides derived from the POMC mRNA. The activities of the melanocortin peptides is discussed in the section following ACTH.
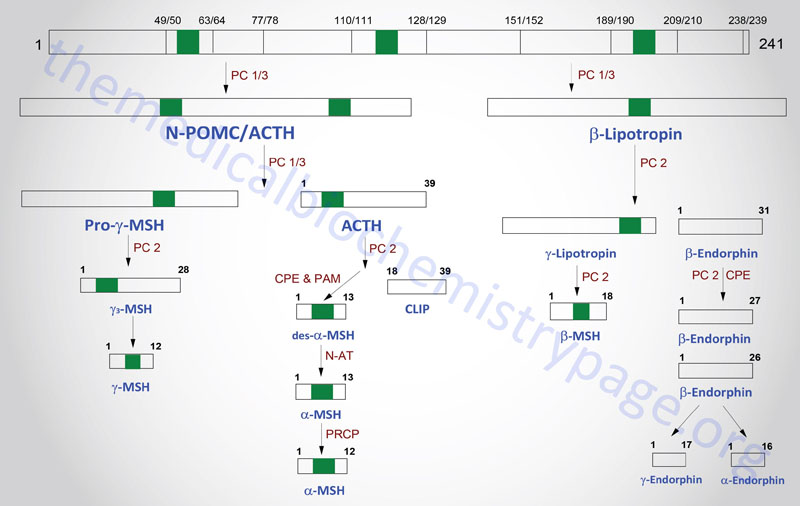
Many of the other POMC products are synthesized in other neural tissues that contain proteases with appropriate specificity. In human embryos and in pregnant women, the intermediate lobe is active and leads to the production of endorphins and enkephalins. These same endorphin-producing pathways are active in other neural tissues, and since they bind to the opioid receptors in other parts of the brain they are assumed to represent natural opioid-like analgesic compounds.
Adrenocorticotropic Hormone, ACTH
ACTH is a 39 amino acid peptide that is derived by post-translational modification from the 241 amino acid preproprotein, POMC. ACTH is the main physiologically active product of the actions of the hypothalamic releasing hormone, CRH, on the anterior pituitary. Although CRH is the primary stimulus for ACTH release, other hormones also exert effects on ACTH release. CRH stimulates a pulsatile secretion of ACTH with peak levels seen before waking and declining as the day progresses. Negative feedback on ACTH secretion is exerted by cortisol at both the hypothalamic and anterior pituitary levels. Thus, the primary product of the systemic actions of ACTH regulates the further actions of this corticotropic hormone. Additional factors that influence ACTH secretion include physical, emotional, and chemical stresses. These stressors include pain, cold exposure, acute hypoglycemia, trauma, depression, and surgery. The stress-mediated increases in ACTH secretion are the result of the actions of vasopressin and CRH.
The biological role of ACTH is to stimulate the production of adrenal cortex steroids, principally the glucocorticoids cortisol and corticosterone. ACTH also stimulates the adrenal cortex to produce the mineralocorticoid, aldosterone as well as the androgen, androstenedione. ACTH exerts its effects on the adrenal cortex by binding to a specific receptor that is a member of the melanocortin receptor family. The ACTH receptor is identified as MC2R for melanocortin-2 receptor. The ACTH receptor is a Gs-type G-protein coupled receptor (GPCR). The activity of the MC2R is dependent upon a a small accessory protein, called melanocortin receptor accessory protein (MRAP). The function of MRAP is to regulate trafficking of MC2R to the plasma membrane and to enhance ACTH binding and activation of MC2R. The binding of ACTH to MC2R binding triggers activation of adenylate cyclase, elevation of cAMP, and increased PKA activity of adrenal cortex tissue.
Several targets of the ACTH activated PKA are hormone sensitive lipase, steroidogenic acute regulatory protein (StAR), and CYP11A1 (also called P450-linked side chain-cleaving enzyme, P450ssc, 20,22-desmolase, or cholesterol desmolase). Activation of HSL increases the de-esterification of cholesterol esters generating free cholesterol. The activation of StAR results of increased transport of free cholesterol into the mitochondria where steroid hormone synthesis is initiated. The activation of CYP11A1 results in increased conversion of cholesterol to pregnenolone during steroid hormone synthesis.
Secondary adrenal insufficiency occurs in patients with deficiencies in pituitary ACTH production or secretion. Whereas, primary adrenal insufficiency (adrenal hypoplasia) is characteristic of Addison disease which was originally diagnosed as the result of lesions in the adrenal glands caused by tuberculosis. Secondary adrenal insufficiency is characterized by weakness, fatigue, nausea, vomiting, and anorexia. On the opposite side of the abnormal ACTH spectrum are the adrenal hyperplasias. These include the congenital adrenal hyperplasias (CAH) and Cushing syndrome. The CAH are a family of inherited disorders that result from loss-of-function mutations in one of several genes involved in adrenal steroid hormone synthesis. Endogenous causes of Cushing syndrome are pituitary corticotroph adenomas resulting in excess ACTH production and secretion.
The characteristic features of Cushing syndrome are psychiatric disturbances (depression, mania, and psychoses), central obesity, hypertension, diabetes, moon-shaped face, thin fragile skin, easy bruising, and purple striae (stretch marks). In addition, Cushing syndrome patients manifest with gonadal dysfunction that is characteristic of hyperandrogenism with excess body and facial hair (hirsutism) and acne.
POMC-Derived Melanocortins and Feeding Behavior
The POMC-derived melanocortin peptides include α-MSH, β-MSH, γ-MSH, ACTH1-24, and ACTH1-13–NH2 (desacetyl-α-MSH; indicated as des-α-MSH in above Figure). The POMC-derived melanocortins belong to a family of peptides referred to as the melanocortin system. This system includes the POMC-derived melanocortins which exhibit agonist activities, the antagonist peptide agouti-related peptide (AgRP), the melanocortin receptors (MCR), and the melanocortin receptor accessory proteins (MRAPs). The MCR family of receptors consists of five identified members termed MC1R through MC5R.
The melanocortin system has been shown to be critical in the regulation of food intake and energy expenditure via a number of different assay systems involving both humans and animals. The details of the role of melanocortins in appetite regulation are discussed in the Gut-Brain Interrelationships and the Control of Feeding Behavior page.
Vasopressin and Oxytocin
The principal hormones of the posterior pituitary are the nonapeptides oxytocin and vasopressin (mammalian form is called arginine vasopressin, AVP). Vasopressin is also known as antidiuretic hormone (ADH). The amino acid sequences of vasopressin and oxytocin differ by only two amino acids. Both of these hormones are synthesized as prohormones in neural cell bodies of the hypothalamus and mature as they pass down axons in association with carrier proteins termed neurophysins. The axons terminate in the posterior pituitary, and the hormones are secreted directly into the systemic circulation.
The neurophysins themselves are derived from the oxytocin and vasopressin preproproteins. The oxytocin preproprotein contains neurophysin 1 and the vasopressin preproprotein contains neurophysin 2. The human genes coding for pre-provasopressin-neurophysin 2 (prepro-AVP-NP2) and prepro-oxytocin-neurophysin 1 (prepro-OT-NP1) are similar in their intron-exon structure and they are linked together with 12kb of intervening DNA. Interestingly, the two genes are transcribed from opposite DNA strands.
The vasopressin preproprotein is derived from the AVP gene which is located on chromosome 20p13 and is composed of 4 exons that encode the 164 amino acid preproprotein. In addition to Arg-vasopressin and neurophysin 2, the AVP encoded preproprotein contains a peptide called copeptin. The oxytocin preproprotein is derived from the OXT gene which is also located on chromosome 20p13 and is composed of 4 exons that encode the 125 amino acid preproprotein.
Vasopressin is known as antidiuretic hormone (ADH), because it is the main regulator of body fluid osmolarity through induced renal reabsorption of water. The designation arginine vasopressin (AVP) is used when discussing vasopressins from different mammals. Marsupials and pigs produce a vasopressin peptide where the arginine is replaced by a lysine and is thus, referred to as lysine vasopressin. The secretion of vasopressin is regulated in the hypothalamus by osmoreceptors which sense water and Na+ concentration and stimulate increased vasopressin secretion when plasma osmolarity increases. The secreted vasopressin increases the reabsorption rate of water in principal cells of the collecting ducts of the kidney tubule, causing the excretion of urine that is concentrated in Na+ and thus yielding a net drop in osmolarity of body fluids. Vasopressin deficiency leads to production of large volumes of watery urine and to polydipsia (increased desire for fluid intake). These symptoms are diagnostic of a condition known as diabetes insipidus. Diabetes insipidus has numerous causes that include effects on the hypothalamus and/or pituitary (central diabetes insipidus) or the kidneys (nephrogenic diabetes insipidus).
Vasopressin Receptors
Vasopressin binds plasma membrane receptors are that G-protein coupled receptors (GPCR) that activate signaling events through their associated G-proteins that are either coupled to the cAMP second messenger system or through the PLCβ pathway. There are three kinds of vasopressin receptors designated V1A (V1A or just V1), V1B (V1B: also known as the V3 receptor), and V2 (V2).
Although there are three vasopressin receptors, the major actions of vasopressin are elicited through activation of the V1A and V2 receptors. The V1A receptor is encoded by the AVPR1A gene which is located on chromosome 12q14.2 and is composed of 2 exons that encode a 418 amino acid protein. The V1B receptor is encoded by the AVPR1B gene which is located on chromosome 1q32.1 and is composed of 2 exons that encode a 424 amino acid protein. The V2 receptor is encoded by the AVPR2 gene which is located on the X chromosome (Xq28) and is composed of 5 exons that generate two alternatively spliced mRNAs encoding V2 receptor isoform 1 (371 amino acids) and V2 receptor isoform 2 (309 amino acids).
The V1A and V1B receptors are GPCR that activate Gq-type G-proteins leading to the activation of PLCβ and the subsequent hydrolysis of PIP2 which results in increased intracellular Ca2+ concentration and the activation of PKC. The V2 receptor is a GPCR that activates a Gs-type G-protein leading to the activation of adenylate cyclase resulting in increased cAMP levels and activation of PKA. The V1A (V1) receptors are found in vascular smooth muscle cells and vasopressin binding to these receptors triggers vascular contraction resulting in increased blood pressure. The V1A receptor is also expressed in smooth muscle cells of the uterus (myometrium), in hepatocytes, and in platelets. The V1B (V3) receptor is expressed in the anterior pituitary.
The V2 receptor is found primarily in the basolateral membranes (facing the blood) of principal cells of the collecting ducts of the kidney tubules where its activation is responsible for triggering vasopressin-mediated water retention, thereby, affecting osmolarity. Activation of the V2 receptor results in increased PKA activity which phosphorylates and triggers the mobilization of vesicles containing the water transporter, aquaporin 2 (AQP2), to the apical (luminal) membranes of principal cells. Mutations in the gene encoding the V2 receptor are responsible for X-linked nephrogenic diabetes insipidus. The V2 receptor is also expressed in vascular endothelial and vascular smooth muscle cells.
Oxytocin
Oxytocin is produced in the magnocellular neurosecretory cells of the paraventricular nucleus and supraoptic nucleus of the hypothalamus and is then stored in axon terminals of the anterior pituitary. While stored in the pituitary, oxytocin is bound to neurophysin I in Herring bodies. Secretion of oxytocin is stimulated by electrical activity of the oxytocin cells of the hypothalamus. The actions of oxytocin are elicited via the interaction of the hormone with high affinity receptors.
The oxytocin receptor is encoded by the OXTR gene. The OXTR gene is located on chromosome 3p25.3 and is composed of 6 exons that generate five alternatively spliced mRNAs, all of which encode the same 389 amino acid protein. The oxytocin receptor is a GPCR that is coupled to a Gq–type G-protein leading to activation of PLCβ and, thus the hydrolysis of PIP2 resulting in increased intracellular Ca2+ concentration and the activation of PKC. The affinity for the oxytocin receptor for oxytocin is dependent upon Mg2+ and cholesterol, both of which act as positive allosteric regulators.
Oxytocin secretion in nursing women is stimulated by direct neural feedback obtained by stimulation of the nipple during suckling. This response to oxytocin is referred to as the “let-down response”. Its physiological effects include the contraction of mammary gland myoepithelial cells, which induces the ejection of milk from mammary glands.
Human milk is enriched in fat, predominantly in the form of triglycerides. Indeed, triglycerides constitute roughly 98% of all the fat in human milk. The triglycerides in milk originate from dietary intake of fat, lipolysis occurring in adipose tissue and liver, and from de novo synthesis in mammary epithelial cells. The oxytocin receptor is expressed at high levels in the plasma membranes of adipocytes. The activation of the oxytocin receptor on adipocytes induces adipose tissue lipolysis, thereby providing a source of fatty acids for mammary epithelial cell triglyceride synthesis.
Another primary action of oxytocin is the stimulation of uterine smooth muscle contraction leading to childbirth. The uterine effect of oxytocin is due, in part, to increased production and release of the prostaglandin PGF2α from the myometrium and to a lesser extent from the decidua.
In males the circulating levels of oxytocin increase at the time of ejaculation. It is believed that the increase in oxytocin levels causes increased contraction of the smooth muscle cells of the vas deferens thereby propelling the sperm toward the urethra.
Growth Hormone
Growth hormone (GH; also known as somatotropin), human chorionic somatomammotropin, hCS (also called human placental lactogen, hPL), and prolactin (PRL) comprise the growth hormone family. All have about 200 amino acids, 2 disulfide bonds, and no sites of glycosylation. Although each has special receptors and unique characteristics to their activity, they all possess growth-promoting and lactogenic activity. Mature GH (22,000 daltons) is synthesized in acidophilic pituitary somatotrophs as a single polypeptide chain. Because of alternate RNA splicing, a small amounts of smaller molecular forms are also secreted. Growth hormone and hCS are proteins encoded by genes in a cluster of five genes termed the human growth hormone/human chorionic somatomammotropin (GH/CS) gene cluster that spans 6.6kbp on chromosome 17q23.3.
In addition to the GH gene (gene symbol: GH1) the GH/CS gene cluster contains the chorionic somatomammotropin 1 and 2 genes (gene symbols: CSH1 and CSH2), the chorionic somatomammotropin-like 1 gene (gene symbol: CSHL1), and the growth hormone 2 gene (gene symbol: GH2). Expression of the GH1 gene occurs only in the pituitary whereas, the other four genes in the GH/CS gene cluster are expressed only in placental tissues. The GH1 gene is composed of 5 exons that generate three alternatively spliced mRNAs, each of which encode a unique protein isoform.
Humans respond to natural or recombinant human or primate growth hormone with appropriate secretion of IGF-1, but growth hormone of other species has no normal biological effect in man. The latter is puzzling because interspecies GH homologies are quite high in many cases, and most other species respond well to human growth hormone. In humans, growth hormone promotes gluconeogenesis and is consequently hyperglycemic. Growth hormone promotes amino acid uptake by cells, with the result that GH therapy puts an organism into positive nitrogen balance, similar to that seen in growing children. Growth hormone is also lipolytic, inducing the breakdown of adipose tissue lipids and thus, providing energy supplies that are used to support the stimulated protein synthesis induced by increased amino acid uptake. In bone GH stimulates bone length and bone mineral density.
Growth Hormone Receptor
Growth hormone elicits its effects by binding to a plasma membrane localized receptor (GHR) that is a member of the class I cytokine receptor family, which includes more than 30 receptors such as the prolactin receptor (PRLR) and the interleukin-6 (IL-6) receptor.
The GHR was the first member of the class I cytokine receptor family to be cloned. The GHR protein is encoded by the GHR gene that is located on chromosome 5p13.1–p12 and is composed of 18 exons that generate 11 alternatively spliced mRNAs. These mRNAs encode five distinct protein isoforms. The predominant form of the GHR is a 638 amino acid protein that is a single-pass transmembrane protein that contains one cytokine receptor homology domain (CRH) and a cytoplasmic intracellular domain (ICD).
The cytokine receptor family proteins lack an intrinsic enzymatic activity and, therefore, utilize the binding of non-receptor protein tyrosine kinases (PTK) for their signal transduction. There are two conserved domains in the ICD of the cytokine receptor family termed “Box 1” and “Box 2” that coordinate activities of the GHR. The Box 2 motif in these receptors acts as a binding site for members of the Janus kinase (JAK) family of kinases of which there are four family members, JAK1, JAK2, JAK3, and TYK2 that can bind to specific receptors of this family. The growth hormone receptor only interacts with JAK2 and the interaction activates JAK2 to phosphorylate multiple tyrosine residues on the ICD of the GHR. Upon phosphorylation of the GHR by JAK2, proteins of the STAT (signal transducer and activator of transcription) family dock to the GHR and they are then phosphorylated by JAK2. The STAT proteins that interact with the GHR are STAT5a and STAT5b. Although the GHR also activates STAT1 and STAT3 via JAK2, this does not require their binding to the tyrosine phosphorylated GHR. In addition to the JAK/STAT signal transduction pathway, the GHR activates the RAS/ERK (extracellular signal-regulated kinase) and PI3K (phosphatidylinositol-3-kinase)/AKT signal transduction pathways.
There are a number of genetic deficiencies associated with GH. GH-deficient dwarfs lack the ability to synthesize or secrete GH, and these short-statured individuals respond well to GH therapy. Pygmies lack the IGF-1 response to GH but not its metabolic effects; thus in pygmies the deficiency is post-receptor in nature. Finally, Laron dwarfs have normal or excess plasma GH, but lack liver GH receptors and have low levels of circulating IGF-1. The defect in these individuals is clearly related to an inability to respond to GH by the production of IGF-1. The production of excessive amounts of GH before epiphyseal closure of the long bones leads to gigantism, and when GH becomes excessive after epiphyseal closure, sacral bone growth leads to the characteristic features of acromegaly.
Human Chorionic Somatomammotropin (hCS)
Human chorionic somatomammotropin is produced by the placenta late in gestation. This hormone has also been called human placental lactogen (hPL) and chorionic growth hormone-prolactin (CGP). The hCS gene is found in the human growth hormone/human chorionic somatomammotropin (GH/CS) gene cluster as described above in the Growth Hormone section. The amino acid composition of hCS is similar to human growth hormone. Evidence suggests that due to the similarities between growth hormone, hCS and prolactin they likely evolved from a single progenitor hormone gene.
At its height the hormone is secreted at a rate of about 1 g/day, the highest secretory rate of any known human hormone. However, little hCS reaches the fetal circulation. The amount of hCS that is secreted is proportional to the size of the placenta. Low levels of hCS during pregnancy are a sign of placental insufficiency. The biological actions of hCS are similar to those of growth hormone suggesting that it functions as a maternal growth hormone of pregnancy. The hormone induces the retention of potassium, calcium, and nitrogen, decreases glucose utilization and increases lipolysis.
Prolactin (PRL)
Prolactin is produced by acidophilic pituitary lactotrophs. The PRL gene is located on chromosome 6p22.3 and is composed of 7 exons that generate two alternatively spliced mRNAs, both of which encode the same 227 amino acid protein. Two promoter elements have been identified in the PRL gene that are located 5.5kbp apart. The 5′-promoter directs the expression of prolactin in decidualized endometrium as well as in lymphoblastoid cells. The more proximal promoter directs pituitary lactotroph expression and this promoter is controlled by the activity of the POU-domain transcription factor, PIT1.
Although the PRL gene is thought to have evolved from an ancestral hormone gene common to growth hormone (GH) and human chorionic somatomammotropin (hCS), prolactin shares only 16% amino acid homology to these other two hormones. The prolactin protein is 198 amino acids in length with a molecular weight of 22,000 Daltons. Prolactin is known to bind zinc (Zn2+) and the binding of this metal stabilizes prolactin in the secretory pathway.
Prolactin is the lone trophic hormone of the pituitary that is routinely under negative control by prolactin release-inhibiting hormone (PIH or PIF), which is, in fact, the catecholamine neurotransmitter, dopamine. Decreased hypophyseal dopamine production, or damage to the hypophyseal stalk, leads to rapid up-regulation of PRL secretion. A number of other hypothalamic releasing hormones induce increased prolactin secretion; as a result, it is unclear whether a specific prolactin-releasing hormone (PRH) exists for up-regulating PRL secretion. Prolactin does not appear to play a role in normal gonadal function yet hyperprolactinemia in humans results in hypogonadism.
Prolactin Receptor
Prolactin elicits its effects by binding to a plasma membrane localized receptor (PRLR) that is a member of the class I cytokine receptor family, which includes more than 30 receptors such as the growth hormone receptor (GHR) and the interleukin-6 (IL-6) receptor.
The prolactin receptor is encoded by the PRLR gene that is located on chromosome 5p13.2 and is composed of 16 exons that generate six alternatively spliced mRNAs, each of which encode a distinct protein isoform. The PRLR is a single-pass transmembrane protein that contains one cytokine receptor homology domain (CRH) and a cytoplasmic intracellular domain (ICD).
The cytokine receptor family proteins lack an intrinsic enzymatic activity and, therefore, utilize the binding of non-receptor protein tyrosine kinases (PTK) for their signal transduction. There are two conserved domains in the ICD of the cytokine receptor family termed “Box 1” and “Box 2” that coordinate activities of the PRLR. The Box 2 motif in these receptors acts as a binding site for members of the Janus kinase (JAK) family of kinases, of which there are four family members, JAK1, JAK2, JAK3, and TYK2 that can bind to specific receptors of this family. Similar to the GHR, the PRLR only interacts with JAK2 and the interaction activates JAK2 to phosphorylate multiple tyrosine residues on the ICD of the PRLR. Upon phosphorylation of the PRLR by JAK2, proteins of the STAT (signal transducer and activator of transcription) family dock to the PRLR and they are then phosphorylated by JAK2. The STAT proteins that interact with the PRLR are STAT5a and STAT5b.
Prolactin secretion increases during pregnancy and promotes breast development in preparation for the production of milk and lactation. Although prolactin serves this important function in breast development during pregnancy there is no evidence to indicate that it functions during normal breast tissue development before or during puberty. During pregnancy the increased production of estrogen enhances breast development but it also suppresses the effects of prolactin on lactation. Following parturition (birth) estrogen (as well as progesterone) levels fall allowing lactation to occur. This is to ensure that lactation is not induced until the baby is born.
The Pancreatic Polypeptide Family
The pancreatic polypeptide (PP) family of hormones comprises two gut hormones, pancreatic polypeptide (PP) and peptide tyrosine-tyrosine (PYY), as well as the central nervous system hormone neuropeptide Y (NPY). Each of these peptide hormones contains 36 amino acids consisting of numerous tyrosines (hence the Y peptides nomenclature) and an α-amidation at the C-terminus. The three-dimensional structure of these hormones includes a hairpin-like motif referred to as the pancreatic polypeptide fold (PP-fold). The PP-fold is required for interaction of the hormones with specific G-protein coupled receptors (GPCRs).
Details of the actions of PP, PYY, and NPY are discussed in the Gut-Brain Interrelationships and the Control of Feeding Behavior page.
Melanin-Concentrating Hormone, MCH
Melanin-concentrating hormone (MCH) was originally identified as a 19 amino acid cyclic peptide that induced the lightening of the skin in fish. Subsequently the peptide was identified in rodents to be over-expressed in response to fasting and also elevated in genetically obese mice (ob/ob mice).
Details of the actions of MCH are discussed in the Gut-Brain Interrelationships and the Control of Feeding Behavior page.
The Orexins
The orexins constitute two neuroendocrine peptides derived from the same gene. These peptides are designated orexin A and orexin B. Details of the actions of the orexins are discussed in the Gut-Brain Interrelationships and the Control of Feeding Behavior page.
Gastrointestinal Hormones and Peptides
There are more than 30 peptides currently identified as being expressed within the digestive tract, making the gut the largest endocrine organ in the body. The regulatory peptides synthesized by the gut include hormones, peptide neurotransmitters and growth factors. Indeed, several hormones and neurotransmitters first identified in the central nervous system and other endocrine organs have subsequently been found in enteroendocrine cells and/or neurons of the gut.
Visit the Table of Major Human Hormones page to see a more complete list of gastrointestinal peptides and hormones.
Many of the gut-derived hormones listed on the following Table are discussed in detail in the Gut-Brain Interrelationships and Control of Feeding Behavior page.
Table of Gastrointestinal Hormones
| Hormone | Location | Major Action |
| Cholecystokinin (CCK) | enteroendocrine I cells predominantly of the duodenum, jejunum | stimulates gallbladder contraction and bile flow; increases secretion of digestive enzymes from pancreas; vagal nerves in the gut express CCK1 (CCKA) receptors |
| Cholesin | NPC1L1 expressing enterocytes | expression and release stimulated by NPC1L1-mediated uptake of dietary cholesterol; when released it binds to GPR146 on hepatocytes leading to repression of SREBP2-mediated activation of cholesterol biosynthesis genes |
| Enterostatin | derived from N-terminal end of pancreatic colipase; pentapeptide human enterostatin contains the sequence: APGPR | regulates fat intake; peripheral or central administration inhibits consumption of a high-fat diet but not a low-fat diet |
| Famsin | 178 amino acids; cleaved from the membrane bound protein encoded by the C17orf78 gene | release is induced by fasting and promotes fasting-induced metabolism upon binding to the olfactory family receptor encoded by the OR10P1 (olfactory receptor family 10 subfamily P member 1) gene |
| FGF19 | gallbladder, duodenum, ileum | member of the large FGF family of growth factors; expression of FGF19 gene activated by transcription factor FXR, FXR is activated when ileal enterocytes absorb bile acids, when released to the portal circulation FGF19 stimulates hepatic glycogen and protein synthesis while inhibiting glucose production; reduces the expression and activity of CYP7A1 which is the rate-limiting enzyme in bile acid synthesis; acts in the gallbladder to induce relaxation and refilling with bile acids |
| Gastrin | made in enteroendocrine G cells of the gastric antrum and duodenum | gastric acid and pepsin secretion; exists in two major forms: little gastrin (17 amino acids) and big gastrin (34 amino acids), both result from a single precursor protein of 101 amino acid; both species contain a Y residue in the C-terminal portion of the protein that may be either sulfated (gastrin II) or nonsulfated (gastrin I); both forms bind to the CCK2 (CCKB) receptor on stomach and gut parietal cells with an affinity equal to that of CCK; the C-terminal tetrapeptide of both gastrins and CCK are identical and possess all the biological activities of both gastrin and CCK |
| Gastrin-releasing peptide (GRP), is a bombesin-related peptide | stomach, duodenum | released from vagal nerve; stimulates release of gastrin from G cells of the stomach and CCK from small intestinal enteroendocrine I cells; binds to a GPCR identified as gastrin-releasing peptide receptor (GRPR); GRPR is also known as bombesin 2 receptor (BB2 or BB2R); bombesin is an antibacterial peptide that was isolated from the skin of the frog, Bombina bombina; in addition to GRP, the peptide neuromedin B (NMB) is a member of the bombesin-like peptide family |
| Ghrelin | primary site is A (X-like) enteroendocrine cells of the stomach oxyntic (acid secreting) glands, minor synthesis in intestine, pancreas and hypothalamus | regulation of appetite (increases desire for food intake); energy homeostasis; glucose metabolism; gastric secretion and emptying, insulin secretion |
| Glucagon-like peptide 1 (GLP-1) | enteroendocrine L cells predominantly in the ileum and colon | potentiates glucose-dependent insulin secretion; inhibits glucagon secretion; inhibits gastric emptying |
| Glucagon-like peptide 2 (GLP-2) | enteroendocrine L cells predominantly in the ileum and colon | enhances digestion and food absorption; inhibits gastric secretions; promotes intestinal mucosal growth |
| Glucose-dependent insulinotropic polypeptide (GIP), originally called gastric inhibitory polypeptide | enteroendocrine K cells of the duodenum and proximal jejunum | inhibits secretion of gastric acid; enhances insulin secretion |
| Motilin | proximal small intestine | initiates inter-digestive intestinal motility; stimulates release of PP; stimulates gallbladder contractions |
| Nesfatin-1 | primarily expressed in enteroendocrine X/A-like cells in stomach and in white adipose tissue | proteolytic product of the 420 amino acid precursor protein encoded by the NUCB2 (nucleobindin 2) gene; following removal of the 24 amino acid signal peptide the 396 amino acid peptide is cleaved by proprotein convertase subtilisin/kexin type 1 (PCSK1) and PCSK2; nesfatin-1 consists of amino acids 1–82; proteolytic processing also generates nesfatin-2 (amino acids 85–163) and nesfatin-3 (amino acids 166–396); stimulates reduced feeding via actions in the hypothalamus |
| Neurotensin | enteroendocrine N cells along the small intestine and proximal large intestine; also in hypothalamus | a 13-amino acid peptide derived from a precursor that also produces neuromedin N (NMN); involved in satiety responses and slows gastric emptying; also involved in nociception (sensation of pain) |
| Obestatin | primary site is stomach, minor synthesis in intestine | derived from pro-ghrelin protein; acts in opposition to ghrelin action on appetite |
| Oxyntomodulin | enteroendocrine L cells predominantly in the ileum and colon | contains all of the amino acids of glucagon (see Figure below); inhibits meal-stimulated gastric acid secretion similar to GLP-1 and GLP-2 action; induces satiety, decreases weight gain, and increases energy consumption; has weak affinity for GLP-1 receptor as well as the glucagon receptor, may mimic glucagon actions in liver and pancreas |
| Pancreatic polypeptide: PP | pancreas F cells, colon and rectum | inhibits pancreatic bicarbonate and protein secretion |
| Peptide tyrosine tyrosine (PYY) | enteroendocrine L cells predominantly in the ileum and colon | reduced gut motility; delays in gastric emptying, and an inhibition of gallbladder contraction; exerts effects on satiety via actions in the hypothalamus |
| Secretin | enteroendocrine S cells of the duodenum and to a lesser extent the jejunum | secreted in response to pH decrease in lumen of duodenum as a result of gastric emptying; increases pancreatic bicarbonate secretion; inhibits gastric secretions; stimulates PP secretion; induces satiety through actions in the ventromedial hypothalamus (VMH) |
| Somatostatin | made by D cells of the gut and δ-cells of the pancreas, also produced in hypothalamus and other organ systems | inhibits release and action of numerous gut peptides, e.g. CCK, OXM, PP, gastrin, secretin, motilin, GIP; also inhibits insulin and glucagon secretion from pancreas |
| Substance P, a member of the tachykinin family that includes neurokinin A (NKA) and neurokinin B (NKB) | entire gastrointestinal tract | CNS function in pain (nociception); involved in vomit reflex; stimulates salivary secretions; induces vasodilation antagonists; have anti-depressant properties |
| Vasoactive intestinal peptide (VIP) | gut, pancreas, brain | 28 amino acid peptide that belongs to the glucagon and secretin superfamily; VIP is closely related to a protein identified as pituitary adenylate cyclase-activating polypeptide type 1 (PACAP); initiates smooth muscle relaxation; stimulates pancreatic bicarbonate secretion; increases glycogen breakdown; within the brain VIP exerts effects on circadian rhythms; exerts effects by binding to three different GPCR identified as VPAC1 [VIP/PACAP (pituitary adenylate cyclase activating peptide 1) receptor type1], VPAC2, and PAC1 (pituitary adenylate cyclase-activating polypeptide type 1 receptor); VPAC1 and VPAC2 are encoded by the VIPR1 and VIPR2 genes, respectively; PAC1 is encoded by the ADCYAP1R1 gene |
Adipose Tissue Hormones and Cytokines
Adipose tissue is not merely an organ designed to passively store excess carbon in the form of fatty acids esterified to glycerol (triglycerides). More complete information on the roles of adipose tissue in overall metabolism and inflammation can be found in the Adipose Tissue: Not Just Fat page.
Mature adipocytes synthesize and secrete numerous enzymes, growth factors, cytokines and hormones that are involved in overall energy homeostasis. Many of the factors that influence adipogenesis are also involved in diverse processes in the body including lipid homeostasis and modulation of inflammatory responses. In addition, a number of proteins secreted by adipocytes play important roles in these same processes. In fact recent evidence has demonstrated that many factors secreted from adipocytes are proinflammatory mediators and these proteins have been termed adipocytokines or adipokines. Members of this class of protein secreted from adipocytes include tumor necrosis factor-alpha (TNFα), interleukin-6 (IL-6) and leptin. Listed in the Table below is only a subset of proteins known to be secreted by adipose tissue and the focus is on those that effect overall metabolic homeostasis and modulate inflammatory processes. As is clear from the Table, not all the proteins are unique to adipose tissue. Details of the structure and function of several proteins follows the Table.
Table of Adipose Tissue Hormones and Adipokines
| Factor | Principal Source | Major Action |
| Adiponectin also called adipocyte complement factor 1q-related protein (ACRP30), and adipoQ | adipocytes | see Adipose Tissue: Not Just Fat page |
| Adipsin (also called complement factor D) | adipocytes, liver, monocytes, macrophages | rate limiting enzyme in complement activation |
| C-reactive protein (CRP) | hepatocytes, adipocytes | is a member of the pentraxin family of calcium-dependent ligand binding proteins; assists complement interaction with foreign and damaged cells; enhances phagocytosis by macrophages; levels of expression regulated by circulating IL-6; modulates endothelial cell functions by inducing expression of various cell adhesion molecules, e.g. ICAM-1, VCAM-1, and selectins; induces MCP-1 expression in endothelium; attenuates NO production by downregulating NOS expression; increase expression and activity of PAI-1 |
| IL-6 | adipocytes, hepatocytes, activated Th2 cells, and antigen-presenting cells (APCs) | acute phase response, B cell proliferation, thrombopoiesis, synergistic with IL-1 and TNF on T cells |
| Leptin | predominantly adipocytes, mammary gland, intestine, muscle, placenta | see Adipose Tissue: Not Just Fat page |
| monocyte chemotactic protein-1 (MCP-1) | leukocytes, adipocytes | is a chemokine defined as CCL2 (C-C motif, ligand 2); recruits monocytes, T cells, and dendritic cells to sites of infection and tissue injury |
| plasminogen-activator inhibitor-1 (PAI-1) | adipocytes, monocytes, placenta, platelets, endometrium | see the Hemostasis: Biochemistry of Blood Coagulation page for more details |
| Resistin | adipocytes, spleen, monocytes, macrophages, lung, kidney, bone marrow, placenta | see Adipose Tissue: Not Just Fat page |
| TNFα | primarily activated macrophages, adipocytes | induces expression of other autocrine growth factors, increases cellular responsiveness to growth factors and induces signaling pathways that lead to proliferation |
Irisin: Exercise-Induced Skeletal Muscle Hormone (Myokine)
Irisin, named after the Greek Goddess Iris, messenger of the Olympian Gods, was discovered in studies aimed at defining mechanisms by which exercise results in improvement in metabolic status in obesity and type 2 diabetes. Exercise has been shown to increase whole body energy expenditure in excess of the calories that are required for the actual work performed. Related studies in mice demonstrated that muscle-specific expression of the transcriptional co-activator PGC-1α results in resistance to age-related obesity and diabetes. PGC-1α was initially identified as a peroxisome proliferator-activated receptor-gamma (PPARγ)-interacting protein from brown adipose tissue (BAT) that was involved in regulating the process of adaptive thermogenesis in response to cold. Subsequent studies determined that the primary function of PGC-1α was to stimulate mitochondrial biogenesis and oxidative metabolism.
The role of exercise in improving metabolic status in tissues other than skeletal muscle suggested that a protein was secreted from exercising muscle and acting on tissues such as adipose tissue and liver. In studies in mice it was found that PGC-1α expression in muscle stimulates an increase in expression of a membrane protein called FNDC5, fibronectin type III domain-containing protein 5. When FNDC5 is expressed in skeletal muscle, in response to PGC-1α activation, it is cleaved and secreted as the hormone (myokine), irisin.
The FNDC5 gene is located on chromosome 1p35.1 and is composed of 7 exons that generate three alternatively spliced mRNAs, two of which encode proteins that likely initiate translation at AUA codons. Evidence indicates that the preproprotein that encodes irisin initiates translation at an in-frame ATG codon that is upstream of the purported alternative AUA start codon, given that deletion of the AUA codon did not reduce the production of irisin.
When cleaved from FNDC5, irisin contains 110 amino acids. The amino acid sequences of irisin are highly conserved with human and mouse proteins being 100% identical. This high amino acid conservation implies a highly conserved function that is likely to be mediated by irisin binding to a cell surface receptor. The identity of an irisin receptor is as yet unknown. The highest basal levels of expression of FNDC5 are seen in brain and heart, with low basal expression in liver, lung, skeletal muscle, and testis.
Irisin expression and secretion is induced in response to exercise and activates profound changes in the subcutaneous white adipose tissue (WAT), stimulating expression of uncoupling protein 1 (UCP1) and results in a broad program of brown fat-like development. These BAT-like cells induced in WAT are most commonly referred to as beige or brite fat cells. Importantly, this causes a significant increase in total body energy expenditure and resistance to obesity-linked insulin resistance. Even moderate increases in serum levels of irisin result in increases in energy expenditure in mice with no changes in physical activity or food intake. Similarly, irisin is induced with exercise in humans. The actions of irisin recapitulate many of the most important benefits of exercise and muscle activity.
The activity of irisin at inducing brown thermogenesis in WAT is quite profound being able to result in increased levels of UCP1 mRNA ranging from 7-500 fold. In addition to induction of UCP1, irisin activates the expression of several other BAT genes such as elongation of very long-chain fatty acids 3 (ELOVL3), cytochrome c oxidase polypeptide 7A1 (COX7A1), and otopetrin 1 (OTOP1). Conversely, genes such as leptin, that are characteristic of white fat development, are downregulated in response to irisin. In addition to the induction of BAT-like cells in WAT, irisin activity is associated with a large increase in oxygen consumption, improvement in glucose tolerance, and a reduction in fasting insulin. These activities of irisin illustrate that the hormone is a significant effector of increased energy expenditure, improved diet-induced insulin resistance, and reduced body weight.
The therapeutic potential of irisin is obvious. Exogenously administered irisin can induce the browning of subcutaneous fat and thermogenesis, and it presumably could be prepared and delivered as an injectable polypeptide. Increased formation of brown or beige/brite fat has been shown to have anti-obesity and antidiabetic effects and adult humans have significant deposits of UCP1-positive brown fat.
Natriuretic Hormones
Natriuresis refers to enhanced urinary excretion of sodium. This can occur in response to specific hormonal signals, in certain disease states, and through the action of diuretic drugs. At least three peptides of the natriuretic hormone family have been identified and are referred to as ANP, BNP, and CNP.
Atrial natriuretic peptide (ANP: also called atrial natriuretic factor, ANF) was the first cardiac natriuretic hormone identified. This hormone is secreted by cardiac myocytes when sodium chloride intake is increased and when the volume of the extracellular fluid expands. Specifically, ANP is released from myocytes in the wall of the right atrium of the heart in response to increased venous volume returning to the heart via the inferior and superior vena cava. Active ANP is a 28-amino acid peptide containing a 17-amino acid ring formed by intrachain disulfide bonding. Two smaller forms of ANP have also been isolated from the brain.
A second natriuretic peptide (originally called BNP since it was first isolated from porcine brain) has been identified and found in human heart and blood (but not human brain). BNP is a 32-amino acid peptide and has different amino acids in its 17-amino acid ring as well as being encoded for by a different gene. Although the acronym BNP is still commonly used, the protein is now known as B-type natriuretic peptide or also ventricular natriuretic peptide (but still with the BNP acronym) since it is secreted by cardiac ventricular myocytes.
In humans, a third natriuretic peptide (CNP) is present in the brain but not in the heart.
The action of ANP is to cause natriuresis presumably by increasing glomerular filtration rate (its exact mechanism of action remains unclear). ANP induces relaxation of the mesangial cells of the glomeruli and thus may increase the surface area of these cells so that filtration is increased. Alternatively, ANP might act on tubule cells to increase sodium excretion. Other effects of ANP include reducing blood pressure, decreasing the responsiveness of adrenal glomerulosa cells to stimuli that result in aldosterone production and secretion, inhibit secretion of vasopressin, and decreasing vascular smooth muscle cell responses to vasoconstrictive agents. These latter actions of ANP are counter to the effects of angiotensin II. In fact, ANP also lowers renin (see next section) secretion by the kidneys thus, lowering circulating angiotensin II levels.
ANP is encoded by the NPPA (natriuretic peptide A) gene. The NPPA gene is located on chromosome 1p36.21 and is composed of 3 exons that encode a 151 amino acid preproprotein. Expression of the NPPA gene occurs primarily in cardiac atrial myocytes but lower levels of expression also occur in the brain, kidney, uterus, and lung. Following synthesis and removal of the signal peptide, the pro-ANP protein is stored in secretory vesicles. Upon stimulated release, the cardiac transmembrane serine protease, corin, cleaves pro-ANP releasing the mature form of ANP (28 amino acids) from the C-terminus of pro-ANP.
BNP is encoded by the NPPB (natriuretic peptide B) gene. The NPPB is located on chromosome 1p36.2 and is composed of 3 exons that encode a 134 amino acid preproprotein. Expression of the NPPB gene occurs primarily in cardiac ventricular myocytes. Like the processing of prepro-ANP, following synthesis of prepro-BNP the signal peptide (25 amino acids) is removed and pro-BNP is stored intracellularly. BNP is carbohydrate modified by O-glycosylation. Biologically active BNP (32 amino acids and referred to as BNP-32) is cleaved from pro-BNP by either corin (as for ANP) or by furin. Furin is a protease that is a member of the proprotein convertase subtilisin/kexin type (PCSK) family of proteases and as such is also identified as PCSK3.
Natriuretic Peptide Receptors
Three different natriuretic peptide receptors have been identified. These receptors are ANPR-A (encoded by the NPR1 gene), ANPR-B (encoded by the NPR2 gene), and ANPR-C (encoded by the NPR3 gene). The natriuretic peptide receptors are members of the larger family of single transmembrane spanning guanylate cyclase enzymes. As such the NPR1 encoded protein is also known as GUCY2A or GC-A and the NPR2 encoded protein is also known as GUCY2B or GC-B.
The NPR1 gene is located on chromosome 1q21.3 and is composed of 22 exons that encode a 1061 amino acid precursor protein.
The NPR2 gene is located on chromosome 9p13.3 and is composed of 27 exons that encode a 1047 amino acid precursor protein.
The NPR3 gene is located on chromosome 5p13.3 and is composed of 13 exons that generate three alternatively spliced mRNAs, each of which encode a distinct protein isoform.
Expression of the NPR1 gene is highest in adipocytes of adipose tissue but is also high in kidney, adrenal glands, vascular endothelium, and heart. Expression of the NPR2 gene is highest smooth muscle with the next highest levels of expression being found in the brain (predominantly the pituitary), and endometrial tissue. ANP and BNP are the natural ligands for the ANPR-A receptor and bind with relatively equivalent affinity and elicit the same responses. CNP is the ligand for the ANPR-B receptor. The function of the ANPR-C protein is to serve as a clearance receptor removing natriuretic peptides from the blood via receptor-mediated endocytosis.
When ANP or BNP bind to receptor, an increase in the intrinsic guanylate cyclase activity results leading to production of cyclic GMP (cGMP). Within vascular smooth muscle cells the increased cGMP exerts numerous effects resulting in smooth muscle relaxation and vasodilation. Activation of cGMP-dependent protein kinase (specifically PKGIα) leads to phosphorylation of regulatory subunits of myosin light chain phosphatases (specifically protein phosphatase 1 regulatory subunit 12A which is encoded by the PPP1R12A gene) leading to enhanced removal of phosphates from myosin light chains and reduced contractile activity. The activity of cGMP is also direct in that it inhibits smooth muscle plasma membrane L-type Ca2+ channels resulting in reduced intake of Ca2+ and, as a consequence, reduced activation of myosin light chain kinases furthering vasodilation.
Renin-Angiotensin System
The renin-angiotensin system (RAS; also commonly called the renin-angiotensin-aldosterone system, RAAS) is responsible for regulation of blood pressure. Bioactive angiotensin (angiotensin II) is derived from the precursor protein, angiotensinogen, predominantly produced by the liver. Angiotensinogen is derived from the AGT gene which is located on chromosome 1q42.2 and is composed of 5 exons that encode a 485 amino acid preproprotein. Renin is a protease/hormone produced by the kidneys and is responsible for cleavage of angiotensinogen initiating the production of bioactive angiotensin II. The renin gene (symbol: REN) is located on chromosome 1q32.1 and is composed of 10 exons that encode a 406 amino acid preproprotein. Following removal of the leader peptide from preprorenin, functional renin is released from prorenin by an as yet unidentified renal protease. Given that the circulating levels of angiotensinogen and angiotensin converting enzyme (ACE, see below) are in excess, the release of renin from the kidney represents the rate limiting step in the RAAS.
The intra-renal baroreceptor system is a key mechanism for regulating renin secretion. A drop in blood pressure results in the release of renin from juxtaglomerular cells (JG cells; also called granular cells) which are specialized smooth muscle cells in the wall of the afferent arterioles at the base of the glomerulus in the juxtaglomerular apparatus. These are the only cells in the human body that synthesize and secrete renin. Renin secretion is also regulated by the rate of Na+ and Cl– transport across the apical (tubular lumen side) membrane of epithelial cells of the macula densa. The kidney macula densa is a cluster of specialized epithelial cells found in the nephron just distal to the loop of Henle.
The transporter responsible for the sensing of Na+ and Cl– flux is the apical (tubular lumen side) membrane-localized NKCC2 (Na+-K+-Cl– cotransporter 2) transporter which is encoded by the SLC12A1 gene. The higher the rate of transport of these ions the lower the rate of renin secretion. The only enzymatic function for renin (an aspartyl protease) is to cleave a 10-amino acid peptide from the N-terminal end of angiotensinogen. This cleaved decapeptide is called angiotensin I. Recent evidence has demonstrated that prorenin and renin can exert effects, unrelated to the enzymatic activity of renin, through binding to a specific receptor expressed within the vasculature and the glomerulus of the kidney (see below).
Angiotensin I is then cleaved by the action of angiotensin-converting enzyme (ACE: a membrane-bound Zn2+-dependent dicarboxypeptidase) generating the bioactive octapeptide hormone, angiotensin II (Asp-Arg-Val-Tyr-Ile-His-Pro-Phe). The function of ACE is to remove two amino acids from the C-terminal end of angiotensin I. Angiotensin-converting enzyme is highly expressed by vascular endothelial cells, renal proximal tubular epithelial cells, ciliated intestinal epithelial cells, lung epithelia, and developing male germ cells. Expression is also found in several areas of the brain and in the choroid plexus. When monocytes differentiate to macrophages, and when dendritic cells are immunologically activated, expression of ACE is induced. Unlike renin which has a single substrate, ACE exhibits activity to a wide range of substrates ranging in size from tripeptides to proteins of 42 amino acids. The major substrates for ACE are angiotensin I and bradykinin, both of which are involved in the regulation of blood pressure.
Angiotensin II can be cleaved by another membrane-bound Zn2+-dependent peptidase (glutamyl aminopeptidase, also known as aminopeptidase A), which removes one amino acid from the N-terminal end generating the heptapeptide hormone angiotensin III (Arg-Val-Tyr-Ile-His-Pro-Phe). The vasopressive activity of angiotensin III is similar, but less potent, than angiotensin II and is exerted by binding to the same receptors to which angiotensin II binds. Like angiotensin II, angiotensin III exerts effects leading to increased blood pressure. Whereas, angiotensin II exerts its effects equivalently on several target tissues, angiotensin III is most potent via actions within the central nervous system. Indeed, evidence indicates that angiotensin III is the primary effector of the renin-angiotensin system in the control of blood pressure via the central sympathetic nervous system.
As indicated above, the renin gene encodes a 406 amino acid preproprotein. Removal of the signal peptide (20 amino acids) and the pro portion (46 amino acids) of prorenin results in the production of the 340 amino acid active form of the enzyme. Several alternative mRNAs are generated from the REN gene via alternative promoter usage and via alternative splicing. However, the function of these alternative mRNAs is not fully defined. The angiotensin-converting enzyme gene (symbol: ACE) is located on chromosome 17q23.3 and is composed of 26 exons that generate three alternatively spliced mRNAs, each of which generate a distinct ACE isoform. ACE isoform 1 is referred to as the endothelial or somatic form and is a 1306 amino acid precursor. The ACE isoform 2 is referred to as the testicular form and is a 732 amino acid precursor.
Angiotensin II was originally identified as hypertensin and angiotonin. It is one of the most potent naturally occurring vasoconstrictors. In addition to its effects on the vasculature, angiotensin II (and its derivative angiotensin III) acts on the kidneys to increase renal tubular Na+ and Cl– reabsorption, water retention, and K+ excretion, on the posterior pituitary to induce the release of vasopressin (anti-diuretic hormone, ADH), stimulates the zona glomerulosa cells of the adrenal cortex to secrete aldosterone, and exerts effects in the central nervous system resulting in increased sympathetic outflow from the rostral ventrolateral medulla of the brain stem.
The vasoconstrictive action of angiotensin II is primarily exerted on the arterioles and leads to a rise in both systolic and diastolic blood pressure. It is this action of angiotensin II on blood pressure that led to the development of a class of drugs called the ACE inhibitors for use as anti-hypertensive drugs. As the name implies, ACE inhibitors prevent ACE from converting angiotensin I to angiotensin II. All of the drugs that inhibit the activity of ACE contain the suffix “–pril“, e.g. captopril. One side-effect of the use of ACE inhibitors is a persistent dry cough leading to lack of compliance with the drug in a number of patients. The cause of the dry cough is due to reduced degradation of bradykinin, a known substrate for ACE. Although bradykinin is primarily involved in the contractile activity of vascular smooth muscle, it also induces constriction of the bronchioles in the lungs and it is this latter activity that contributes to the dry cough with the use of ACE inhibitors.
In individuals that are depleted of sodium or who have liver disease (e.g. cirrhosis), the pressive actions of angiotensin II are greatly reduced. These conditions lead to increased circulating levels of angiotensin II which in turn leads to a down-regulation in the numbers of angiotensin II receptors on smooth muscle cells. As a consequence, administration of exogenous angiotensin II to these individuals has little effect. In addition to the other physiological responses to angiotensin II indicated, angiotensin II affects the contractility of the mesangial cells of the kidney leading to decreased glomerular filtration rate. One additional effect of angiotensin II is to potentiate the release of norepinephrine from adrenal medullary chromaffin cells.
Angiotensin Receptors
Two distinct types of angiotensin II receptors have been identified, AT1R and AT2R. The AT1R is a classical serpentine G-protein coupled receptor (GPCR). The AT1R is coupled to both a Gq-protein that leads to activation of PLCβ, and a Gi-protein that inhibits adenylate cyclase. The AT1R is expressed in the heart, blood vessels, kidney, adrenal cortex, lung and brain. The AT1R protein is encoded by the AGTR1 gene located on chromosome 3q24. The AT2 receptor is also a GPCR and is coupled to activation of Gi-proteins. Expression of the AT2R is limited, in the adult, to the brain (predominantly the cerebellum), adrenal gland, and myometrium. Within the brain the primary AT2R ligand is angiotensin III.
Due primarily to the development of a dry hacking cough with the use of ACE inhibitors, drugs that block the activity of the AT1R were developed for the treatment of hypertension. This class of drug is called the angiotensin receptor blocker (ARB) class. All of the ARB drugs are AT1R antagonists and thus, exert their anti-hypertensive effects at the level of the receptor itself. All drugs in the ARB class contain the suffix “–sartan“. In addition to their use in the treatment of hypertension the ARBs are used to treat diabetic nephropathy and congestive heart failure.
Non-Enzymatic Functions of Renin
As indicated above, renin (as well as prorenin) can exert biological effects that are unrelated to its role as a protease. A distinct receptor for prorenin and renin has been identified that upon renin or prorenin binding elicits a range of signal transduction events. The prorenin/renin receptor is encoded by the ATP6AP2 (ATPase H+ transporting accessory protein 2) gene. The renin receptor is a truncated form of the ATP6AP2 encoded protein that co-localizes with the V-ATPase (vacuolar H+-ATPase) present in several cells types including cardiac myocytes and kidney epithelial cells.
In addition to the regulation of V-ATPase activity in the heart and kidney, recent evidence has shown that the prorenin/renin receptor is coupled to signaling events of both the canonical and non-canonical Wnt pathways. Both of these Wnt signaling pathways are essential for adult and embryonic stem cell functions and for embryonic development. The role of the prorenin/renin receptor in the canonical Wnt/β-catenin pathway is as an adapter between the LRP5/6 protein and the frizzled protein that constitute the Wnt receptor complex. The precise linking mechanism involves the V-ATPase in the endosomal membrane whose activity is required for phosphorylation of LRP5/6 and activation of β-catenin. In addition, the prorenin receptor binds to the frizzled protein controlling its asymmetrical subcellular distribution, and as a result, the polarization of cells tissues which represents the non-canonical Wnt pathway.
Angiotensin Converting Enzyme 2 (ACE2) and Vasodilation
Another ACE-related enzyme, identified as angiotensin converting enzyme 2 is involved in the RAAS but the consequences of its activity are vasodilation, not vasoconstriction. The ACE2 gene is located on the X chromosome (Xp22.2) and is composed of 21 exons that encode a 805 amino acid precursor protein. Like ACE, ACE2 is a protease (a monocarboxyl peptidase) whose substrates include angiotensin II and other angiotensin peptides. With respect to angiotensin II, the activity of ACE2 represents a metabolic pathway responsible for limiting the extent of angiotensin II activity. When ACE2 cleaves a single amino acid from the C-terminus of angiotensin II the resulting peptide is referred to as angiotensin-(1-7). Angiotensin-(1-7) exerts vasodilating effects by binding to, and activating, the MAS1 receptor. The MAS1 receptor is a member of the G-protein coupled receptor (GPCR) superfamily of receptors. In addition to vasodilation, the activation of MAS1 receptor by angiotensin-(1-7) results in natriuresis which contributes to reduction in blood pressure. The ACE2-mediated degradation of angiotensin II also leads to the production of angiotensin-(1-9) which is inactive and thus, results in reduction in blood pressure independent of the actions of angiotensin-(1-7) acting via the MAS1 receptor.
ACE2 exerts effects in tissues other than the kidney and heart which are involved in the regulation of homeostatic and/or immune surveillance mechanisms. ACE2 activity has been shown to be required for normal gestational development given that elevations in angiotensin II levels in the placenta result in defects in fetal growth. ACE2 functions in the CNS as well by regulating oxidative stress in the paraventricular nucleus and rostral ventrolateral medulla, thereby, regulating sympathetic nervous system activity. Disturbances in ACE2 activity have been implicated in a variety of pathologies including diabetic nephropathy, hypertension, atherosclerosis, and heart failure.
Bradykinin
Bradykinin is a 9-amino acid vasoactive peptide that induces vasodilation and increases vascular permeability. Activated tissue kallikrein cleaves lysyl-bradykinin (also called kallidin) from LMWK. Lysyl-bradykinin is bradykinin with a lysine residue at the N-terminus making it a 10-amino acid vasoactive peptide.
The details of bradykinin synthesis and biological activity are covered in the Hemostasis: Biochemistry of Blood Coagulation page.
Endothelins
The human endothelins are a family of three 21 amino acid peptides that exert an array of effects within and outside of the cardiovascular system. The original activity, that is now known as endothelin-1 (ET-1), was initially referred to as an endothelium-derived constricting factor. This activity was discovered after is was found that a peptide-like substance, produced in cultured bovine aortic endothelial cells, could induce constriction of isolated blood vessels. The endothelin peptide purified from cultured porcine aortic endothelial cells was used for amino acid sequencing and subsequent isolation of cDNA clones. Following the characterization of the original endothelin cDNA several related sequences were characterized.
Endothelin Genes and Proteins
In humans there are three primary genes that encode the functional 21 amino acid endothelins that are identified as ET-1, ET-2, and ET-3. All three endothelin proteins form two intramolecular disulfide bonds between four cysteine residues. ET-1 represents that predominant endothelin expressed in humans as well as the predominant endothelial cell produced endothelin. Endothelin-1 is the most potent naturally produced vasoconstricting molecule exerting dramatic contractile actions in both arteries and veins. Endothelin-2 was originally identified by its ability to contract isolated mouse ileum and so was referred to as vasoactive intestinal contractor, VIC.
The ET-1 protein is encoded by the EDN1 gene that is located on chromosome 6p24.1 and is composed of 9 exons that generate two alternatively spliced mRNAs encoding preproproteins of 212 amino acids (isoform 1) and 211 amino acids (isoform 2). The ET-2 protein is encoded by the EDN2 gene that is located on chromosome 1p34.2 and is composed of 6 exons that generate two alternatively spliced mRNAs encoding preproproteins of 178 amino acids (isoform 1) and 145 amino acids (isoform 2). The ET-3 protein is encoded by the EDN3 gene that is located on chromosome 20q13.32 and is composed of 6 exons that generate five alternatively spliced mRNAs, each of which encode a distinct preproprotein isoform. The mature 21 amino acid forms of ET-1 and ET-2 differ by two amino acids, positions 6 and 7. In ET-1 amino acid 6 is Leu and 7 is Met, whereas in ET-2 those positions are Trp and Leu, respectively. The mature ET-3 protein differs from ET-1 at six amino acids, positions 2, 4, 5, 6, 7, and 14.
The endothelins are all synthesized as precursor preproproteins requiring proteolytic cleavage to the active 21 amino acid forms. The signal sequences are removed in the ER via the action of signal peptidase. The primary intermediate protein of 39 amino acids, called “big endothelin”, is produced from all three proendothelins via the action of members of the furin family of proteases which are members of the proprotein convertase subtilisin/kexin type (PCSK) family.
The big-endothelins are then cleaved to their active 21 amino acid forms by one of two endothelin converting enzymes identified as ECE-1 and ECE-2. Both ECE-1 and ECE-2 belong to the type II membrane-bound zinc metalloprotease family of enzymes. Big endothelin-3 has also been shown to be a substrate for the protease identified as Kell metallo-endopeptidase. The ECE-1 enzyme is encoded by the ECE1 gene which is located on chromosome 1p36.12 and is composed of 23 exons that generate four alternatively spliced mRNAs, each o which encode a distinct protein isoform The ECE-2 enzyme is encoded by the ECE2 gene which is located on chromosome 3q27.1 and is composed of 19 exons that generate three alternatively spliced mRNAs, each of which encode a distinct protein isoform.
Endothelin Receptors
The endothelins exert their effects by interacting with specific receptors that are members of the G-protein coupled receptor (GPCR) family. There are two human endothelin receptors identified as ETA (also designated ETA) and ETB (also designated ETB) which are encoded by the EDNRA and ENDRB genes, respectively. The ETA and ETB receptors share 63% amino acid sequence identity. Endothelin-1 (ET-1) and ET-2 bind to the ETA receptor with equal affinity which is greater than the affinity for ET-3. All three endothelins bind to the ETB receptor with equal affinity. The ETA and ETB receptors are both coupled to Gq-type G-proteins such that following endothelin-1 binding there is activation of PLCγ and subsequent increases in DAG, IP3, and intracellular Ca2+.
The EDNRA gene is located on chromosome 4q31.22–q31.23 and is composed of 9 exons that generate three alternatively spliced mRNAs, each of which encode a distinct protein isoform.
The EDNRB gene is located on chromosome 13q22.3 and is composed of 9 exons that generate four mRNAs through the use of alternative promoters and alternative splicing. The ETB isoform 1 precursor protein is 442 amino acids and is encoded by two distinct mRNAs that utilize different promoters, one of which is specific to the placenta.
Although the big-endothelins are found in the blood, and they contain the 21 amino acids of the mature endothelins, they do not bind to either endothelin receptor at physiological concentration. Both the ETA and ETB receptors are subject to various post-translational modifications that include palmitoylation, phosphorylation, and glycosylation. Hirschsprung disease (absence of ganglionic nerve fibers in the colon) is a multigenic disorder where mutations in the EDNRB gene play a causative role. Waardenburg syndrome, which is associated with sensorineural hearing loss, hypopigmentation of skin and hair, and pigmentary disturbances of the irises, is also linked to mutations in the EDNRB gene as well as with mutations in the EDN3 gene which encoded endothelin-3 (ET-3).
Expression of the endothelin receptor genes is ubiquitous having been found in virtually all tissues and organs receiving a blood supply. Expression of ETA is highest in vascular smooth muscle and expression of ETB is highest in endothelial cells. The highest level of ETA expression is in the heart and lungs while expression in the brain is quite low. In contrast, the highest level of expression of ETB is in the brain with the lungs also having high ETB expression. ETB receptors are also found in the heart, with abundant levels seen in the conduction system of the atrioventricular node (AV node).
Endothelin Expression and Functions
Expression of ET-1 predominates in vascular endothelial cells and represents the most abundant isoform of endothelin in the human cardiovascular system. Additional sites of ET-1 expression include epithelial cells in the lungs, kidney, and colon, in leukocytes such as macrophages and monocytes, in peripheral enteric glia cells, and cells of the central nervous system such as certain neurons and glial cells. Expression of ET-1 in vascular endothelial cells occurs in all types of vessels including large conduit arteries, resistance arteries, large veins, and venules. Synthesis and release of ET-1 occurs continuously from vascular endothelial cells with changes in the levels of released protein being primarily regulated at the level of transcriptional control.
In contrast to the ubiquitous expression of ET-1, the expression of ET-2 is primarily found the vasculature, heart, pituitary, medulla oblongata, lung, kidney, intestine, and ovaries. Pharmacological investigations have demonstrated that ET-1 and ET-2 are highly similar and both peptides are produced and released from some of the same cell types, however, ET-1 and ET-2 exhibit distinct physiological and potentially pathophysiological roles. These differences are believed to be due to differential gene expression as well as to differences in peptide synthesis.
Expression of endothelin-3 (ET-3) is mainly localized to neurons and glia of the neostriatum, hypothalamic nuclei, hippocampus, and Purkinje cells of the cerebellum and medulla oblongata. Human endothelial cells do not expression the EDN3 gene. As indicated above in the discussion of endothelin receptors, ET-3 exhibits differential receptor affinity being highest for ETB. Since the expression of ETB is highest in the brain, and because of the predominant expression of the EDN3 gene in the brain, it has been proposed that ET-3 is the brain endothelin.
Regulated expression of the EDN1 gene has been the most extensively studied of the three endothelin genes. Transcriptional regulation of the EDN1 gene has been shown to be exerted by several different transcription factors which includes hypoxia-induced factor 1 (HIF-1), GATA-binding factor 2 (GATA2), activator protein 1 (AP-1), nuclear factor-kappa B (NF-κB), forkhead box protein O1 (FOXO1), and vascular endothelial zinc finger 1 (VEZF1). Numerous stimuli can result in alterations in the expression level of the EDN1 gene including both mechanical (e.g. shear stress) and metabolic (chemical) processes. Insulin signaling in endothelial cells leads to repressed EDN1 expression via PKB/AKT-mediated effects on the activity of VEZF1. Conversely glucose effects on EDN1 expression are stimulatory via effects on the activity of NF-κB. Hypoxic conditions, as well as ethanol, stimulate EDN1 expression through activation of HIF-1 activity. Additional mediators of EDN1 expression include various steroids (e.g. glucocorticoids) and peptides such as angiotensin II, cytokines, leptin, and transforming growth factor beta (TGFβ), and numerous additional metabolic molecules such as fatty acids (e.g. oleic acid).
Parathyroid Hormone (PTH)
Parathyroid hormone (PTH) is synthesized and secreted by chief cells of the parathyroid glands in response to systemic Ca2+ levels. There are four parathyroid glands that lie adjacent to the thyroid gland and consist of two superior glands and two inferior glands. The PTH gene is located on chromosome 11p15.3 and consists of 4 exons that generate two alternatively spliced mRNAs. The isoform 1 encoding mRNA directs the synthesis of a 115 amino acid preproprotein. In this PTH mRNA exon 1 is untranslated, exon 2 encodes a 25-amino acid signal peptide and part of the prohormone, and exon 3 encodes the remaining part of the prohormone (6 amino acids) and the whole biologically active 84 amino acid (molecular weight of 9300 Daltons) PTH protein. The isoform 2 encoding mRNA directs the synthesis of a protein starting from an alternate 5′ exon and, therefore, an alternate translation start codon. Whether the isoform 2 PTH protein is processed to a functional hormone is unclear. As PTH is synthesized it is processed through the ER and Golgi apparatus where first the signal peptide is removed and then the propeptide, with the active PTH molecule, is stored in dense neuroendocrine-type granules.
There exists a related protein identified as PTH-related protein (PTHrP). PTHrP was identified originally as a protein causing severe hypercalcemia in patients with pheochromocytoma (PCC) which is a rare malignancy of the chromaffin cells of the adrenal medulla. The normal functions of PTHrP include roles in fetal bone development where it suppresses the maturation of chondrocytes so that the onset of hypertrophic differentiation during endochondral bone growth is delayed. In addition PTHrP exhibits antiproliferative effects in adults by regulating epidermal and hair follicle cell growth as well as inhibiting angiogenesis.
CaSR Role in the Secretion of PTH
Secretion of PTH from the parathyroid gland is controlled via the activity of the Ca2+ sensing receptor (CaSR). The CaSR is expressed in the parathyroid glands, renal tubule cells, bone marrow, thyroid gland C-cells, gastrin-secreting cells in the stomach, several areas of the brain, as well as in several other tissues.
The CaSR is a G-protein coupled receptor (GPCR) that responds to small changes in circulating Ca2+ concentrations. When initially characterized, the CaSR was the first receptor shown to be activated by a ligand that was an ion. The CaSR belongs to the class C GPCR family.
The Ca2+ sensing receptor is encoded by the CASR gene which is located on chromosome 3q13.33–q21.1 and is composed of 11 exons that generate two alternatively spliced mRNAs that encode two protein isoforms. The CaSR is coupled to the activation of Gi, Gq, and G12-type G-proteins resulting in decreased production of cAMP and increased production of DAG and and IP3. Expression of the CASR gene predominates in the parathyroid gland and the kidneys but is also expressed in the intestines, the brain, the pancreas, and bone.
The rate of PTH secretion is controlled by the interaction of Ca2+ with the CaSR. The synthesis and secretion of PTH from chief cells is constitutive, but Ca2+ regulates the level of PTH in chief cells (and thus its secretion) by increasing the rate of PTH proteolysis when plasma Ca2+ levels rise and by decreasing the proteolysis of PTH when Ca2+ levels fall.
The role of PTH is to regulate Ca2+ concentration in extracellular fluids. The feedback loop that regulates PTH secretion therefore involves the parathyroid gland, extracellular Ca2+, and the PTH target tissues.
Role of the CaSR in Protein Sensing and Glucose Tolerance
PTH Receptors
PTH acts by binding to cAMP- and PLCβ-activating plasma membrane receptors, initiating a cascade of reactions that culminates in the biological response. There are two receptors that recognize PTH identified as the PTH-1 and PTH-2 receptors (PTH1R and PTH2R). The PTH1R gene is located on chromosome 3p21.31 spanning 32kbp and composed of 20 exons generate two alternatively spliced mRNAs, both of which encode the same 585 amino acid protein. The PTH2R gene is located on chromosome 2q34 and is composed of 14 exons that generate two alternatively spliced mRNAs both of which encode distinct protein isoforms. Both receptors are related to a small sub-family of peptide hormone receptors that includes the receptors for ACTH, calcitonin, vasoactive intestinal peptide (VIP), and secretin.
The PTH receptors are coupled to Gs-type G-proteins that activate adenylate cyclase resulting in increased cAMP and consequent activation of PKA, as well as Gq-type G-proteins that activate PLCβ resulting in hydrolysis of membrane PIP2 releasing IP3 and DAG. The IP3 stimulates release of intracellular Ca2+ stores and DAG activates PKC. The PTH-1 receptor is activated by both PTH and PTHrP, whereas, the PTH-2 receptor is activated only by PTH. The PTH-1 receptor is found predominantly in bone and kidney. The body response to PTH is complex but is aimed in all tissues at increasing Ca2+ levels in extracellular fluids. PTH induces the dissolution of bone by indirectly stimulating osteoclast activity (through initial activation of osteoblasts which express the PTH-1 receptor), which leads to elevated plasma Ca2+ and phosphate. In the kidney, PTH reduces renal Ca2+ clearance by stimulating its reabsorption. At the same time, PTH reduces the reabsorption of phosphate and thereby increases its clearance. Finally, PTH acts on the liver, kidney, and intestine to stimulate the production of the steroid hormone 1,25-dihydroxycholecalciferol (calcitriol), which is responsible for enhancing Ca2+ absorption in the intestine.
PTH Functions in the Kidney
With respect to the kidneys and overall calcium homeostasis, the major regulator is PTH. Within the nephron of the kidney calcium reabsorption from the glomerular filtrate occurs in several locations including the proximal tubule, the thick ascending limb (TAL) of the loop of Henle, the distal convoluted tubule (DCT), and in the connecting tubule (CNT). Within the proximal tubule and the TAL the reabsorption of calcium occurs via passive paracellular (between cells) transport mechanisms due to concentration gradients and electrochemical gradients established by various ion transporters. In these regions of the nephron PTH has no direct effects on calcium movement from the tubular lumen to the blood. However, hyperparathyroidism results in decreased sodium transport in the proximal tubule and TAL which exerts a secondary negative effect on calcium transport in these segments. The result of these effects of hyperparathyroidism is hypercalciuria and nephrolithiasis (kidney stones).
The primary sites for direct effects of PTH, within the kidney, are the DCT and the CNT. The ability of PTH to exert an increase in calcium reabsorption in these regions of the distal nephron is due to altered activity of the major calcium reabsorption transporter in these segments identified as transient receptor potential vanilloid 5, TRPV5. Reabsorption of calcium via TRPV5 leads to activation of the calcium-binding protein calmodulin. Calmodulin then binds to cytosolic sites in the C-terminus of TRPV5 which results in inactivation of the transporter.
PTH activation of the PTH-1 receptor (PTH1R) in cells of the DCT and CNT results in activation of PKA through the associated Gs-type G-protein. PKA activation results in the phosphorylation of numerous sites on TRPV5 resulting in a loss of calmodulin binding and a consequent increase in calcium reabsorption by TRPV5. In addition, PTH action in the DCT and CNT leads to increased TRPV5 abundance in the apical membrane allowing for more calcium reabsorption. This latter effect of PTH is exerted through the action of PKC which is activated by the Gq-type G-protein associated with the PTHR1.
One of the major intracellular calcium-binding proteins in the duodenum of humans that is involved in movement of calcium from the apical to the basolateral membrane for eventual transport to the blood is calbindin-D9K (encoded by the S100G gene; also called CALB3). A related protein, calbindin-D28K (encoded by the CALB1 gene) is expressed in the DCT of the nephron. The action of PTH in the DCT results in increased levels of calbindin-D28K allowing for more reabsorbed calcium to be transported to the basolateral membrane for efflux to the blood.
Within the kidneys PTH is also critical for the regulation of phosphate homeostasis. Phosphate reabsorption occurs exclusively in the proximal tubule where approximately 80% of the phosphate in the glomerular filtrate is reabsorbed. What is not reabsorbed appears in the urine indicating that the distal regions of the nephron do not contribute to phosphate reabsorption. The major phosphate transporters of the proximal tubule are encoded by the SLC34A1 and SLC34A3 genes which encode the Na+-phosphate cotransporter 2a (NPT2a) and NPT2c proteins.
Within the proximal tubule the PTH-1 receptor is expressed on both the apical and basolateral membranes of the epithelial cells of the tubule. The basolateral membrane PTH-1 receptor binds the PTH present in the blood circulating through the peritubular capillaries. The apical membrane PTH-1 receptor has been shown to bind small fragments of PTH, that are still biologically active, filtered in the glomerulus. The signal transduction cascade initiated by basolateral membrane PTH-1 receptors involves a Gs-type G-protein and the activation of PKA. The apical membrane PTH-1 receptor activates a signaling cascade involving a Gq-type G-protein and the activation of PKC.
The ability of the PTH-1 receptor to activate both PKA and PKC pathways is controlled by its interaction with the proteins identified as Na+-H+exchanger regulator factor 1 (NHERF1) and NHERF2. When the PTH-1 receptor is associated with either of these proteins it activates the PKC pathway and when these factors are not associated with the receptor it activates the PKA pathway. Both NHERF1 and NHERF2 are exclusively localized to the apical membrane which explains the difference in PKC versus PKA activation at the different membranes. Regardless of basolateral or apical membrane localization, PTH-1 receptor activation, and the resultant increase in kinase activity, results in inhibition of Na+-dependent phosphate reabsorption in the proximal tubule.
The necessity for PTH to simultaneously activate Ca2+ reabsorption and inhibit phosphate (principally the HPO42– form) reabsorption is to ensure that the Ca2+ remains in the ionized state. In the blood, calcium is distributed between the protein-bound form (35%-50%, primary protein is albumin), that complexed with phosphate and organic acids (10%), and the ionized (free Ca2+) form. During hypocalcemic conditions the level of ionized calcium in the blood can drop significantly before the other bound forms. In the presence of adequate phosphate ion in the blood, any increased uptake of calcium into the blood would most likely form insoluble salts with the phosphate, thereby, restricting cellular access to the ionized Ca2+that is needed in the hypocalcemic state. For this reason the PTH-mediated inhibition of renal reabsorption of phosphate, in conjunction with increased Ca2+ reabsorption, ensures that the absorbed calcium remains in the ionized state in the blood.
PTH Functions in Bone
PTH exerts both catabolic and anabolic effects in bone which are dependent upon the type of bone and the specific cell types responding to PTH binding its receptor. Parathyroid hormone (PTH) stimulates osteoclast formation by binding to its receptor on stromal/osteoblastic cells and stimulating the production of Receptor Activator of NFKappaB Ligand (RANKL) and inhibiting the expression of RANKL decoy receptor, osteoprotegerin (OPG).
PTH binds to PTH1R on cells of the osteoblastic lineage including osteoprogenitor cells, lining cells, immature osteoblasts, mature osteoblasts, and osteocytes. The binding of PTH stimulates a variety of factors resulting in increased proliferation of mesenchymal stem cells, such that these cells are committed into the osteoblast lineage. In addition, the actions of PTH result in enhanced osteoblast differentiation leading to new bone matrix production and ultimately mineralization of bone tissue. Osteoblastic cells also produce RANKL whose function is to stimulate osteoclast production and function. Osteoblasts also produce the soluble RANKL decoy receptor, osteoprotegerin (OPG). PTH enhances production of RANKL and inhibits production of OPG leading to increased osteoclastic bone resorption. Bone resorption leads to the release of Ca2+ and Pi as a result of the degradation of hydroxyapatite, the major mineral component of bone. The actions of PTH can result in the stimulation of bone turnover leading to either a net increase in bone formation or a net increase in bone resorption.
The primary function of PTH is to respond to reduced circulating Ca2+ levels and to stimulate resorption of bone Ca2+. However, recombinant PTH has proven to be of significant benefit in the treatment of osteoporosis. Although PTH increases bone resorption it also increases the formation of new bone which is the more pronounced response to PTH. Intermittent infusion of recombinant PTH (teriparatide) results in new bone formation and has shown efficacy in the treatment of the bone loss associated with osteoporosis. This PTH-induced phenomenon occurs as a result of the laying down of protein matrix and mineralization that occurs not only in the previous existing matrix, but in the new matrix that is formed. Patients receiving teriparatide show an increase in bone density as well as a decrease in fractures when compared to other forms of treatment. Whereas, continuous elevation of PTH levels, as occurs in humans due to abnormal secretion from the parathyroid glands, results in loss of bone mass leading to osteoporosis, intermittent elevation of PTH that occurs with the daily injections of teriparatide has the opposite effect of building bone.
Calcitonin Family
Calcitonin
Calcitonin is a 32-amino acid peptide secreted by C cells of the thyroid gland. Calcitonin is a hypocalcemia-inducing peptide that exerts its effects in numerous species by antagonizing the effects of PTH. In humans, however, the role of calcitonin in calcium homeostasis is of limited physiological significance. The circulating levels of calcitonin are low and extreme variations in these levels have not been associated with disruptions in calcium or phosphate homeostasis in humans.
There are two human calcitonin genes identified as α and β. The α gene (symbol: CALCA) is located on chromosome 11p15.2 spanning 6.5kbp and is composed of 5 exons. The β gene (symbol: CALCB) is located on the same chromosome location as the CALCA gene and it is also composed of 5 exons. Transcription of the CALCA gene yields three different mRNAs as a result of alternative splicing, two of which encode the same protein and the third encodes a distinct protein with a distinct biological activity.
The two different CALCA gene derived proteins are calcitonin and the neuropeptide, calcitonin gene-related protein (CGRP, also identified as α-CGRP). Exon 1 of the CALCA gene is a non-coding exon and exons 2–4 encode calcitonin. The CGRP mRNA is composed of exons 1–3, 5 and 6. The CALCB gene encodes β-CGRP (also identified as CGRP-2) mRNA which is translated into CGRP in the central nervous system. The CGRP produced from the CALCB gene exhibits cardiovascular effects, neurotransmitter functions, and may serve a role in early development.
In non-human species, calcitonin exerts its hypocalcemia-inducing effects primarily through inhibition of osteoclast-mediated bone resorption. Calcitonin has been shown to reduce the synthesis of osteopontin (OPN, also referred to as secreted phosphoprotein 1, SPP1), a protein made by osteoclasts and responsible for attaching osteoclasts to bone. Secondarily, calcitonin affects serum Ca2+ levels by the stimulation of renal Ca2+ clearance. These effects of calcitonin are the result of the interaction of the hormone with a specific receptor identified as the CTR. The CTR is closely related to the PTHR and the secretin receptors which together, as described above for PTH, form a distinct subfamily of GPCR. The CTR gene (symbol: CALCR) is located on chromosome 7q21.3.
In humans the major benefits of calcitonin are its use in the treatment of osteoporosis and to suppress bone resorption in Paget disease. Paget disease (osteitis deformans) is a disorder of bone remodeling that results in accelerated rates of bone turnover and disruption of normal bone architecture. The naturally occurring calcitonins vary in amino acid sequence between species. The salmon calcitonin is 10–100 times more potent than mammalian calcitonins in lowering serum calcium levels and because of this activity it is used therapeutically such as in the treatment of Paget disease. The other medically significant fact related to human calcitonin is its use as a biomarker for sporadic and inherited forms of thyroid medullary cancers in humans.
Calcitonin Gene-Related Protein: CGRP
As indicated above, CGRP (α-CGRP) is encoded by one of the alternatively spliced mRNAs derived from the CALCA gene. A second CGRP (β-CGRP or CGRP-2) is derived from the CALCB gene. CGRP has recently been shown to play a role in the metabolic changes associated with aging. These effects are exerted via an interactive pathway involving pain receptors of the transient receptor potential cation channel subfamily V (vanilloid) member 1 (TRPV1) pain receptors. TRPV1 expressing neurons form a dense network innervating the pancreas. Stimulation of these receptor induces the release of substance P and CGRP. Within the pancreas, substance P induces neurogenic inflammation while CGRP antagonizes insulin release. When the TRPV1 gene is knocked out in mice there is a significant decrease in CGRP release from neurons in the pancreas resulting in insulin secretion and the promotion of metabolic health. These TRPV1 knock-out mice are long-lived and display the metabolic profiles of more youthful mice even when they are older. These results suggest that inhibition of specific pain response pathways and/or antagonism of CGRP signaling within the pancreas could hold promise in the treatment of age related metabolic disturbances.
Antagonism of the CGRP receptor has also been found to ameliorate the symptoms of migraine headaches. Several mechanisms for CGRP in migraine headaches have been proposed including dilation of cerebral and dural blood vessels, release of inflammatory mediators from mast cells, and transmission of nociceptive information from intracranial blood vessels to the nervous system. Several small molecule receptors antagonists and monoclonal antibodies against the CGRP receptor have been developed for the treatment of migraines. Currently approved monoclonal antibodies against the CGRP receptor that can be prescribed to migraine sufferers are erenumab (Aimovig™), fremanezumab (Ajovy™), and galcanezumab (Emgality™). Small molecule CGRP receptor antagonists that are approved for use include rimegepant (Nurtec ODT™) and ubrogepant (Ubrelvy™)
Calcitonin Peptide Family Receptors
In addition to calcitonin and CGRP, the calcitonin family of peptides includes amylin, adrenomedullin (AM), and adrenomedullin 2 (AM2, commonly known as intermedin). These peptides all interact with receptor complexes that contain the calcitonin receptor at their core. The two G protein-coupled receptors (GPCR) which are receptors for these peptides are the calcitonin receptor (encoded by the CALCR gene) and the calcitonin receptor-like receptor (CRLR; encoded by the CALCRL gene). These belong to the sub-family of GPCR known as the secretin-like or class B family of GPCR (see the Signal Transduction Pathways: G-Proteins and GPCR page for more information on GPCR classes).
CALCR and CRLR proteins can form complexes with members of the membrane protein family called the receptor activity-modifying proteins (RAMP), which consists of RAMP1, RAMP2, and RAMP3 in humans. RAMP are type I transmembrane proteins composed of a large N-terminal extracellular domain, a single α-helical transmembrane domain, and a short intracellular domain. The RAMP regulate receptor pharmacology, receptor signaling, and receptor trafficking.
RAMP association with CALCR or with CRLR generates multiple distinct receptor subtypes with different specificities for the calcitonin peptide family. CALCR together with RAMP1 forms the CGRP receptor. In contrast, the two adrenomedullin (AM) receptors, AM1 and AM2, are formed by CALCR and RAMP2 or RAMP3, respectively. Interaction of each of the three RAMPs with CALCR form the amylin receptors described above.