
Last Updated: May 24, 2026
Introduction to the Opioids
Opioids are any compounds that resemble the naturally occurring alkaloids found in the resin of the opium poppy. The term opiate refers only to the alkaloids found in poppy resin whereas, opioid refers to opiates, opioid peptides, and synthetic substances that elicit opiate-like effects. The primary opiates in opium poppy resin are morphine and codeine. Preparations from the opium poppy have been used for thousands of years for a variety of medicinal purposes. The earliest uses were for the treatment of the diarrhea associated with dysentery. The widespread use of opium preparations led to the identification of their euphoric and addictive properties. Several other important side effects are associated with opioid use, such as sedation and respiratory depression.
Currently, the major pharmacologic use for opiates and opioids is in the management of chronic and/or intense pain. The ability for humans to sense pain is due to the encoding and processing of harmful stimuli through afferent nerve signals in both the peripheral and central nervous systems. The specific types of nerve endings that allow for pain sensation are referred to as nociceptors (pain receptors). When stimulated by either mechanical, chemical, or thermal mechanisms, these nerve endings trigger autonomic responses that are subjectively recognized as painful. Humans possess three distinct types of nociceptors called visceral, somatic, and cutaneous pain receptors. The analgesic effects of opiate and opioid compounds results from their ability to decrease nociception (perception of pain), decrease reaction to pain, and to increase the level of tolerance for pain. Opioids can also suppress coughing which is an indication for their pharmacologic administration. Opioids exert all of these effects by binding to a specific class of receptors called the opioid receptors (see below).
There are several (in excess of 150) synthetic opioids (e.g. heroin, dihydromorphine, oxycodone, hydrocodone, hydromorphone, fentanyl) that exert effects that in many cases are hundreds of times more powerfully than morphine. An additional important synthetic opioid is methadone, used in the treatment of opiate addiction. The effects of opioids can be reversed with opioid antagonists such as naloxone or naltrexone. Indeed, these “antidotes” are quite effective in the treatment of opioid overdose. Both naloxone and naltrexone function as competitive inhibitors of the opioid receptors by binding to the receptors with higher affinity than natural or synthetic opioids, but without receptor activation. Both naloxone and the anesthetic, ketamine, are known to have unfavorable side effects that includes hallucinations, which precludes their widespread use. The hallucinogenic effects of these drugs are most likely due to their elicited responses at one of the opioid receptors, the κ (kappa) opioid receptor, KOR (see below).
Although the most well studied pharmacological applications of opiates and opioids are those involved in the modulation of nociception and sedation, this page will focus principally on the effects of these compounds on feeding behaviors and the consequent regulation of metabolic processes.
Endogenous Opioids
Subsequent to the pharmacologic characterization of the receptors to which opioids, such as morphine, bind and exert their analgesic effects, naturally occurring opioids were identified. The first endogenous opioids with morphine-like action were discovered in the brain in 1974 and subsequently characterized and given the name enkephalin. The endogenous opioid family refers to a network of genes coding for neuropeptide ligands and their cell surface receptors. Each of the characterized endogenous opioids is a peptide. There are several endogenous opioids that are divided into three major families: the enkephalins, the endorphins, and the dynorphins (see Table below). All of the peptides in these three families are collectively referred to as endorphins which is a termed derived from a contraction of “endogenous morphine”.
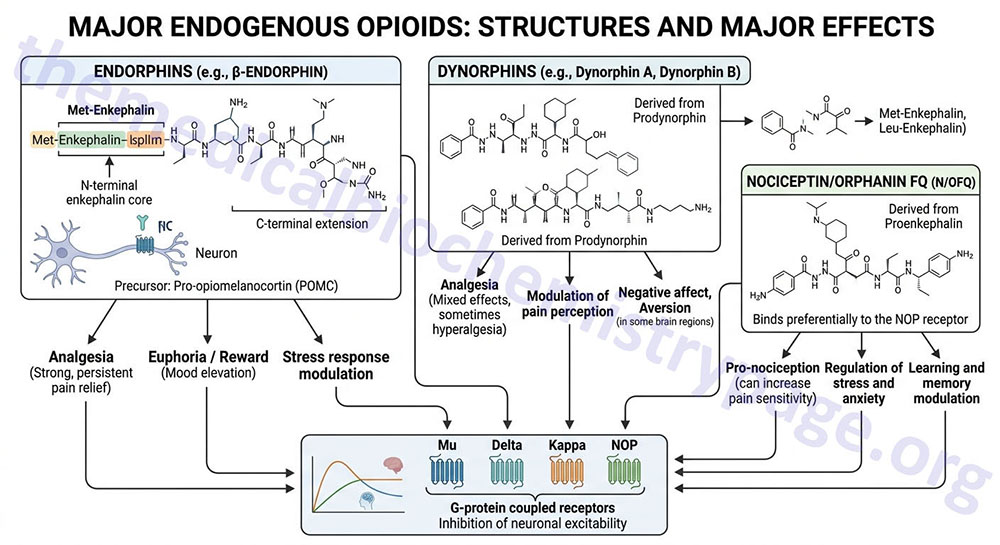
All of the endogenous opioid peptides (excluding nociceptin) share the enkephalin sequence (YGGFL or YGGFM) at the N terminus, with differing extensions at the C terminus (see below for details). Collectively, the endogenous opioids exert effects that are important in nociception (sensation of pain), analgesia, general activity and locomotion, gastrointestinal, renal, hepatic, and cardiovascular functions, respiration and thermoregulation, electrical-related activity and neurophysiology, seizures and neurologic disorders, immunological responses, and numerous behavioral processes. These behavioral processes include, but are not limited to, eating and drinking, learning and memory, sexual activity, mental illness and mood, tolerance and dependence, stress responses, and alcohol and drug abuse.
All of the endogenous opioids exert their effects by binding to one or more of the three subtypes of opioid receptor or the opioid receptor-like 1 receptor (see next section). Studies on the biological activities of the enkephalins led to the identification of the δ (delta) receptor (DOR). Subsequently it was shown that β-endorphin preferentially bound the μ (mu) receptor (MOR), while dynorphin A exhibited preference for the KOR.
An additional opioid receptor, identified as opioid receptor-like 1 (OPRL1), was instrumental in the identification of an additional member of the endogenous opioid family, a protein called nociceptin. Nociceptin is also known as orphanin FQ. The name nociceptin was coined for this peptide because it showed pain-like activity in certain assays and its classification as an opioid peptide was based entirely on its structural similarity to other members of the family listed in the following Table. Unlike the members of the endorphin families of endogenous opioids, nociceptin does not contain the enkephalin sequence at its N-terminus. Whereas, the opioids are potently analgesic (reduce pain sensation), nociceptin is a potent anti-analgesic. The nociceptin peptide receptor (NOP; OPRL1) is a member of the GPCR family of receptors as are the other opioid receptors. The NOP was at one point misclassified as the κ3 opioid receptor subtype. Unlike the other opioid receptors, NOP is not sensitive to the antagonist naloxone.
Table of the Major Endogenous Opioids
| Precursor Protein | Major Opioid | Opioid Peptide Sequence | Preferred Receptor |
| Preproenkephalin | Leu-enkephalin | YGGFL | MOR (OPRM1), DOR (OPRD1) |
| Preproenkephalin | Met-enkephalin | YGGFM | MOR (OPRM1), DOR (OPRD1) |
| Proopiomelanocortin (POMC) | β-endorphin | YGGFMTSEKSQTPLVTLFKNAIIKNVHKKGQ | MOR (OPRM1), DOR (OPRD1) |
| Preprodynorphin | Dynorphin A(1-17) | YGGFLRRIRPKLKWDNQ | KOR (OPRK1) |
| Preprodynorphin | Dynorphin B(1-13) | YGGFLRRQPKVVT | KOR (OPRK1) |
| Prepronociceptin | Nociceptin | FGGFTGARKSARKLANQ | Opioid receptor-like 1 (OPRL1) |
Numerous additional naturally occurring opioid peptides have been isolated and characterized but are not discussed in detail in this page. The opioid peptide known as opiorphin was originally isolated from saliva and subsequently discovered to function via its ability to inhibit several proteases known to inactivate the enkephalins. Several opioid peptides are also derived via protease action on proenkephalin, such as the octapeptide known as adrenorphin. Adrenorphin is a potent opioid agonist that functions by binding to both the MOR and the DOR. As discussed in the last section of this page, the casomorphins are peptides derived from the milk protein casein that function as modulators of MOR signaling in the gut. The hemorphins are a related family of opioid peptides derived from the β-chain of hemoglobin and function by binding to all three opioid receptor classes. The hemorphins include hemorphin-4, spinorphoin, and valorphin.
Enkephalins
The enkephalins are pentapeptide opioids, and they were the first endogenous opioids to be identified. There are two enkephalins that differ only in the C-terminal amino acid. Leu-enkephalin contains the amino acids YGGFL (Tyr-Gly-Gly-Phe-Leu), whereas Met-enkephalin contains YGGFM. The term enkephalin is derived from the Greek word for brain: enkephalos. Both enkephalins are derived from the proenkephalin gene, PENK located on chromosome 8q12.1. The PENK gene is composed of 4 exons that encode a 267 amino acid preproprotein.
Both enkephalins exert their biological effects primarily by binding to the delta (δ) opioid receptor (DOR). Leu-enkephalin exerts some of its effects via binding and activating the mu (μ) opioid receptor (MOR). Both enkephalins are considered to be the primary endogenous ligands for the DOR. Synthesis of both met-enkephalin and leu-enkephalin occurs primarily within the central nervous system (CNS) and the adrenal medulla.
Within the CNS the basal ganglia contain the highest concentrations of the enkephalins. In the periphery the enkephalins are produced by sympathetic ganglia and post-ganglionic neurons. In addition to the adrenal glands, the enkephalins are found in endocrine cells of the gastrointestinal mucosa. The major effects of the enkephalins, exerted through activation of the DOR, are the alleviation of pain and a reduction in nociception.
Met-enkephalin is also involved in the regulation of memory and emotions, feeding behaviors, immune functions, gastrointestinal motility, gastric and pancreatic secretions, and carbohydrate metabolism. In addition to the modulation of nociception, Leu-enkephalin modulates gonadal functions.
Dynorphins
The term dynorphin is derived from the contraction of “dyn” from the Greek, dynamis = power and “orphin” for endogenous morphine peptide. The dynorphins, dynorphin A and B, are encoded by the prodynorphin gene, PDYN located on chromosome 20p13. The PDYN gene is composed of 10 exons that generate five alternatively spliced mRNAs all of which encode the same 254 amino acid preproprotein.
Contained within the preprodynorphin peptide are three opioid domains containing the YGGFL enkephalin pentapeptide sequence. Each of these three opioid domains is followed by three different, highly basic C-terminal extensions. The protease, proprotein convertase subtilisin/kexin type 2 (PCSK2) has been shown to be responsible for the processing of preprodynorphin within the brain. PCSK2 is historically referred to as prohormone convertase 2 (PC2).
There are several prodynorphin opioids derived from the precursor peptide. These opioids include large peptides (big-dynorphin and leumorphin), the intermediate-sized kappa opioid receptor selective peptides dynorphin A and dynorphin B (as well as α-neoendorphin), and the shorter peptides dynorphin A(1-8) and Leu-enkephalin. All of these preprodynorphin-derived peptides are able to activate opioid receptors, however, the preproprotein is also processed into several peptides that do not exhibit opioid receptor activation capabilities.
The major opioid receptor to which the dynorphins bind is the κ-opioid receptor (KOR). Indeed, the dynorphins are considered the primary endogenous ligands for the KOR. Dynorphins are primarily localized within the hippocampus, amygdala, hypothalamus, striatum and spinal cord. The functions of the dynorphins are related to learning and memory, emotional control, stress response and nociception. The non-opioid receptor actions of dynorphin peptides are exerted principally through direct effects on NMDA receptors.
β-Endorphin
The term β-endorphin was given to this endogenous opioid because it is derived from a proopiomelanocortin (POMC) precursor protein called β-lipotropin and because it is an endogenous morphine-like peptide. The POMC preproprotein is derived from the POMC gene located on chromosome 2p23.3 which is composed of 4 exons that generate four alternatively spliced mRNAs, all of which are translated into the same 267 amino acid preproprotein.
Given that fact that β-endorphin is derived from the POMC precursor protein it is understandable that the majority of the peptide originates from, and is stored within, cells of the anterior pituitary. Synthesis of β-endorphin is also seen in immune cells such as T- and B-lymphocytes, monocytes, and macrophages.
The principal effects of β-endorphin are exerted within the peripheral and central nervous systems (PNS and CNS, respectively). Within the PNS β-endorphin produces an analgesic effect as a result of binding to opioid receptors at both pre- and post-synaptic nerve terminals. The major opioid receptor to which β-endorphin binds is the μ-opioid receptor (MOR). Activation of the MOR by β-endorphin results in the inhibition of tachykinin release (particularly substance P) which are key modulators of pain transmission.
Within the CNS, β-endorphin also binds to the MOR primarily at presynaptic nerve terminals. In the case of the CNS, the analgesic effects of β-endorphin is exerted by the inhibited release of the inhibitory neurotransmitter, GABA, leading to excess production and release of dopamine. In this capacity the released dopamine is associated with pleasure sensations.
Humans produce a family of five endorphin peptides, all of which possess the YGGFL or YGGFM enkephalin sequence at their N-termini. The five endorphins are β-endorphin (31 amino acids), α-endorphin (16 amino acids), γ-endorphin (17 amino acids), α-neoendorphin (10 amino acids), and β-neoendorphin (9 amino acids). The α-endorphin and γ-endorphin peptides are identical except for the presence of a Leu residue at the C-terminus of the γ-endorphin peptide making it a 17-amino acid peptide.
Nociceptin
Nociceptin was simultaneously characterized in two different laboratories and therefore, is also known as orphanin FQ. The identification of nociceptin was the result of trying to identify the ligand for a previously characterized orphan G-protein coupled receptor (GPCR) called opioid receptor-like 1 (ORL1). The term orphanin FQ is derived from the fact that the N-terminus of the pro-protein contains the amino acids phenylalanine (F) and glutamine (Q). Nociceptin exhibits a high degree of sequence similarity to the dynorphins. Nociceptin is a 17 amino acid peptide that is derived from the prepronociceptin protein encoded by the PNOC gene. The PNOC gene is located on chromosome 8p21.1 and is composed of 5 exons that generate two alternatively spliced mRNAs encoding two isoforms of prepronociceptin. Prepronociceptin isoform 1 is a 176 amino acid protein and isoform 2 is a 112 amino acid protein.
The original biological activity attributed to nociceptin was an anti-opioid hyperalgesic effect in supraspinal pain pathways. However, in spinal pain pathways nociceptin exerts analgesic properties. Since its initial isolation and characterization, nociceptin has been demonstrated to modulate numerous other physiological processes. These other functions of nociceptin include effects related to CNS pathophysiology such as anxiety, depression, hyperphagia and obesity, addiction, Parkinson disease, and cognition.
Outside the CNS nociceptin effects are antitussive (suppress coughing), they exert negative chronotropic and inotropic functions on the heart, lead to vasodilation, result in inhibition of gastrointestinal motility, the inhibition of sepsis, and inhibition of inflammatory processes.
Opioid Receptors
Three different opioid receptor families have been identified. These three receptor families were initially characterized by their pharmacologic properties and were subsequently demonstrated to be encoded by three distinct genes. The three opioid receptor families are identified as the μ (mu) opioid receptor (MOR), the δ (delta) opioid receptor (DOR), and the κ (kappa) opioid receptor (KOR). All members of these three receptor families are G-protein coupled receptors (GPCR). The DOR cDNA was the first of the opioid receptors to be cloned. Following isolation and characterization of cDNA and genomic clones for each of the opioid receptors it was discovered that all three genes generate multiple mRNA variants, primarily as a result of alternative promoter usage or alternative splicing events. All of the opioid receptors cause hyperpolarization (inhibition) of neurons in response to ligand binding. In most assays, opioid activation of MOR or DOR results in very similar effects. Indeed these two receptors are often found co-existing in the same neurons.
The Mu Opioid Receptors
The μ opioid receptor (MOR) was originally so-called due to the fact that it was identified on the basis of its affinity and responsiveness to morphine. The MOR is the primary site of action for the most commonly used opioids (morphine, heroin, fentanyl, and methadone). The single MOR gene (gene symbol: OPRM1) is located on chromosome 6q25.2 and is composed of 17 exons.
As a result of alternative promoter usage and alternative splicing, transcription from the OPRM1 gene yields 19 different mRNAs. Ten of these various MOR encoding mRNAs result from alternative promoter usage. Three of the 19 OPMR1 mRNAs encode the same protein.
When the single OPRM1 gene is knocked out in mice, all μ opioid receptor activity is lost verifying that the various pharmacologically definable subtypes of MOR are the result of this single gene. Several of the alternative forms of MOR generated as a result of the alternatively mRNA splicing events contain only six transmembrane spanning domains as opposed to the canonical seven transmembrane spanning domains of the GPCR superfamily of receptors.
Expression from the OPRM1 gene is complex given that the gene contains two distinct promoter elements. These promoters are identified as the distal (DP) and proximal (PP) promoters. The DP drives expression of a single primary transcript, whereas, the PP drives the expression of multiple transcripts that result from a cluster of transcriptional start sites. Both the DP and PP are highly GC-rich, lack a typical TATA-box, and contain multiple transcriptional regulatory elements.
When assessing the functions of the various MOR subtypes that have been observed to be generated from the OPRM1 gene they have been classified based upon the observed pharmacology. These classifications assign three distinct MOR subtypes identified as μ1, μ2, and μ3. Activation of μ receptors results in the opening of G protein-gated inwardly rectifying K+ (GIRK) channels, inhibition of voltage-gated Ca2+ channels, and reduction of adenylate cyclase-mediated cAMP production. Binding of β-endorphin to μ receptors results in disinhibition of dopaminergic neurons resulting in increased dopamine influx. This effect of β-endorphin plays a role the reward and reinforcement effects of dopamine and, as such, is believed to contribute to the development of drug dependence.
In addition to binding exogenous and endogenous opioids, the μ receptors mediate the action of several non-opioid drugs that elicit characteristics of abuse such as alcohol and nicotine. In addition, the μ receptors are involved in the activities of the stress-responsive hypothalamic-pituitary axis (HPA).
The Delta Opioid Receptors
The δ opioid receptor (DOR) was originally so-called due to the fact that it was identified on the basis of the responsiveness of the vas deferens to opioids. The endogenous ligands that bind to the opioid receptors are the enkephalins. Activation of the DOR is involved in the modulation of nociception, analgesia, reward, addiction, and affective state. The single DOR gene (gene symbol: OPRD1) is located on chromosome 1p35.3 and is composed of 3 exons. Two major transcription initiation sites have been identified in the OPRD1 gene. The promoter region of the gene is also TATA-less and GC-rich similar to the organization of the OPRM1 gene. When the single OPRD1 gene is knocked out in mice, all δ opioid receptor activity is lost verifying that the various pharmacologically definable subtypes of DOR are the result of this single gene.
When assessing the functions of the various DOR subtypes that have been observed to be generated from the OPRD1 gene they have been classified based upon the observed pharmacology. These classifications assign two distinct DOR subtypes identified as δ1, and δ2.
The Kappa Opioid Receptors
The κ opioid receptor (KOR) was originally so-called due to the fact that it was identified on the basis of its affinity and responsiveness to ketocyclazocine. The single KOR gene (gene symbol: OPRK1) is located on chromosome 8q11.23 and is composed of 5 exons that generate three alternatively spliced mRNAs due to the use of alternative promoter elements. The OPRK1 gene contains a non-coding exon upstream of the initiating codon, and two functional promoters termed promoter 1 (P1) and promoter 2 (P2).
Promoter P1 initiates transcription from a cluster of sites 700 to 1100 nucleotides upstream of the ATG initiation codon. Promoter P1 can, therefore, drive the expression of two distinct KOR mRNAs that differ in their 5′-UTR sequences but which contain the same protein coding capacity encoded in the four exons within the mRNAs.
Promoter P2 is located within intron 1 and initiates transcription from a single site located at position –93 relative to the ATG initiation codon located in exon 2. Promoter P2, therefore, drives expression of a single mRNA that contains only three protein coding exons.
P1 is the major promoter used in most tissues, whereas P2 is active only in certain brain areas. Both P1 and P2 are GC-rich and do not contain canonical TATA-boxes. When the single OPRK1 gene is knocked out in mice, all κ receptor activity is lost verifying that the various pharmacologically definable subtypes of KOR are the result of this single gene.
When assessing the functions of the various KOR subtypes that have been observed to be generated from the OPRK1 gene they have been classified based upon the observed pharmacology. These classifications assign five distinct KOR subtypes identified as κ1a, κ1b, κ2a, κ2b, and κ3. The dynorphins are considered the primary endogenous ligands for the KOR. The KOR subtypes are widely distributed throughout the central nervous system. Activation of these receptors is involved in the regulation of a wide array of activities including nociception, addiction to drugs of abuse, neuroendocrine function, cardiovascular function, respiration, temperature control, feeding behavior, and stress. In addition, κ opioid receptor activation modulates reward pathways elicited by opioids, cocaine, and other such stimuli. These latter effects are thought to be exerted via the ability of KOR activation to modulate dopaminergic tone.
Nociceptin Receptor (Opioid Receptor-Like 1)
The nociceptin/orphanin FQ receptor is identified as the opioid (opiate) receptor-like 1 protein (OPRL1). The nociceptin receptor is also identified as the nociceptin/orphanin FQ (N/OFQ) peptide receptor, NOP. Like the three other opioid receptor families, the nociceptin receptor belongs to the GPCR family of receptors. The opioid receptor-like 1 protein (nociceptin receptor) exhibits a high degree of sequence similarity to the kappa opioid receptors (KOR).
The opioid receptor-like 1 protein is encoded by the OPRL1 gene that is located on chromosome 20q13.33 and is composed of 8 exons that generate six alternatively spliced mRNAs. Three of the alternative mRNAs encode the same 370 amino acid protein.
A particularly interesting feature of the OPRL1 gene is that a promoter for this gene also functions as a promoter for another gene whose direction of transcription is from the opposite strand as that for the transcription of OPRL1. This other gene is known as regulator of G-protein signaling 19 (RGS19).
Opioids and Appetite Modulation
A substantial amount of scientific evidence demonstrates that the action of various opioids, especially via the μ-opioid receptor (MOR), is a major contributor to both the motivation to seek food and the pleasure of consuming food. These effects were originally observed in laboratory rodents in response to the administration of the opioid receptor antagonist, naloxone. Administration of naloxone results in a significant reduction in short-term food intake in rodents. These effects of naloxone were also observed in many other animal species in addition to rodents. Research into the effects of opioids on feeding behavior also demonstrated that this class of molecule also contributes to the hedonistic aspects of the reward processes associated with feeding behavior.
One interesting observation on the effects of naloxone was that there is a much higher level of reduced food intake following naloxone administration if the presented diet is highly palatable to the animal as opposed to the effects when the presented diet was less palatable. The desire to consume a sucrose-rich meal is much more reduced following naloxone administration than the change in desire for a starch-rich meal. Not only does naloxone exhibit these differences in effect with palatable versus non-palatable foods, it is also observed when examining the effects on drinking behavior. Naloxone administration causes a more significant reduction in fluid intake if the fluid is more palatable than when not or is just plain water.
Given the findings of opioids on feeding behavior in laboratory animals it should not be difficult to understand why numerous human studies on the modulation of hedonic behaviors by opioids were undertaken. In particular, studies have been carried out to determine the pharmacologic benefit of opioids and opioid antagonists on normal feeding behaviors, disordered feeding (e.g in anorexia), and on alcohol abuse. Most human studies utilize the opioid antagonists naloxone, naltrexone, and nalmefene.
Feeding behaviors in humans involve two distinct feedback processes. The first behavior is the wanting and liking of food which is a positive feedback process associated with the stimulation of actual appetite. Appetite stimulation is learned process that comes about as a result of an individuals prior exposure to the sight and smell of palatable food. An appreciation for this concept can be made by comparing the foods considered delicious by one person, whereas another individual may find the same food unpalatable due primarily to lack of prior taste and smell. The second feedback process is a negative one associated with the sensations of satiety and satiation. These negative feedback processes involves the stimulation of multiple interacting systems within the brain, principally within the hypothalamus. These latter processes are directly related to the gut-brain axis.
Both the positive and negative feedback processes can be significantly modified by numerous factors. These factors include food texture, energy density of the food, the macronutrient content of the food, and the time of day when the food is presented for consumption. The physiological status of an individual can also exert a profound impact on these feedback mechanisms. The sex of an individual, the age, the hormonal status, the level of hydration, and the level of hunger all contribute to the modification of the level of feedback control over feeding behavior. Regardless of each of these modifiers, the single most intense factor controlling human feeding behavior is the pleasure-reward response.
In human studies, opioid antagonism is most significantly associated with the hedonic (pleasure-reward) aspects of feeding behavior, the so-called palatability of a particular food. Another significant result of opioid antagonism studies on feeding behavior is that humans (and rodents) exhibit the same level of motivation to start eating regardless of whether naloxone or a placebo was administered. However, in spite of a similar level of motivation to eat, the rate of ingestion and the amount of food ingested is decreased in humans given an opioid antagonist. These observations define the opioid palatability hypothesis which states that opioids modulate feeding behaviors via the stimulation of an increased perception of food palatability. This can be seen very distinctly in laboratory animals following direct injection of morphine or opioid antagonists into the hypothalamus. Morphine injection induces hyperphagia, whereas, naloxone injection results in significant reduction s in food intake. The direct role of the MOR in the process of opioid modulation of feeding behavior was demonstrated in MOR knock-out mice. These mice exhibit a decreased motivation to consume not only palatable food but also bland food.
The opioid palatability hypothesis has at its core, the presumption that the overall sensory experiences associated with the consumption of palatable foods will be associated with the release of opioid peptides. Indeed, support for this hypothesis is seen when laboratory animals consume palatable food. In these animals there is an observed increase of β-endorphin levels in the hypothalamus. As pointed out in the Gut-Brain Interrelationships page, the hypothalamus is a major brain region (along with the brain stem) responsible for the control of feeding behaviors.
Mice fed a high-fat diet or a high-carbohydrate diet are observed to have significant increases in the level of dynorphin and enkephalin in specific hypothalamic nuclei, specifically the arcuate nucleus (ARC) and the paraventricular nucleus (PVN). The ARC is a primary nucleus of the hypothalamus involved in the control of feeding behaviors and satiety as it is composed of neurons that produce and secrete the principal orexigenic and anorexigenic neuropeptides. The PVN is a hypothalamic nucleus enriched in secondary neurons whose functions are to transmit feeding behavior signals from the ARC to the nucleus of the solitary tract (nucleus tractus solitarii, NTS).
The NTS is a major nucleus of the brain stem that is associated with the sensation of satiety and thus, results in the termination of the desire for food. In obese humans, the level of β-endorphin in the blood is found to be three times that seen in lean subjects. Given the results of opioid studies in both laboratory animals and in humans, there is ample evidence to support the suggestion that pharmacologic opioid antagonism may be beneficial in the treatment obesity and other eating disorders. Although several human studies have shown that opioid antagonism, in obese individuals, does indeed result in short-term decreases in food intake, long term treatment has been ineffective as a means to control weight.
Nutropoids and Appetite Modulation
As discussed in greater detail in the Gut-Brain Interrelationships page, it is well established that hormonal signals produced by the gastrointestinal system, in response to food intake, exert profound effects on overall feeding behavior in humans. These hormones include ghrelin, cholecystokinin (CCK), glucagon-like peptide 1 (GLP-1), and peptide tyrosine tyrosine (PYY). In addition to the gut-derived hormones, the enteric nervous system of the gut plays a critical role in sensing food intake, or lack thereof, and transmitting signals to the brain that modulate feeding behaviors. Recently it was shown that the small intestine contributes to overall endogenous glucose production (intestinal gluconeogenesis). This intestinal-derived de novo glucose is detected by a sensor system in the portal vein which then transmits this signal to the brain via the enteric nervous system to initiate a decrease in the level of hunger. This role of the small intestine was discovered from studies aimed at understanding the effects of protein-rich diets following gastric bypass surgery. Numerous studies have also demonstrated that protein-rich diets, in normal and overweight individuals, induces the sensation of satiety.
The term nutropoid was coined to refer to any of several opioid-like peptides that are derived from ingested and digested food. Indeed, the satiety inducing effects exerted by protein-enriched diets can, in part, be attributed to the production of nutropioids and to subsequent opioid antagonism at the level of opioid receptor signaling system, in particular via the μ-opioid receptors (MORs). Experimental studies have demonstrated that there is a close proximity of cells expressing MORs and nerve fibers in the walls of the portal vein. As discussed above, it is known that modulation of MOR signaling can interfere with the control of food intake. Opioid agonists enhance food intake, whereas antagonists inhibit it. The distribution of MOR expression occurs at highest levels in regions of the brain that are known to be critical for the control of reward-driven appetite as well as the desire to consume food. Within the small intestine, MOR expression is also high and these receptors control gastrointestinal motility. This latter fact can be best evidenced in individual who take opiate analgesics for chronic and severe pain which results in reduced gut motility resulting in potentially severe constipation.
Protein-derived opioid-like oligopeptides were first shown to be associated with the digestion of caseins from milk. The peptide identified as β-casomorphin1-7 was shown to have MOR agonist activity in vitro. However, it is unlikely though that this peptide could reach the brain following oral ingestion. Indeed, β-casomorphin1-7 is degraded by the liver and is not detected in systemic blood after the ingestion of milk or dairy products. Despite these observations, this does not mean that β-casomorphin, and other protein digestion-derived nutropoids, cannot exert effects via MOR signaling via gastrointestinal or mesenteric portal sites. Evidence for this latter concept comes from studies on the food intake suppressive effects of naloxone in humans. Naloxone, like β-casomorphin1-7, is actively degraded by the liver, yet still suppresses food intake when given orally in humans.
The current research into the ability of digested proteins to produce nutropoids that act at the MOR demonstrates that the effects do indeed occur via a gut-brain neural circuitry. The nutropoid-MOR signaling from the gut involves the process of intestinal gluconeogenesis and the resultant intestinal glucose induced satiety responses indicated earlier. In laboratory mice that have had their MOR gene knocked out, as well as in mice with an ablated intestinal gluconeogenesis capacity, there is no longer any satiety response to the consumption of a protein-rich diet nor to the direct portal vein administration of MOR antagonists. These result further confirm that the modulation of feeding behavior via intestinal MOR signaling pathways involves the activation of intestinal gluconeogenesis.
Another potential source of gastrointestinal nutropoids are gut microbiota. Several studies have shown a direct correlation between various gut microbiota and the etiology of obesity. Evidence suggests that certain species of gut bacteria may either produce microbial opioids, or facilitate conversion of dietary peptides into potent MOR ligands. Although no direct link has yet been found in the human gastrointestinal system the potential for the interaction of human gut flora and the nutropioid gut-brain circuitry is intriguing.