
Last Updated: May 26, 2026
Introduction to Oxidative Phosphorylation
The large quantity of NADH that results from cytosolic glycolysis and the NADH and FADH2 that results from mitochondrial fatty acid oxidation and the TCA cycle is used to supply the energy for ATP synthesis via oxidative phosphorylation. The oxidation of NADH and FADH2, with phosphorylation of ADP to form ATP, are processes supported by the mitochondrial electron transport assembly and ATP synthase, which are integral protein complexes of the inner mitochondrial membrane (IMM).
The electron transport assembly is comprised of a series of protein complexes that catalyze sequential oxidation-reduction reactions; some of these reactions are thermodynamically competent to support ATP production via ATP synthase provided a coupling mechanism, such as a common intermediate, is available. Proton translocation and the development of a transmembrane proton gradient provides the required coupling mechanism.
Principles of Reduction/Oxidation (Redox) Reactions
Redox reactions involve the transfer of electrons from one chemical species to another. The oxidized plus the reduced form of each chemical species is referred to as an electrochemical half cell. Two half cells having at least one common intermediate comprise a complete, coupled, redox reaction. Coupled electrochemical half cells have the thermodynamic properties of other coupled chemical reactions. If one half cell is far from electrochemical equilibrium, its tendency to achieve equilibrium (i.e., to gain or lose electrons) can be used to alter the equilibrium position of a coupled half cell. An example of a coupled redox reaction is the oxidation of NADH by the electron transport chain:
NADH + ½O2 + H+ → NAD+ + H2O
The thermodynamic potential of a chemical reaction is calculated from equilibrium constants and concentrations of reactants and products. Because it is not practical to measure electron concentrations directly, the electron energy potential of a redox system is determined from the electrical potential or voltage of the individual half cells, relative to a standard half cell. When the reactants and products of a half cell are in their standard state and the voltage is determined relative to a standard hydrogen half cell (whose voltage, by convention, is zero), the potential observed is defined as the standard electrode potential, Eo. If the pH of a standard cell is in the biological range, pH 7, its potential is defined as the standard biological electrode potential and designated Eo′. By convention, standard electrode potentials are written as potentials for reduction reactions of half cells. The free energy of a typical reaction is calculated directly from its Eo′ by the Nernst equation as shown below, where n is the number of electrons involved in the reaction and F is the Faraday constant (23.06 kcal/volt/mol or 94.4 kJ/volt/mol):
ΔG°′ = –nFΔEo′
For the oxidation of NADH, the standard biological reduction potential is –52.6 kcal/mole. With a free energy change of –52.6 kcal/mole, it is clear that NADH oxidation has the potential for driving the synthesis of a number of molecules of ATP since the standard free energy for the reaction below is +7.3kcal/mole:
ADP + Pi → ATP
Classically, the description of ATP synthesis through oxidation of reduced electron carriers indicated three moles of ATP could be generated for every mole of NADH and two moles for every mole of FADH2. However, direct chemical analysis has shown that for every two electrons transferred from NADH to oxygen, 2.5 equivalents of ATP are synthesized and 1.5 for FADH2. Despite these new measurements many texts still use the historical values of 3 moles of ATP per mole of NADH and 2 moles of ATP per mole of FADH2. As such these values will be utilized in this page.
Complexes of the Electron Transport Chain
NADH and FADH2 are oxidized by a series of catalytic redox carriers that are integral protein complexes of the inner mitochondrial membrane, IMM. The free energy change in several of these steps is very exergonic. Coupled to these oxidation-reduction steps is a transport process in which protons (H+) from the mitochondrial matrix are translocated to the space between the inner and outer mitochondrial membranes. Specifically, the organization of the electron transport chain (ETC) complexes and ATP synthase is within the portions of the IMM that form cristae (specialized folded domains of the IMM) such that the H+ are not free to diffuse throughout the whole of the inner membrane space.
The redistribution of protons leads to formation of a proton gradient (ΔpH) across the inner mitochondrial membrane. The magnitude (“size”) of the gradient is proportional to the free energy change of the electron transfer reactions. The result of these reactions is that the redox energy of NADH and FADH2 is converted to the energy of the proton gradient. In the presence of ADP, protons flow down their thermodynamic gradient from the inner membrane space of the mitochondrion back into the mitochondrial matrix. This process is facilitated by a proton carrier in the inner mitochondrial membrane known as ATP synthase (also called complex V). As its name implies, this carrier is coupled to ATP synthesis.
Electron flow through the mitochondrial electron transport assembly is carried out through several enzyme complexes identified as complexes I, II, III, and IV. The ATP synthase complex is sometimes also called complex V. Electrons enter the transport chain primarily from mitochondrial NADH and FADH2 generated during the oxidation of pyruvate, fatty acids, and amino acids. The origin of cytosolic NADH is from the pathway of glycolysis and the electrons in cytosolic NADH are transported into the mitochondria via the actions of the malate-aspartate shuttle (to mitochondrial NADH) or by the glycerol phosphate shuttle (to mitochondrial FADH2). The liver, heart, and kidney predominantly utilize the malate-aspartate shuttle, whereas, adipose tissue, skeletal muscle, and brain predominantly utilize the glycerol phosphate shuttle.
Diagrammatic Representation of Mitochondrial Electron FLow
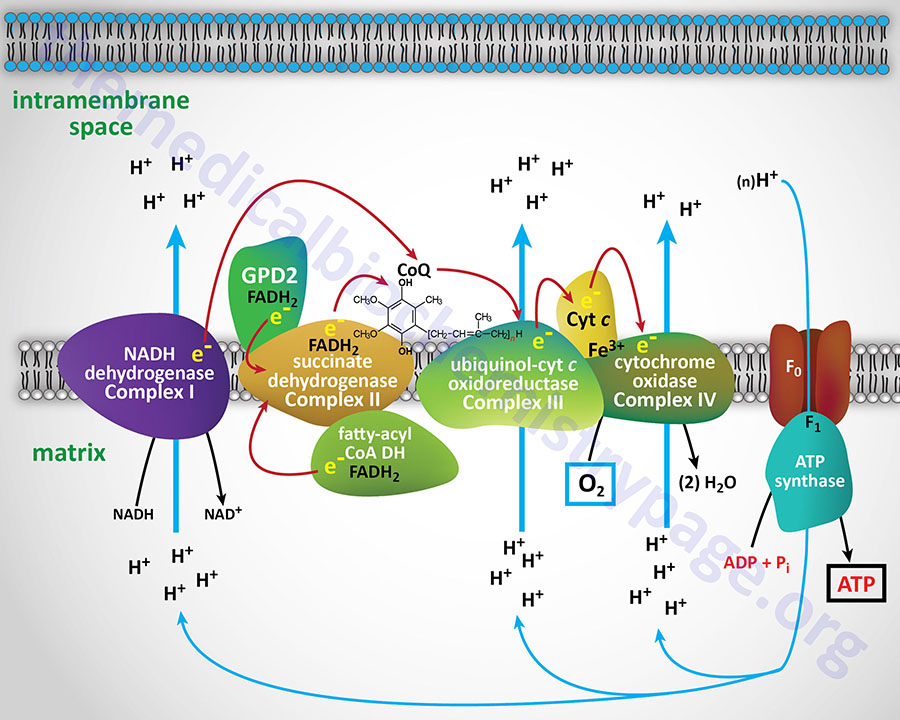
With the exception of NADH, succinate, and CoQ, all of the components of the pathway are integral proteins of the inner mitochondrial membrane whose cofactors undergo redox reactions. NADH and succinate are soluble in the mitochondrial matrix, while CoQ is a small, mobile carrier that transfers electrons between the primary dehydrogenases of complexes I and II and cytochrome b. Although possessing mobility, CoQ is also restricted to the membrane phase because of the highly hydrophobic character of the polyisoprene portion of the molecule.
As shown in the Figure above, the mitochondrial electron transport proteins are clustered into multiprotein complexes known as complexes I, II, III, and IV. Within complex I the seven protein subunits that comprise the NADH dehydrogenase activity are all encoded by the mitochondrial genome. The remaining subunits are all encoded within the nuclear genome. All of the subunits of complex II are encoded by the nuclear genome. The cytochrome b of complex III is the only mitochondrial genome encoded subunit of this complex. Within complex IV the three subunits comprising cytochrome c oxidase are each encoded by the mitochondrial genome with the remaining subunits encoded within the nuclear genome. All but two of the subunits of ATP synthase are encoded by the nuclear genome while the ATP synthase 6 and ATP synthase 8 subunits are encoded by the mitochondrial genome.
In addition to the core protein subunits of each of the complexes of oxidative phosphorylation, there are numerous assembly factors required to ensure correct formation of each complex. The importance of the assembly factors in functional formation of these complexes can be demonstrated by the mitochondrial encephalomyopathies that result due to mutations in several genes. For example, GRACILE syndrome (Growth Retardation, Amino aciduria, Cholestasis, Iron overload, Lactic acidosis, and Early death) is caused by mutations in the BCS1L gene which is required for proper assembly of complex III. The BCS1L (BCS1-like) gene is so-called because it is the human homolog of the yeast bsc1 (biogenesis of cytochrome sc1 complex) gene. The BCS1L encoded protein is a member of the AAA family of ATPases.
Complex I
Complex I, most commonly called NADH dehydrogenase (also known as NADH:CoQ oxidoreductase or NADH-ubiquinone oxidoreductase), is composed of NADH dehydrogenase with FMN as cofactor, plus non-heme iron proteins having at least one iron sulfur (Fe-S) center. Complex I is responsible for transferring electrons from NADH to CoQ. The ΔEo′ for the latter transfer is 0.42V, corresponding to a ΔGo′ of –19 kcal/mol of electrons transferred. With its highly exergonic free energy change, the flow of electrons through complex I is more than adequate to drive ATP synthesis.
There are a total of 45 protein subunits in complex I. Of the 45 proteins 38 are derived from the nuclear genome and 7 are derived from the mitochondrial genome (mtDNA). The organization of complex I is such that a portion of the complex is embedded in the inner mitochondrial membrane and the rest of the complex protrudes into the mitochondrial matrix.
The dehydrogenase activity of the complex is responsible for the oxidation of NADH to NAD+ and contains the NDUFV1, NDUFV2, and NDUFS1 subunits (where the ND designation refers to NADH Dehydrogenase). The dehydrogenase activity of the complex transfers electrons to ubiquinone (CoQ10) and is composed of the NDUFS2, NDUFS3, NDUFS7, and NDUFS8 subunits. The proton translocation module of complex I is composed of, at a minimum, all seven of the mtDNA encoded proteins.
Deficiencies in complex I proteins are the most common causes of mitochondrial diseases in humans and these disorders encompass a wide degree of heterogeneity in pathological presentation. The infantile presenting complex I disorders include Leigh syndrome, fatal infantile lactic acidosis (FILA), neonatal cardiomyopathy, leukoencephalopathy, pure myopathy, and combined hepatopathy and tubulopathy.
Due to the composition of complex I being the result of both mtDNA and nuclear DNA encoded proteins, disorders that are associated with defects in complex I proteins are divided accordingly. The complexity and heterogeneity of complex I disorders can be emphasized by the constellation of disorders termed MELAS, mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes. MELAS is not the result of a single gene defect and as such represents a constellation of related disorders that can result from defects in several of the mitochondrial genome encoded proteins of complex I.
Complex II
Complex II which is most commonly called succinate dehydrogenase (the TCA cycle enzyme) is also known as succinate:CoQ oxidoreductase or succinate:ubiquinone oxidoreductase). Complex II is composed of four protein subunits and all four subunits are integral membrane proteins in the inner mitochondrial membrane. All four subunits of complex II are encoded by the nuclear genome and the genes are SDHA, SDHB, SDHC, and SDHD.
The ΔEo′ for electron flow through complex II is about 0.05 V, corresponding to a ΔGo′ of –2.3 kcal/mol of electrons transferred, which is insufficient to drive ATP synthesis. The difference in free energy, of electron flow through complexes I and II, accounts for the fact that a pair of electrons originating from NADH and passing to oxygen supports production of 2.5 (more commonly reported as 3) equivalents of ATP, while two electrons from FADH2 (at complex II) support the production of only 1.5 (more commonly reported as 2) equivalents of ATP.
Complex III
Reduced CoQ (CoQH2) diffuses in the lipid phase of the membrane and donates its electrons to complex III, whose principal components are the heme proteins known as cytochromes b (encoded by the MT-CYB gene) and c1 (encoded by the CYC1 gene) and a non-heme-iron protein, known as the Rieske iron sulfur protein (encoded by the UQCRFS1 gene). Complex III is known as ubiquinol-cytochrome c oxidoreductase or as CoQ-cytochrome reductase and often as the cytochrome bc1 complex. Complex III is composed of 11 protein subunits, all of which, excepting cytochrome b, are encoded by the nuclear genome.
The MT-CYB, CYC1, and UQCRFS1 encoded proteins constitute the electron transfer center of complex III. In contrast to the heme of hemoglobin and myoglobin, the heme iron of all cytochromes participate in the cyclic redox reactions of electron transport, alternating between the oxidized (Fe3+) and reduced (Fe2+) forms.
The electron carrier from complex III to complex IV is the smallest of the cytochromes, cytochrome c (molecular weight 12,000; encoded by the CYCS gene). The release of cytochrome c from the mitochondria to the cytosol is the major trigger of the mitochondria-induced apoptosis pathway.
Another important protein in the functioning of complex III is the assembly factor encoded by the BCS1L gene (BCS1-like: the human homolog of a yeast gene identified as BCS1). Mutations in the BCS1L gene result in the lethal disorder called GRACILE syndrome (GRACILE: Growth Retardation, Aminoacidurina, Cholestasis, Iron overload, Lactic acidosis, Early death). Another disorder that results from mutations in the BCS1L gene and the MT-CYB gene is known as Björnstad syndrome. One form of Leigh syndrome also results from mutations in the BCS1L gene.
Complex IV
Complex IV, also known as cytochrome c oxidase (COX), contains the hemeproteins known as cytochrome a and cytochrome a3, as well as two copper-containing proteins in which the copper undergoes a transition from Cu+ (cuprous) to Cu2+ (cupric) during the transfer of electrons through the complex to molecular oxygen. The copper centers of complex IV are termed CuA and CuB. The copper centers of complex IV are critical in the reduction of the oxidized iron (ferric iron: Fe3+) of cytochrome c which was oxidized by the electrons from complex III. The Fe3+ iron of cytochrome c is the site of cyanide (CN–) inhibition of complex IV.
Complex IV is composed of a total of 13 protein subunits. Oxygen is the final electron acceptor, with water being the final product of oxygen reduction. The catalytic core of complex IV is composed of the mitochondrial genome (mtDNA) encoded proteins MT-CO1 and MT-CO2 as well as cytochrome a and cytochrome a3. Another mtDNA encoded protein, MT-CO3, is a structural protein in the complex. The other 10 subunits of complex IV are encoded by the nuclear genome and are identified as COX genes (COX4, COX5A, COX5B, COX6A, COX6B, COX6C, COX7A, COX7B, COX7C, and COX8). The COX6A and COX7A genes encode tissue-specific isoforms (COX6A1 and COX6A2; COX7A1 and COX7A2) with the H (heart)-type being expressed in heart and skeletal muscle (COX6A2 and COX7A1) and the L (liver)-type being expressed in non-muscle tissues (COX6A1 and COX7A2). There are two isoforms of the COX4 protein encoded by the COX4I1 and COX4I2 genes. There is a testis-specific isoform of COX6B (identified as COX6B2). Mutations in the COX genes are the most common causes of defects in oxidative-phosphorylation.
Normal oxidation of NADH or succinate is always a 2-electron reaction, with the transfer of two hydride ions to a flavin. A hydride ion is composed of one proton and one electron. Unlike NADH and succinate, flavins can participate in either 1-electron or 2-electron reactions; thus, flavin that is fully reduced by the dehydrogenase reactions can subsequently be oxidized by two sequential 1-hydride reactions. The fully reduced form of a flavin is known as the quinol form and the fully oxidized form is known as the quinone form; the intermediate containing a single electron is known as the semiquinone or semiquinol form.
Like flavins, CoQ (also known as ubiquinone) can undergo either 1- or 2-electron reactions leading to formation of the reduced quinol, the oxidized quinone, and the semiquinone intermediate. The ability of flavins and CoQ to form semiquinone intermediates is a key feature of the mitochondrial electron transport systems, since these cofactors link the obligatory 2-electron reactions of NADH and succinate with the obligatory 1-electron reactions of the cytochromes.
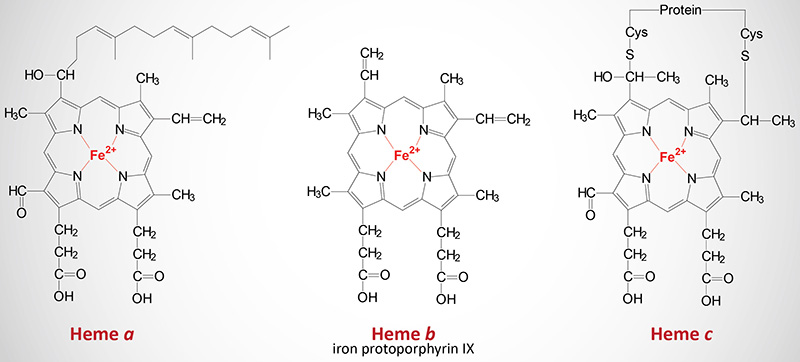
The cytochromes are heme proteins. Like hemoglobin and myoglobin, the cytochromes generally contain one heme group per polypeptide, except for cytochrome b which has two heme residues in one polypeptide chain. There are three forms of heme found in heme proteins, each of which are derived from iron protoporphyrin IX. Cytochromes vary in the structure of the heme and in its binding to apoprotein. Cytochromes of the c type contain a modified iron protoporphyrin IX known as heme c. In heme c the two vinyl (–C=C–) side chains are covalently bonded to cysteine sulfhydryl residues of the apoprotein. Only cytochromes of the c type contain covalently bound heme. Heme a is also a modified iron protoporphyrin IX. Heme a is found in cytochromes of the a type and in the chlorophyll of green plants.
Oxidative Phosphorylation and ATP Synthesis
The free energy available as a consequence of transferring two electrons from NADH or FADH2 to molecular oxygen is –57 kcal/mol and –36 kcal/mol, respectively. Oxidative phosphorylation captures this energy via the synthesis of the high-energy phosphate of ATP. In order for oxidative phosphorylation to proceed, two principal conditions must be met. First, the inner mitochondrial membrane (IMM) must be physically intact so that protons can only re-enter the matrix of the mitochondrion by a process coupled to ATP synthesis. Second, a proton gradient must be developed across the inner mitochondrial membrane.
The energy of the proton gradient is known as the chemiosmotic potential, or proton motive force (PMF). This potential is the sum of the concentration difference of protons (ΔpH) across the IMM and the difference in electrical charge (ΔΨ) across this membrane. Recent studies have found that complex I transports sodium ions (Na+) into the cristae formed by the IMM and that these Na+ contribute to the generation of the ΔΨ component of the PMF.
The two electrons from NADH generate a 6-proton gradient. Thus, oxidation of one mole of NADH leads to the availability of a PMF with a free energy of about –31.2 kcal (6 x –5.2 kcal). The energy of the gradient is used to drive ATP synthesis as the protons are transported back down their thermodynamic gradient into the matrix of the mitochondrion.
ATP Synthase (Complex V)
Protons return to the mitochondrion through the integral membrane and matrix -associated protein complex known as ATP synthase (or complex V). ATP synthase is a member of the F-type ATPase family of transporters.
ATP synthase is a multiple subunit complex that forms two functional domains identified as the F0 and the F1 domains. The F1 domain resides in the matrix of the mitochondria and the F0 domain is located in the inner mitochondrial membrane. The F0 complex is composed of a ring structure called the c-ring that consists of at least eight copies of the same protein (the C proteins).
Additional components of the F0 complex (one copy each) are the subunits identified as A(a), B1(b1), D(d), F6, and O (commonly identified as oligomycin sensitivity-conferring protein, OSCP) that collectively form the peripheral stalk of the complex. Additional protein subunits of the F0 complex include the E(e), F2(f), G(g), and A6L proteins that each span the inner mitochondrial membrane. All of the nuclear genes that encode components of the ATP synthase complex are members of the ATPase 5 family.
There are three genes in humans that encode c-ring C proteins. The ATP5G1 (also known as ATP5MC1) gene encodes the C1 protein, the ATP5MC2 (also known as ATP5G2) gene encodes the C2 protein, and the ATP5MC3 (also known as ATP5G3) gene encodes the C3 protein.
The B1 subunit is encoded by the ATP5PB (also known as ATP5F1) gene.
The D subunit is encoded by the ATP5PD (also known as ATP5H) gene.
The F6 subunit is encoded by the ATP5PF (also known as ATP5J) gene.
The O subunit (OSCP) is encoded by the ATP5PO (also known as ATP5O) gene.
The E subunit is encoded by the ATP5ME (also known as ATP5I) gene.
The F2 subunit (sometimes identified as the f subunit) is encoded by the ATP5MF (also known as ATP5J2) gene.
The G subunit is encoded by the ATP5MG (also known as ATP5L) gene. Another gene, ATP5MGL (also known as ATP5L2), encodes a G subunit related protein identified as G2.
Two of the subunits of the Fo complex are encoded by mitochondrial genes, the A and A6L subunits. The A subunit is encoded by the MT-ATP6 gene and the A6L subunit is encoded by the MT-ATP8 gene.
The F1 complex is composed of three copies of both the α and β subunits and one copy of the γ, δ, and ε subunits.
The α subunit is encoded by the ATP5F1A (also known as ATP5A1) gene.
The β subunit is encoded by the ATP5F1B (also known as ATP5B) gene.
The γ subunit is encoded by the ATP5F1C (also known as ATP5C1) gene.
The δ subunit is encoded by the ATP5F1D (also known as ATP5D) gene.
The ε subunit is encoded by the ATP5F1E (also known as ATP5E) gene.
In addition to the F0/F1 core complex proteins there are several regulatory, assembly, and associated proteins that are fundamental to the synthesis of ATP by ATP synthase. During the normal process of mitochondrial respiration (oxidative phosphorylation) there is a high proton-motive force, PMF. The PMF is composed of a strong proton (ΔpH) and electrical (ΔΨ) gradient across the inner mitochondrial membrane that favors ATP synthesis. Under conditions of compromised mitochondrial function, the PMF falls below the threshold required for ATP synthesis and the activity of ATP synthase can actually reverse such that it hydrolyzes ATP to pump protons back into the inner membrane space.
Since depletion of ATP can result in cell death there must be a mechanism to prevent excess reverse activity of ATP synthase. The regulatory function is carried out by the associated protein identified as inhibitor protein, IF1. The function of IF1 to inhibit ATP synthase activity operates in a pH-dependent manner, being most active at a pH below 6.5. The activity of IF1 protects cells during periods of ischemia where O2 levels fall leading to a reduced proton gradient. IF1 functions as a homodimer which binds to two ATP synthase complexes through interactions with the F1 β and γ subunits.
Another important ATP synthase regulatory protein is called factor B. Factor B interacts with the F0 complex on the matrix side of the inner mitochondrial membrane. The function of factor B is to block leaking of protons across the inner mitochondrial membrane. The ATP synthase assembly factors ATP11 and ATP12 are required to protect the hydrophobic surfaces of the unassembled α and γ subunits of the F1 complex, preventing them from aggregating within the mitochondrial matrix. Another ATP synthase associated protein is called TMEM70 (transmembrane protein 70) which maintains the normal activity level of ATP synthase.
As described earlier, the process of electron transport through complexes I–IV is coupled to the transport of protons (H+) from the matrix to the intermembrane space. This proton transport results in the establishment of an energy gradient (proton motive force, PMF) which is used to power the synthesis of ATP from ADP and inorganic phosphate (Pi) as the protons flow down the gradient back into the matrix through the ATP synthase complex.
In order to synthesize ATP, the ATP synthase complex must have access to both ADP and Pi within the matrix. Once synthesized the ATP must be transported from the matrix of the mitochondrion to the cytosol in exchange for more ADP. The transport of ADP and ATP across the inner mitochondrial membrane occurs through the action of the ADP/ATP carriers (AAC) which are typical antiporters. The required Pi is transported into the matrix via the action of a phosphate transporter.
Humans express three ADP/ATP carriers which are all members of the SLC family of membrane transporters, specifically SLA25A4, SLC25A5, and SLC25A6. The mitochondrial phosphate transporter is also a member of the SLC family of transporters, specifically SLC25A3.
The F1 domain of the ATP synthase complex binds ADP and Pi at its catalytic site inside the matrix. The energy of the proton gradient flowing in the forward direction from the intermembrane space through the Fo domain of the complex is coupled to the synthesis of ATP. The approximate number of protons (H+) required for ATP synthase to produce one mole of ATP is three. Due to the fact that the energy of one mole of H+ is consumed in the course of ADP and Pi transport, the net ratio of H+ to ATP is four to one (4:1).
The rate of ATP synthesis is controlled by the level of ADP within the matrix of the mitochondrion. Since the conversion of ADP to ATP requires O2 as the terminal electron acceptor of electron transport, there is a direct association between ADP concentration and O2 consumption. For this reason ADP is said to exert a direct effect on the control of respiration.
Stoichiometry of Oxidative Phosphorylation
For each pair of electrons originating from NADH, three (precise measurements demonstrate that it is approximately 2.5) moles of ATP are synthesized, requiring 22.4 kcal of energy. Thus, with 31.2 kcal of available energy, it is clear that the proton gradient generated by electron transport contains sufficient energy to drive normal ATP synthesis.
Electrons from FADH2 (e.g. succinate oxidation) have about ⅔ the energy of NADH electrons: they generate a PMF that is about ⅔ as great as NADH electrons and lead to the synthesis of only two (precise measurements demonstrate that it is approximately 1.5) moles of ATP per mole of FADH2 oxidized.
The total energy available in the electrons carried by NADH and FADH2 is not utilized for the synthesis of ATP with the residual energy being released as heat. This heat energy from oxidative metabolism is what ensures that warm blooded animals, such as humans, remain at the same internal temperature, regardless of the external temperature.
Mitochondrial Lactate Metabolism: The Lactate Shuttle
Lactate, regardless of where it is generated, can be taken up by numerous other tissues where it can be metabolized. The use of lactate for energy has been proposed to occur within the mitochondria where it can be oxidized to pyruvate by mitochondrial LDH which localizes to the inner mitochondrial membrane. Under conditions of normal metabolism, mitochondrial LDH slowly reduces pyruvate to lactate which allows for electrons in NADH to be transferred to pyruvate, forming lactate. This process removes electrons from the matrix, and possibly from the cell via lactate efflux to the blood via the action of plasma membrane localized monocarboxylate transporters (MCT). Under conditions of hypoxia, the low levels of oxygen lead to accumulation of NADH and under these conditions mitochondria can increase the oxidation of NADH via the LDH-mediated reduction of pyruvate.
The SLC16A1 gene, that encodes MCT1, is expressed in multiple tissues. MCT1 is responsible for both influx and efflux of lactate as well as for transport of lactate into the mitochondria and the peroxisomes. When present in the inner mitochondrial membrane, MCT1 colocalizes with the chaperone protein identified as CD147 which is encoded by the BSG [basigin (Ok blood group)] gene. CD147 is also called EMMPRIN (extracellular matrix metalloproteinase inducer).
The SLC16A7 gene, that encodes MCT2, is expressed primarily in skeletal muscle, brain, and blood. MCT2 is responsible for efflux of lactate. MCT2 is also responsible for transport of lactate into the peroxisomes.
The SLC16A8 gene, that encodes MCT3, is expressed predominantly in the retina and epithelial cells of the eye. MCT3 is responsible for efflux of lactate.
The SLC16A3 gene, that encodes MCT4, is expressed primarily in the intestines, kidney, skeletal muscle, heart, and lungs. MCT4 is responsible for efflux of lactate.
Metabolism of lactate has been proposed to support mitochondrial respiration even in the absence of mitochondrial import of cytoplasmic pyruvate. Lactate was originally thought to be transported into mitochondria primarily via the actions of the MCT1 transporter. However, recent studies (2026) have shown that the mitochondrial pyruvate carrier (MPC) complex is involved in the mitochondrial export of lactate. The MPC is a heterotetrameric complex composed of subunits encoded by the MPC1 and MPC2 genes. The MPC transporter proteins are members of the large superfamily of solute carrier (SLC) proteins and as such MPC1 is also identified as SLC54A1 and MPC2 is also identified as SLC54A2.
Within the mitochondria, lactate can be oxidized to pyruvate by a complex referred to as the mitochondrial lactate oxidation complex, mLOC. The mLOC is composed of the monocarboxylate transporters, MCT1 and MCT4 (and possibly MCT2), the lactate dehydrogenase isoforms, LDHA and LDHB, cytochrome oxidase (complex IV of oxidative phosphorylation), and a scaffolding chaperone basignin (previously identified as CD147). The presence, and the function, of the mLOC reflects what is referred to as the lactate shuttle, a process whereby the generation and metabolism of lactate is not restricted to the dogmatic paradigm of anaerobic glycolysis.
Regulation of Oxidative Phosphorylation
Since electron transport is directly coupled to proton translocation, the flow of electrons through the electron transport system is regulated by the magnitude of the PMF. The higher the PMF, the lower the rate of electron transport, and vice versa. Under resting conditions, with a high cell energy charge, the demand for new synthesis of ATP is limited and, although the PMF is high, flow of protons back into the mitochondria through ATP synthase is minimal.
When energy demands are increased, such as during vigorous muscle activity, cytosolic ADP rises and is exchanged with intramitochondrial ATP via the transmembrane adenine nucleotide carrier ADP/ATP translocase. Increased intramitochondrial concentrations of ADP cause the PMF to become discharged as protons pour through ATP synthase, regenerating the ATP pool. Thus, while the rate of electron transport is dependent on the PMF, the magnitude of the PMF at any moment simply reflects the energy charge of the cell. In turn the energy charge, or more precisely ADP concentration, normally determines the rate of electron transport by mass action principles.
The rate of electron transport is usually measured by assaying the rate of oxygen consumption and is referred to as the cellular respiratory rate. The respiratory rate is known as the state 4 rate when the energy charge is high, the concentration of ADP is low, and electron transport is limited by ADP. When ADP levels rise and inorganic phosphate is available, the flow of protons through ATP synthase is elevated and higher rates of electron transport are observed; the resultant respiratory rate is known as the state 3 rate. Thus, under physiological conditions mitochondrial respiratory activity cycles between state 3 and state 4 rates.
Inhibitors of Oxidative Phosphorylation
The pathway of electron flow through the electron transport assembly, and the unique properties of the PMF, have been determined through the use of a number of important antimetabolites. Some of these agents are inhibitors of electron transport at specific sites in the electron transport assembly, while others stimulate electron transport by discharging the proton gradient. For example, antimycin A is a specific inhibitor of cytochrome b. In the presence of antimycin A, cytochrome b can be reduced but not oxidized. As expected, therefore, cytochrome c remains oxidized in the presence of antimycin A, as do the downstream cytochromes a and a3.
An important class of antimetabolites are the uncoupling agents exemplified by 2,4-dinitrophenol (DNP). Uncoupling agents act as lipophilic weak acids, associating with protons on the exterior of mitochondria, passing through the membrane with the bound proton, and dissociating the proton on the interior of the mitochondrion. These agents cause maximum respiratory rates but the electron transport generates no ATP, since the translocated protons do not return to the interior through ATP synthase.
Table of Inhibitors of Oxidative Phosphorylation
| Name | Function | Site of Action |
| Rotenone | e– transport inhibitor | Complex I |
| Amytal | e– transport inhibitor | Complex I |
| Antimycin A | e– transport inhibitor | Complex III |
| Cyanide | e– transport inhibitor | Complex IV |
| Carbon Monoxide | e– transport inhibitor | Complex IV |
| Azide | e– transport inhibitor | Complex IV |
| 2,4,-dinitrophenol | Uncoupling agent | transmembrane H+ carrier |
| Pentachlorophenol | Uncoupling agent | transmembrane H+ carrier |
| Oligomycin | Inhibits ATP synthase | OSCP fraction of ATP synthase |
Energy from Cytosolic NADH
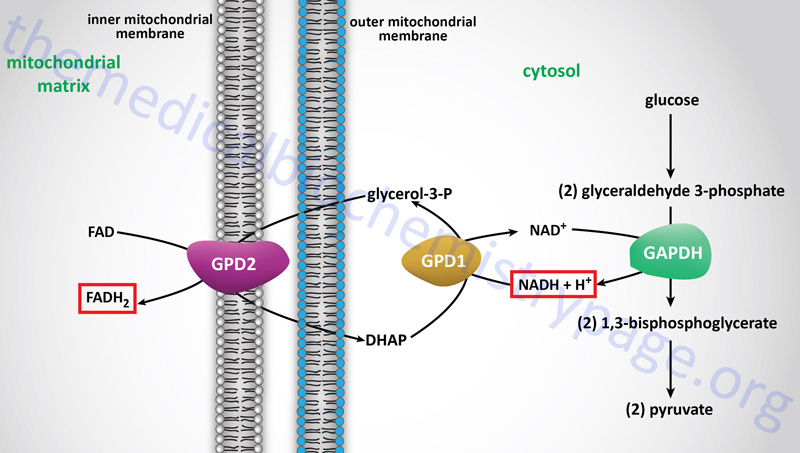
In contrast to oxidation of mitochondrial NADH, cytosolic NADH when oxidized via the electron transport system gives rise to two equivalents of ATP if it is oxidized by the glycerol phosphate shuttle (see above) and three equivalents of ATP if it proceeds via the malate-aspartate shuttle.
The glycerol phosphate shuttle is coupled to an inner mitochondrial membrane, FAD-linked dehydrogenase, of low energy potential like that found associated with Complex II. Thus, cytosolic NADH oxidized by this pathway can generate only approximately 1.5 equivalents of ATP (the ATP yield was originally defined as 2 moles). The shuttle involves two different glycerol-3-phosphate dehydrogenases: one is cytosolic (GPD1), acting to produce glycerol-3-phosphate, and one is an integral protein (GPD2) of the inner mitochondrial membrane that acts to oxidize the glycerol-3-phosphate produced by the cytosolic enzyme. The net result of the process is that reducing equivalents from cytosolic NADH are transferred to the mitochondrial electron transport system. The catalytic site of the mitochondrial glycerol phosphate dehydrogenase is on the outer surface of the inner membrane, allowing ready access to the product of the second, or cytosolic, glycerol-3-phosphate dehydrogenase.
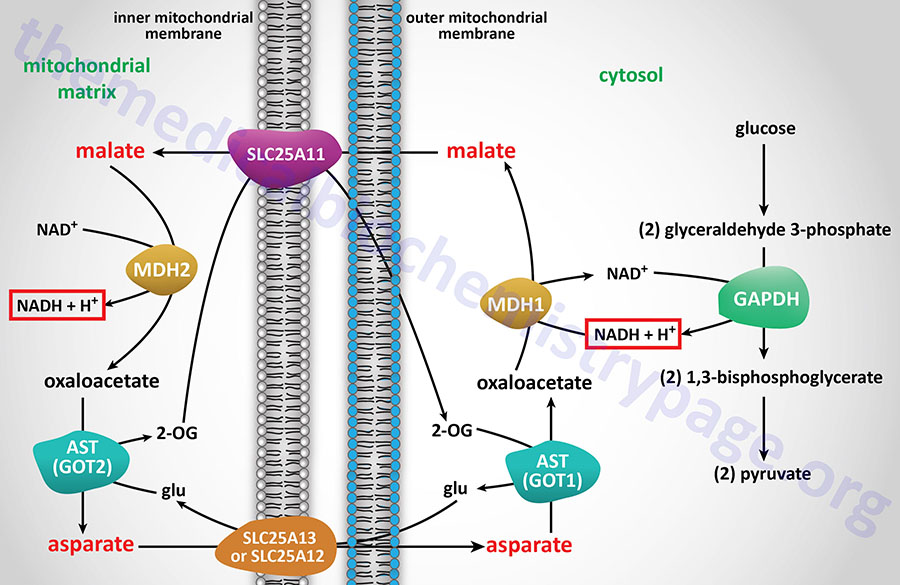
In some tissues, such as that of liver, heart, and kidney, mitochondrial glycerol-3-phosphate dehydrogenase (GPD2) is present in very low amounts, and the malate-aspartate shuttle is the dominant pathway for aerobic oxidation of cytosolic NADH. In contrast to the glycerol phosphate shuttle, the malate-aspartate shuttle allows for the generation of three equivalents of ATP for every cytosolic NADH oxidized.
NADH electrons can be efficiently utilized to reduce oxaloacetate (OAA) to malate via cytosolic malate dehydrogenase (encoded by the MDH1 gene). Malate is transported to the interior of the mitochondrion via the 2-oxoglutarate/malate antiporter. Inside the mitochondrion, malate is oxidized by the MDH2 encoded enzyme of the TCA cycle, producing OAA and NADH. In this step the cytosolic, NADH-derived reducing equivalents become available to the NADH dehydrogenase (complex I) of the inner mitochondrial membrane and are oxidized, giving rise to approximately 2.5 moles of ATP as described earlier. The mitochondrial transaminase uses glutamate to convert membrane-impermeable OAA to aspartate and 2-oxoglutarate. This provides a pool of 2-oxoglutarate for the aforementioned SLC25A11 antiporter. The aspartate which is also produced is translocated out of the mitochondrion, in exchange for glutamate, through the actions of the SLC25A13 antiporter.
Generation of Reactive Oxygen Species, ROS
The mitochondrial electron transport system (electron transport chain: ETC) of oxidative phosphorylation is the major site for the cellular generation of ROS. The ROS produced by cellular processes are superoxide anion (O2●–), hydrogen peroxide (H2O2), and hydroxyl free radical (●OH). Additional ROS include ozone (O3), singlet oxygen (1O2), and peroxy radicals (RO● and RO2●). As electrons pass through the complexes of the ETC some leak out to molecular oxygen (O2) resulting in the formation of superoxide. Approximately 0.15% of the molecular oxygen consumed by mitochondria is reduced to superoxide anion.
The generation of superoxide anion by mitochondria occurs predominantly at complexes I and III. ROS generation also occurs within the plasma membrane and the endoplasmic reticulum (ER) membranes via the action of NADP(H) oxidases. Since superoxide anion cannot diffuse across the inner mitochondrial membrane it remains in the matrix which allows it to react with Fe-S groups in mitochondrial proteins resulting in the liberation of ferrous (Fe2+) iron.
Matrix-associated superoxide anion is rapidly acted upon by superoxide dismutases (SOD). Humans express three SOD genes, SOD1, SOD2, and SOD3. The SOD1 gene encodes a copper-zinc-superoxide dismutase (CuZn-SOD; also designated SOD1). The SOD2 gene encodes a mitochondrial manganese-dependent superoxide dismutase (Mn-SOD; also identified as SOD2). The SOD3 gene encodes an extracellular superoxide dismutase (EC-SOD; also identified as SOD3). All three SOD enzymes dismutate superoxide anion to yield H2O2.
The SOD1 protein is found predominantly in the cytosol but also in the mitochondrial intermembrane space. SOD1 functions as a homodimer. The SOD2 protein is located within the mitochondrial matrix and functions as a homotetramer. The SOD3 protein is alternatively processed into two distinct forms, both of which function as homotetramers. One SOD3 form (termed the type C subunit) interacts tightly with heparin in the extracellular matrix (ECM), the other (termed the type A subunit) does not interact with the extracellular matrix.
Following the generation of H2O2 within the mitochondrial matrix it diffuses into the cytosol where it is eliminated by several antioxidant enzymes that include glutathione peroxidases (GPx1–GPx8), catalase, and peroxiredoxins (PRDX1–PRDX6). Using knock-out mice it has been clearly demonstrated that Mn-SOD (SOD2) is essential for the maintenance of normal mitochondrial function in highly oxidative tissues such as the brain and heart as well as the liver.
If H2O2 remains in the mitochondrial matrix it reacts with the ferrous (Fe2+) iron via the Fenton reaction yielding the hydroxyl free radical which is highly reactive toward DNA and polyunsaturated fatty acids (PUFA). The consequences of hydroxyl free radical action on DNA is the production of a number of oxidative lesions and the release of reactive base aldehydes termed base propenals. The action of hydroxyl free radical on PUFA is the generation of lipid peroxides that represent a major source of electrophilic aldehydes. Electrophilic aldehydes and propenals react with the amino groups of the bases of DNA and with the amino acids cysteine, lysine, arginine, and histidine.
The aldehyde products generated from hydroxyl radical action on PUFA are normally neutralized by glutathione conjugation or via aldehyde dehydrogenase activity in mitochondria. However, these neutralizing reactions can become overwhelmed if the level of mitochondrial ROS production increases.
Cardiolipin is the predominant lipid in the inner mitochondrial membrane. The action of the hydroxyl free radical on cardiolipin releases 4-hydroxynonenal (4-HNE) and on other mitochondrial lipids and mtDNA releases malondialdehyde (MDA), acrolein, and crotonaldehyde. All of these non-radical oxidants can induce protein modifications and mtDNA damage.
Mitochondrial and ER production of ROS contributes to the processes of aging as well as progression of numerous disorders such as type 2 diabetes and Parkinson disease. Dietary constituents can lead to increased ROS production which is evident in obesity and plays a major contributing role in the progression to insulin-resistance and diabetes. Consumption of a high fat diet results in a surplus of NADH and FADH2 that then increases the flux through the ETC with a resultant increase in ROS generation. Indeed, a high fat diet is known to increase the rate of H2O2 production in skeletal muscle mitochondria. Ultimately the increased rate of ROS production by the mitochondria results in mitochondrial dysfunction in skeletal muscle.
ER production of ROS is also a major contributor to disease states such as diabetes. Within the ER, proteins undergo folding into their functional conformations as they transit through to the Golgi and finally to the plasma membrane or secretory vesicles. Proper folding requires intra- and inter-chain disulfide bond formation that involves the oxidation of cysteine residues and the release of electrons. The electrons are passed to protein disulfide isomerase then to ER oxidoreductase 1 alpha and finally to O2 generating superoxide anion.
It is suggested that the nutrient excess seen in obesity and diabetes may play a role in overloading the ER protein folding capacity resulting in increased ROS production. An increase in ER ROS production results in ER stress and the induction of the ER stress response pathways that in turn activate pro-inflammatory pathways. Of significance to diabetes is that ER stress-induced inflammation activates the kinases JNK (JUN N-terminal kinase) and IKKβ (inhibitor of nuclear factor-kappa B kinase subunit beta) that phosphorylate insulin receptor substrates (e.g. IRS1 and IRS2) resulting in impairment of insulin signaling. Nuclear factor-kappa B (NF-κB) is one of the most important transcription factors regulating the expression of proinflammatory genes.
Generation of Reactive Nitrogen Species
Like reactive oxygen species (ROS), reactive nitrogen species (RNS) can be toxic if allowed to accumulate in cells. The major source of various RNS is the synthesis of nitric oxide, NO (correctly identified as NO●). NO reacts with superoxide anion (O2●–) to form a stronger oxidant identified as peroxynitrite anion (ONOO–). The peroxynitrite anion will then react with other molecules to generate additional RNS that includes as nitrogen dioxide (●NO2) and dinitrogen trioxide (N2O3). The reduction of NO also results in the formation of another RNS identified as the nitroxyl anion (NO–). The oxidation of NO in the presence of ●NO2 can lead to the generation of the RNS identified as the nitrosomium cation (NO+).
Reactive nitrogen species (RNS) are neutralized by the same systems that are responsible for the neutralization of reactive oxygen species, ROS. These systems include the various low molecular weight antioxidants such as reduced glutathione (GSH), vitamin C and vitamin E, bilirubin, and urate. There are also several non-catalytic antioxidant proteins such as the glutaredoxins, the thioredoxins (Trx), and the metallothioneins (MT). The catalytic antioxidant proteins, described in the preceding section, include the superoxide dismutases (SOD), catalase, the peroxiredoxins (Prx), and the glutathione peroxidases (GPx).
Role of Transcription Factor NRF2 in Oxidative Stress Responses
A major response to the accumulation of reactive oxidants, such as ROS and RNS, is the activation of a transcription factor identified as NRF2 [nuclear factor erythroid 2 (NFE2)–related factor 2]. The NRF2 protein is encoded by the NFE2L2 (NFE2 like bZIP transcription factor 2) gene. NRF2 is considered a central regulator of the cellular responses to changes in redox status but is also an important regulator of an array of processes that includes mitochondrial function, xenobiotic metabolism, carbohydrate, lipid, and iron metabolism, and DNA repair.
The activity of NRF2 is regulated by its interaction with the protein identified as KEAP1 (Kelch-like ECH-associated protein 1). The ECH acronym refers to Erythroid-derived CNC homology which is the NRF2 protein. The CNC acronym refers to Cap ‘n‘ Collar domain which is found in a subfamily of basic leucine zipper (bZIP) domain containing transcription factors. The CNC domain was originally identified in a Drosophila segmentation gene. The Kelch (German for chalice) domain is a protein-protein interaction domain. The Kelch domain allows KEAP1 to interact with ubiquitin ligases of the Cullin family, specifically Cul3.
Under normal physiological conditions KEAP1 promotes the ubiquitylation of NRF2 preventing it from entering the nucleus. When cysteine residues in KEAP1 are oxidized by increasing levels of reactive molecules such as ROS and RNS, NRF2 is released allowing it to migrate to the nucleus and activate the expression of genes encoding proteins with cytoprotective functions. To date over 250 genes have been identified as direct targets for transcriptional regulation by NRF2. The NRF2 target genes contain an enhancer element with a common ~41 bp DNA sequence element termed an antioxidant response element (ARE).
Several of the target genes of NRF2, as expected, encode antioxidant proteins. These include the HMOX1 (heme oxygenase 1), NQO1 [NAD(P)H quinone oxidoreductase 1], GCLC (glutamate-cysteine ligase catalytic subunit), GCLM (glutamate-cysteine ligase modifier subunit), PRDX1 (peroxiredoxin 1), and TXNRD1 (thioredoxin reductase 1) genes. NRF2 also actively represses the transcription of pro-inflammatory genes in macrophages. Heme oxygenase 1 catalyzes the degradation of heme to free iron, biliverdin, and carbon monoxide (CO) thereby removing free heme which is pro-oxidant. The function of NQO1 is to catalyze the NADPH-dependent reduction of quinones to hydroquinones, thereby preventing the formation of highly reactive semiquinones and associated reactive oxygen species (ROS). Glutamate-cysteine ligase is the first and rate-limiting enzyme in the synthesis of the peptide antioxidant, glutathione, GSH. Thioredoxin reductase 1 is involved in the reduction of thioredoxin which itself is principally involved in the reduction of oxidized disulfide bonds in proteins. The reduction of these disulfide bonds results in oxidation of thioredoxin which then is reduced by thioredoxin reductase.
Mitochondrial Dysfunction in Type 2 Diabetes and Obesity
Well established data demonstrate that mitochondrial dysfunction, particularly as it relates to the processes of oxidative phosphorylation, is contributory to the development of encephalomyopathy, mitochondrial myopathy, and several age-related disorders that include neurodegenerative diseases, the metabolic syndrome, and diabetes. Indeed, with respect to diabetes, several mitochondrial diseases manifest with diabetic complications such as mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) and maternally inherited diabetes and deafness (MIDD).
Normal biogenesis of mitochondria is triggered in response to changes in the ATP/ADP ratio and to activation of AMPK which in turn results in increased expression of PPARγ co-activator 1α (PGC-1α) and nuclear respiratory factor-1 (NRF1). PGC-1α is a master transcriptional co-activator of numerous genes involved in mitochondrial biogenesis. NRF1 is a transcription factor that regulates the expression of mitochondrial transcription factor A (TFAM, for transcription factor A, mitochondrial; also designated mtTFA) which is a nuclear transcription factor essential for replication, maintenance, and transcription of the mitochondrial genome (mtDNA). NRF1 also controls the expression of nuclear genes required for mitochondrial respiration and heme biosynthesis. Evidence has shown that both PGC-1α and NRF1 expression levels are lower in diabetic patients and in non-diabetic subjects from families with type 2 diabetes. The expression of NRF1 is highest in skeletal muscle which is also the tissue that accounts for the largest percentage of glucose disposal in the body and, therefore, is the tissue that is most responsible for the hyperglycemia resulting from impaired insulin signaling.
Mitochondrial dysfunction results in increased production of ROS which activates stress responses leading to increased activity of MAPK and JNK. Both of these serine/threonine kinases phosphorylate IRS1 and IRS2 resulting in decreased signaling downstream of the insulin receptor. Inhibited IRS1 and IRS2 activity results in decreased activation of PI3K. PI3K activation is involved in the translocation of GLUT4 to the plasma membrane resulting in increased glucose uptake. Therefore, inhibition of PI3K results in reduced glucose uptake in skeletal muscle and adipose tissue. Mitochondrial dysfunction results in a reduction in the level of enzymes involved in β-oxidation leading to increases in intramyocellular lipid content. Indeed, skeletal muscle metabolism of lipids has been shown to be impaired in type 2 diabetics. An increased delivery of fatty acids to skeletal muscle, as well as diminished mitochondrial oxidation, results in increased intracellular content of fatty acid metabolites such as diacylglycerol (DAG), fatty acyl-CoAs, and ceramides. These metabolites of fatty acids are all known to induce the activity of protein kinase C isoforms (PKCβ and PKCδ) that phosphorylate IRS1 and IRS2 on serine residues resulting in impaired insulin signaling downstream of the insulin receptor.
Because skeletal muscle consumes the largest amount of serum glucose, mitochondrial dysfunction in this tissue will have the greatest impact on glucose disposal. However, adipose tissue also plays an important role in glucose homeostasis and mitochondrial dysfunction in this tissue has been shown to result in impaired glucose homeostasis resulting in diabetes. For example, when animals are treated with inhibitors of mitochondrial oxidation insulin-stimulated glucose uptake in adipose tissue is significantly impaired. Adipose tissue secretes a number of proteins classified as adipokines. Adiponectin is an adipokine that promotes insulin-sensitivity in insulin-responsive tissues, such as skeletal muscle. When plasma levels of adiponectin are measured in obese or type 2 diabetic subjects it is found to be significantly lower than in age and sex matched control subjects that are of normal weight or that do not have diabetes. In animal studies, the enhancement of adipocyte mitochondrial biogenesis results in increased adiponectin release from adipose tissue. Conversely, expression of adiponectin expression is decreased in adipocytes with mitochondrial dysfunction.
Given that impaired mitochondrial function is clearly associated with obesity and type 2 diabetes, it is not surprising that there is great interest in the use of pharmacology to augment mitochondrial function in the treatment of these disorders. Of significance is the fact that the thiazolidinedione (TZD) class of drugs used to treat the hyperglycemia of type 2 diabetes (see the next section) activate PPARγ which in turn increases the level of activity of PGC-1α. Although the TZDs were first marketed due to their ability to improve insulin sensitivity, they have since been shown to increase mitochondrial functions both in vitro and in vivo. Antioxidants have also been shown to enhance mitochondrial function by reducing the production of ROS. Resveratrol (found in grape skins and red wine) is a potent antioxidant whose activity is, in part, due to its ability to activate the deacetylase SIRT1 (see below). Activated SIRT1 deacetylates PGC-1α resulting in increased transcriptional activity and thus, enhanced mitochondrial biogenesis.
Brown Adipose Tissue and Heat Generation
The uncoupling of proton flow releases the energy of the electrochemical proton gradient as heat. This process, referred to as adaptive thermogenesis, is a normal physiological function of brown adipose tissue. Brown adipose tissue gets its color from the high density of mitochondria in the individual adipose cells. Newborn babies contain brown fat in their neck and upper back that serves the function of non-shivering thermogenesis.
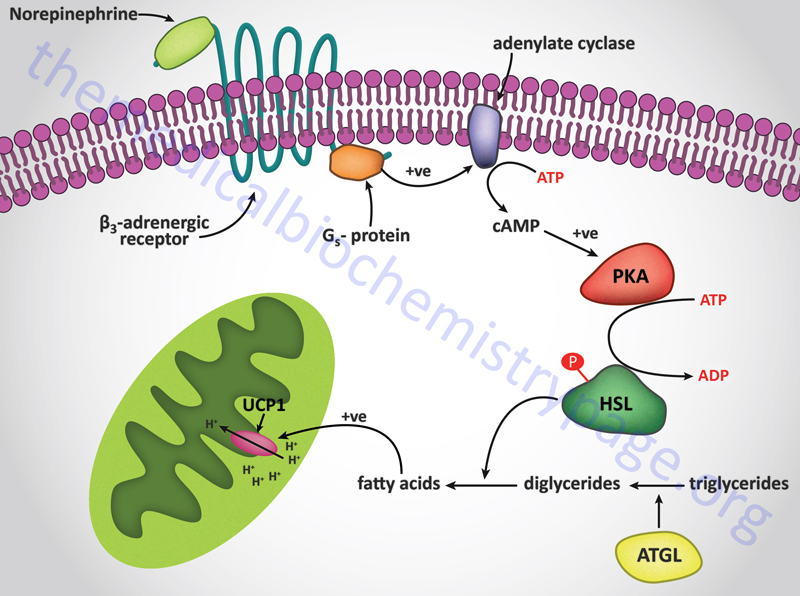
The muscle contractions that take place in the process of shivering not only generates ATP but also produces heat. Non-shivering thermogenesis is a hormonal stimulus for heat generation without the associate muscle contractions of shivering. The process of thermogenesis in brown fat is initiated by the release of free fatty acids from the triglycerides stored in the adipose cells. The hormonal release of fatty acids in brown fat is the same as that described for the mobilization of stored fat in the Lipolysis and the Oxidation of Fatty Acids page. The mitochondria in brown fat contain a protein called uncoupling protein 1, UCP1 (also called thermogenin). There are three uncoupling protein genes in humans identified as UCP1, UCP2, and UCP3. UCP1 is expressed in brown adipose tissue (BAT), UCP2 is expressed in a number of tissues, and UCP3 is expressed in skeletal and heart muscle. Only UCP1 is involved in the process of adaptive thermogenesis.
UCP1 acts as a channel in the inner mitochondrial membrane to control the permeability of the membrane to protons. When norepinephrine is released in response to cold sensation it binds to β-adrenergic receptors on the surface of brown adipocytes triggering the activation of adenylate cyclase. Activated adenylate cyclase leads to increased production of cAMP and the concomitant activation of cAMP-dependent protein kinase (PKA) with the result being phosphorylation and activation of hormone-sensitive lipase. The released free fatty acids bind to UCP1 triggering an uncoupling of the proton gradient and the release of the energy of the gradient as heat.
Other Biological Oxidations
Oxidase complexes, like cytochrome oxidase, transfer electrons directly from NADH and other substrates to oxygen, producing water. Oxygenases, widely localized in membranes of the endoplasmic reticulum, catalyze the addition of molecular oxygen to organic molecules.
There are two kinds of oxygenase complexes, monooxygenases and dioxygenases. Dioxygenases add the two atoms of molecular oxygen (O2) to carbon and nitrogen of organic compounds. Monooxygenase complexes play a key role in detoxifying drugs and other compounds (e.g., PCBs and dioxin) and in the normal metabolism of steroids, fatty acids and fat soluble vitamins. Monooxygenases act by sequentially transferring two electrons from NADH or NADPH to one of the two atoms of oxygen in O2, generating H2O from one oxygen atom and incorporating the other oxygen atom into an organic compound as a hydroxyl group (R-OH). The hydroxylated products are markedly more water-soluble than their precursors and are much more readily excreted from the body. Widely used synonyms for the monooxygenases are: mixed function oxidases, hydroxylases, and mixed function hydroxylases.
The chief components of monooxygenase complexes include cytochrome b5, cytochrome P450, and cytochrome P450 reductase, which contains FAD plus FMN. There are many cytochrome P450 isozymes; for example, up to 50 different P450 gene products can be found in liver, where the bulk of drug metabolism occurs. Some of these same gene products are also found in other tissues, where they are responsible for tissue-specific oxygenase activities. P450 reducing equivalents arise either from NADH via cytochrome b5 or from NADPH via cytochrome P450 reductase, both of which are associated with cytochrome P450 in the membrane-localized complexes.
Enzymatic reactions involving molecular oxygen usually produce water or organic oxygen in well regulated reactions having specific products. However, under some metabolic conditions (e.g., reperfusion of anaerobic tissues) unpaired electrons gain access to molecular oxygen in unregulated, non-enzymatic reactions. The products, called free radicals, are quite toxic. These free radicals, especially hydroxy radical, randomly attack all cell components, including proteins, lipids and nucleic acids, potentially causing extensive cellular damage. Tissues are replete with enzymes to protect against the random chemical reactions that these free radicals initiate. Several free radical scavenging enzymes have been identified.
Superoxide dismutases (SOD) in animals contain either zinc (Zn2+) and copper (Cu2+), known as CuZnSOD, or manganese (Mn2+) as in the case of the mitochondrial SOD. These SOD convert superoxide to peroxide and thereby minimize production of the hydroxy radical, the most potent of the oxygen free radicals. Peroxides produced by SOD are also toxic. They are detoxified by conversion to water via the enzymes of the glutathione peroxidase family as well as by catalase. Mammalian glutathione peroxidases contain the modified amino acid selenocysteine in their reactive centers. Catalase (located in peroxisomes) provides a reductant route for the degradation of hydrogen peroxide. Mammalian catalase has the highest turnover number of any documented enzyme.
Glutathione (see the Pentose Phosphate Pathway page) is important in maintaining the normal reduction potential of cells and provides the reducing equivalents for glutathione peroxidase to convert hydrogen peroxide to water. In red blood cells the lack of glutathione leads to extensive peroxide attack on the plasma membrane, producing fragile red blood cells that readily undergo hemolysis.