
Last Updated: June 19, 2026
Introduction to Nucleotide Biosynthesis and Catabolism
The metabolic requirements for the nucleotides and their cognate bases can be met by both dietary intake or biosynthesis de novo from low molecular weight precursors. However, the ability to salvage nucleotides from sources within the body alleviates any significant nutritional requirement for nucleotides, thus the purine and pyrimidine bases are not required in the diet. The nucleotide salvage pathways are, in part, intertwined with the processes of nucleotide catabolism. Nucleotide salvage represents a major source of nucleotides for synthesis of DNA, RNA and enzyme co-factors.
Within the body the major site of de novo nucleotide biosynthesis, for the replenishment and maintenance of intracellular pools, is the liver. Following their synthesis in the liver the nucleotides are dephosphorylated and in part phosphorolytically cleaved into nucleobases and ribose-1-phosphate for transport to the blood and then subsequent uptake by cells of the other organs.
Extracellular hydrolysis of ingested nucleic acids occurs through the concerted actions of endonucleases, phosphodiesterases, and nucleoside phosphorylases. Endonucleases degrade DNA and RNA at internal sites leading to the production of oligonucleotides. Oligonucleotides are further digested by phosphodiesterases that act from the ends inward yielding free nucleosides. The bases are hydrolyzed from nucleosides by the action of phosphorylases that yield ribose-1-phosphate and free nucleobases. If the nucleosides and/or bases are not re-utilized the purine bases are further degraded to uric acid and the pyrimidines to β-aminoiosobutyrate, NH3 and CO2.
Activation of Ribose-5-Phosphate
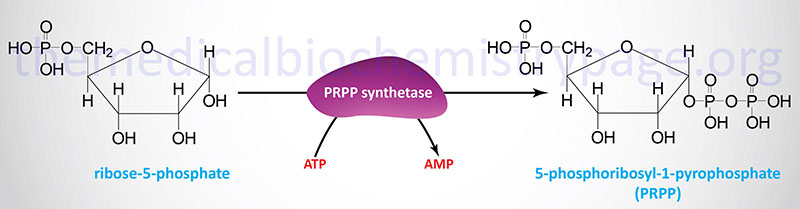
Both the salvage and de novo synthesis pathways of purine and pyrimidine biosynthesis lead to production of nucleoside-5′-phosphates through the utilization of an activated sugar intermediate and a class of enzymes called phosphoribosyltransferases. The activated sugar used is 5-phosphoribosyl-1-pyrophosphate, PRPP. PRPP is generated by the action of PRPP synthetase (also called ribose-phosphate pyrophosphokinase 1) and requires energy in the form of ATP.
At least three different enzymes with PRPP synthetase activity have been identified which are encoded by three distinct genes. These genes are identified as PRPS1, PRPS2, and PRPS1L1 (PRPS1-like 1). The PRPS1 and PRPS2 genes are both located on the X chromosome, PRPS1 is on the q arm (Xq22.3) and PRPS2 is on the p arm (Xp22.2). The PRPS1 gene is composed of 7 exons that generate two alternatively spliced mRNAs encoding isoform 1 (318 amino acids) and isoform 2 (114 amino acids). The PRPS2 gene is also composed of 7 exons that generate two alternatively spliced mRNAs encoding isoform 1 (321 amino acids) and isoform 2 (318 amino acids).
The PRPS1L1 gene is an intronless gene located on chromosome 7p21.1 that encodes a protein of 318 amino acids. The PRPS1L1 gene is expressed exclusively in the testes and translation of the resulting mRNA begins at a non-AUG codon (ACG). Although ACG normally codes for threonine, in the PRPS1L1 mRNA this alternative start codon directs the initiator methionine for the encoded protein.
PRPP Synthetase Mutations
Some mutations in the PRPS1 gene are associated with PRPP synthetase super activity. Because the PRPS1 gene is on the X chromosome the disease is classified as an X-linked recessive disorder. Most affected individuals experience a mild form of the disorder that manifests in late adolescence or early adulthood. Symptoms of the mild form of the disorder are primarily related to excess uric acid production causing uric acid crystalluria and urinary stones, followed by the development of gouty arthritis. Untreated the disorder will result in renal failure from uric acid crystal deposition. The more severe form manifests in infancy or early childhood with symptoms that include hypotonia, ataxia, sensorineural hearing loss, developmental delay, and intellectual impairment,
At the other end of the spectrum are mutations in the PRPS1 gene that result in severe or moderate loss of function of PRPP synthetase. The disorder associated with severe loss of function is called Arts syndrome. Arts syndrome is associated with profound sensorineural hearing loss, hypotonia, ataxia, developmental delay, and intellectual disability predominantly in males. Manifesting females experience much milder symptoms. In early childhood affected males will develop vision loss, peripheral neuropathy, and will have recurrent infections. As a result of the infections and the other complications of Arts syndrome, affected males often do not survive past early childhood.
The moderate loss of PRPP synthetase function results in the disorder called Charcot-Marie-Tooth disease X-linked recessive type 5 (CMTX5). CMTX5 is also known as Rosenberg-Chutorian syndrome. CMTX5 is not really a classical form of CMT disease and most investigators feel the designation is inappropriate for this form of disease which is associated with peripheral nerve problems, deafness, and vision loss.
Purine Nucleotide Biosynthesis
Synthesis of Inosine Monophosphate: IMP
The major site of purine synthesis is in the liver. Synthesis of the purine nucleotides begins with PRPP and leads to the first fully formed nucleotide, inosine 5′-monophosphate (IMP). This pathway is diagrammed below. The purine base without the attached ribose moiety is hypoxanthine. The purine base is built upon the ribose by several amidotransferase and transformylation reactions.
The synthesis of IMP requires five moles of ATP, two moles of glutamine, one mole of glycine, one mole of CO2, one mole of aspartate, and two moles of formate. The formyl moieties are carried on tetrahydrofolate (THF) in the form of N10-formyl-THF (10-formyltetrahydrofolate; 10-formylTHF).
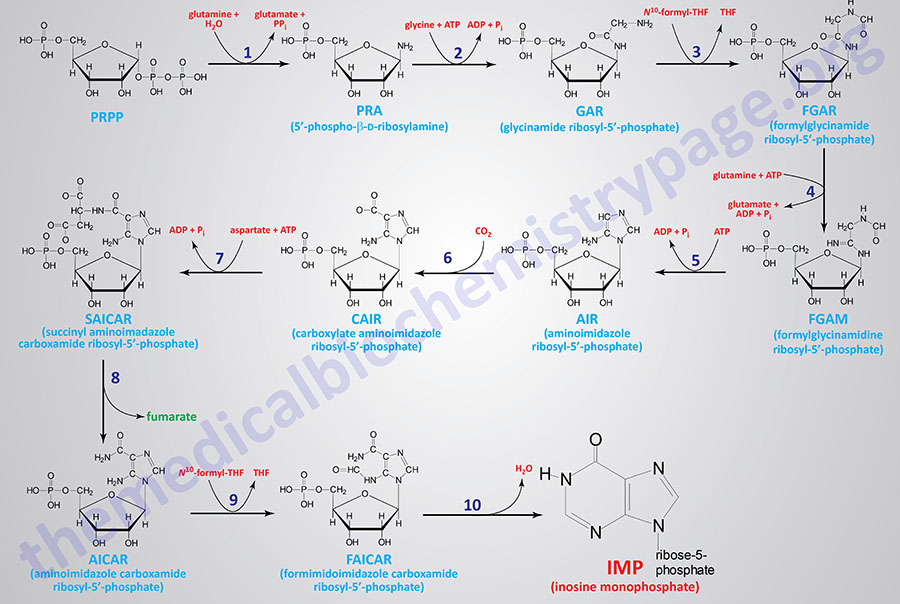
1. glutamine phosphoribosylpyrophosphate amidotransferase (GPAT activity of the PPAT gene)
2. glycinamide ribonucleotide synthetase (GARS activity of the GART gene)
3. glycinamide ribonucleotide formyltransferase (GART activity of the GART gene)
4. phosphoribosylformylglycinamide synthase (PFAS activity of the PFAS gene)
5. aminoimidazole ribonucleotide synthetase (AIRS activity of the GART gene)
6. aminoimidazole ribonucleotide carboxylase (AIRC activity of the PAICS gene)
7. succinylaminoimidazolecarboxamide ribonucleotide synthetase (SAICAR activity of the PAICS gene)
8. adenylosuccinate lyase (ADSL activity of the ADSL gene)
9. 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase (AICARFT activity of the ATIC gene)
10. IMP cyclohydrolase (IMPCH activity of the ATIC gene)
The first reaction (indicated by 1 in the Figure below) of purine synthesis is catalyzed by an enzyme called glutamine phosphoribosylpyrophosphate amidotransferase that is encoded by the PPAT gene (phosphoribosylpyrophosphate amidotransferase: PRPP amidotransferase). This enzyme is often referred to simply as amidotransferase. 5′-Phospho-β-D-ribosylamine (PRA; reaction 1 product) is also identified as 5-phosphoribosylamine. The PPAT gene is located on chromosome 4q12 which is composed of 11 exons that encode a 517 amino acid protein.
The activities that catalyze reactions 2, 3, and 5 are all contained in a single tri-functional enzyme encoded by the GART gene (phosphoribosyl-glycinamide formyltransferase, phosphoribosyl-glycinamide synthetase, phosphoribosyl-aminoimidazole synthetase). Glycinamide ribosyl-5′-phosphate (GAR; reaction 2 product) is also identified as 5′-phosphoribosyl-glycinamide. Formylglycinamide ribosyl-5′-phosphate (FGAR; reaction 3 product) is also identified as formylglycinamide ribonucleotide. Aminoimidazole ribosyl-5′-phosphate (AIR; reaction 5 product) is also identified as 5-aminoimidazole ribonucleotide. The GART gene is located on chromosome 21q22.11 and is composed of 24 exons that generate four alternatively spliced mRNAs. Three of the mRNAs from the GART gene all encode the same 1010 amino acid protein.
Reaction 4 of purine synthesis is catalyzed by phosphoribosyl-formylglycinamide synthase which is encoded by the PFAS gene. Formylglycinamidine ribosyl-5′-phosphate (FGAM; reaction 4 product) is also identified as 5′-phosphoribosylformylglycinamidine. The PFAS gene is located on chromosome 17p13.1 and is composed of 30 exons that encode a protein of 1338 amino acids.
Reactions 6 and 7 are catalyzed by a bi-functional enzyme encoded by the PAICS gene (phosphoribosyl-aminoimidazole carboxylase, phosphoribosyl-aminoimidazole succinocarboxamide synthetase). Carboxylate aminoimidazole ribosyl-5′-phosphate (CAIR; reaction 6 product) is also identified as 4-carboxy-5-aminoimidazole ribonucleotide. Succinyl aminoimidazole carboxamide ribosyl-5′-phosphate (SAICAR; reaction 7 product) is also identified as 5-aminoimidazole-4-(N-succinylcarboxamide) ribonucleotide and also as succinylaminoimidazole carboxamide ribotide. The PAICS gene is located on chromosome 4q12 closely associated with the PPAT gene whose encoded enzyme catalyzes the first step of purine synthesis. The PAICS gene is composed of 21 exons that generate six alternatively spliced mRNAs that collectively encode five different protein isoforms.
Expression of both the PPAT and PAICS genes is coordinately regulated.
Reaction 8 of purine synthesis is catalyzed by adenylosuccinate lyase. Adenylosuccinate lyase is encoded by the ADSL gene. Aminoimidazole carboxamide ribosyl-5′-phosphate (AICAR; reaction 8 product) is also identified as 5-aminoimidazole-4-carboxamide ribonucleotide. The ADSL gene is located on chromosome 22q13.1 and is composed of 14 exons that generate seven alternatively spliced mRNAs, each of which encode a distinct protein isoform.
The last two reactions (9 and 10) are catalyzed by a bi-functional enzyme encoded by the ATIC gene (5-aminoimidazole-4-carboxyamide ribonucleotide formyltransferase, IMP cyclohydrolase). Formimidoimidazole carboxamide ribosyl-5′-phosphate (FAICAR; reaction 9 product) is also identified as 5-formamidoimidazole-4-carboxamide ribotide. The ATIC gene is located on chromosome 2q35 and is composed of 20 exons that encode a protein of 592 amino acids.
Adenylosuccinate Lyase Deficiency, ADSLD
Mutations in the ADSL gene result in the very rare autosomal recessive disorder identified as adenylosuccinate lyase deficiency, ADSLD. ADSLD is a disorder whose symptoms are the result of the accumulation of the toxic intermediates, succinyladenosine (S-Ado) and succinylaminoimidazole carboxamide riboside (SAICAr). SAICAr is the dephosphorylated form of succinylaminoimidazole carboxamide ribotide (SAICAR), the intermediate in de novo purine synthesis. S-Ado is derived from adenylosuccinate, an intermediate in the synthesis of AMP from IMP.
Around 80 cases of ADSLD have been identified. Diagnosis of ADSLD is made by measurement of SAICAr and S-Ado in the blood and urine as well as cerebrospinal fluid. The ratio of S-Ado to SAICAr is diagnostic for the type and severity of the disease.
To date over 50 different mutations in the ADSL gene have been identified in ADSLD patients. Most identified mutations are missense mutations.
Three clinical phenotypes of ADSLD have been defined with the most severe form being a fatal neonatal encephalopathy as well as type I (severe) ADSLD and type II (moderate) ADSLD. The severity of clinical symptoms in ADSLD correlate with the level of residual ADSL activity.
The fatal neonatal form of ADSLD manifests with encephalopathy and a lack of spontaneous movement. In addition these patients experience respiratory failure and intractable seizures that will lead to early death within the first weeks of life. The fatal neonatal form of ADSLD is also associated with prenatal symptoms that include impaired intrauterine growth and microcephaly. The S-Ado/SAICAr ratio in the neonatal fatal form of ADSLD is less than 1.
Type I ADSLD represents is the most commonly identified form of the disease and presents within the first few months of life with severe psychomotor retardation, microcephaly, and early onset of seizures. The S-Ado/SAICAr ratio in type I ADSLD is essentially 1.
Type II ADSLD is associated with symptoms that appear in the first years of life that include slight to moderate psychomotor retardation and transient contact disturbances. Patients with type II ADSLD may not experience seizures but if they do appear it is not until the 2nd to 4th year of life. Additional symptoms include ataxia leading to increased disturbances in gait as well as speech impairment with a minimal use of words. The S-Ado/SAICAr ratio in type II ADSLD is more than 2.
Synthesis of AMP and GMP from IMP
IMP represents a branch point for purine biosynthesis, because it can be converted into either AMP or GMP through two distinct reaction pathways. The pathway leading to AMP requires energy in the form of GTP; that leading to GMP requires energy in the form of ATP. The utilization of GTP in the pathway to AMP synthesis allows the cell to control the proportions of AMP and GMP to near equivalence. The accumulation of excess GTP will lead to accelerated AMP synthesis from IMP instead, at the expense of GMP synthesis. Conversely, since the conversion of IMP to GMP requires ATP, the accumulation of excess ATP leads to accelerated synthesis of GMP over that of AMP.
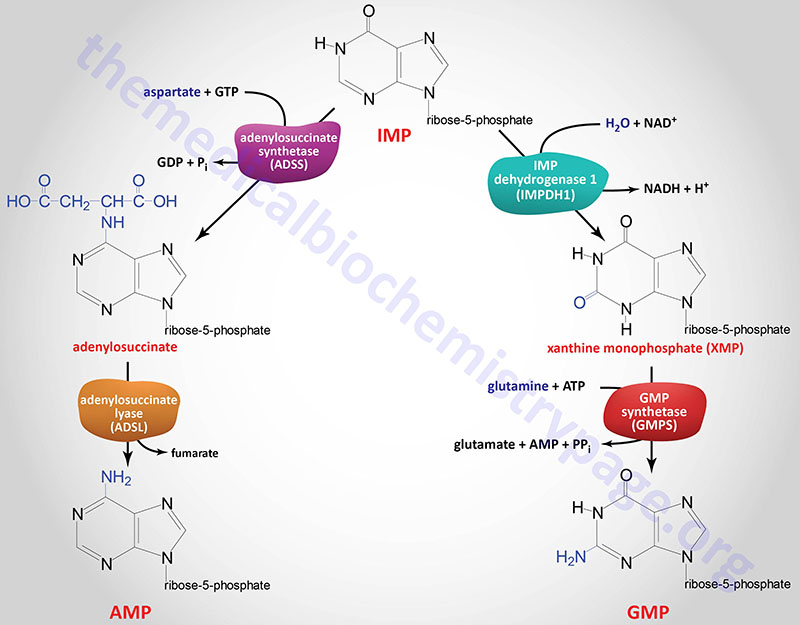
The two enzymes in the IMP to AMP pathway are adenylosuccinate synthetase and adenylosuccinate lyase. Humans express two adenylosuccinate synthetase genes, ADSS1 and ADSS2.
The ADSS1 gene is located on chromosome 14q32.33 and is composed of 16 exons that generate three mRNAs via alternative splicing and alternative promoter usage. All three mRNAs encode different protein isoforms.
The ADSS2 gene is located on chromosome 1q44 and is composed of 15 that generate two alternatively spliced mRNAs, both of which encode distinct protein isoforms.
The adenylosuccinate lyase in this pathway is the same enzyme that catalyzes reaction 8 of de novo purine biosynthesis as described above.
The two enzymes in the IMP to GMP pathway are inosine-5′-monophosphate dehydrogenase (IMP dehydrogenase: IMPDH) and GMP synthetase. Humans express two IMPDH genes identified as IMPDH1 and IMPDH2.
The IMPDH1 gene is located on chromosome 7q32.1 and is composed of 18 exons that generate eight alternatively spliced mRNAs encoding eight protein isoforms.
The IMDPH2 gene is located on chromosome 3p21.31 and is composed of 15 exons that generate four alternatively spliced mRNAs, each of which encode a distinct protein isoform.
Expression of IMPDH1 predominates in the retina, spleen, and resting peripheral blood mononuclear cells but like the IMPDH2 gene is also expressed in most tissues at varying levels. Regardless of the tissue, IMPDH1 is expressed constitutively at low levels. Expression of IMPDH2 is enhanced during proliferation and transformation.
GMP synthetase is encoded by the GMPS gene. The GMPS gene is located on chromosome 3q25.31 and is composed of 19 exons that encode a 693 amino acid protein.
Regulation of Purine Nucleotide Synthesis
The essential rate limiting steps in purine biosynthesis occur at the first two steps of the pathway. The synthesis of PRPP by PRPP synthetase is feed-back inhibited by purine-5′-nucleotides (predominantly AMP and GMP). Combinatorial effects of those two nucleotides are greatest, e.g., inhibition is maximal when the correct concentration of both adenine and guanine nucleotides is achieved.
The amidotransferase reaction catalyzed by PRPP amidotransferase is also feed-back inhibited allosterically by binding ATP, ADP, and AMP at one inhibitory site and GTP, GDP, and GMP at another. Conversely the activity of the enzyme is stimulated by PRPP.
Additionally, purine biosynthesis is regulated in the branch pathways from IMP to AMP and GMP. The accumulation of excess ATP leads to accelerated synthesis of GMP, and excess GTP leads to accelerated synthesis of AMP.
Catabolism of Purine Nucleotides
Catabolism of the purine nucleotides (both ribonucleotides and deoxyribonucleotides) leads ultimately to the production of uric acid which is insoluble and is excreted in the urine. Uric acid excretion and reabsorption occurs within the proximal tubules of the kidney. Elevation in uric acid levels can result in precipitation of urate crystals with monosodium urate crystals being the most commonly occurring in the synovial fluids of the joints.
The catabolism of purine nucleotides involves deamination reaction, phosphate removal from the nucleoside monophosphates, phosphorylytic removal of the ribose yielding ribose-1-phosphate, and finally oxidation of the nucleobases to uric acid.
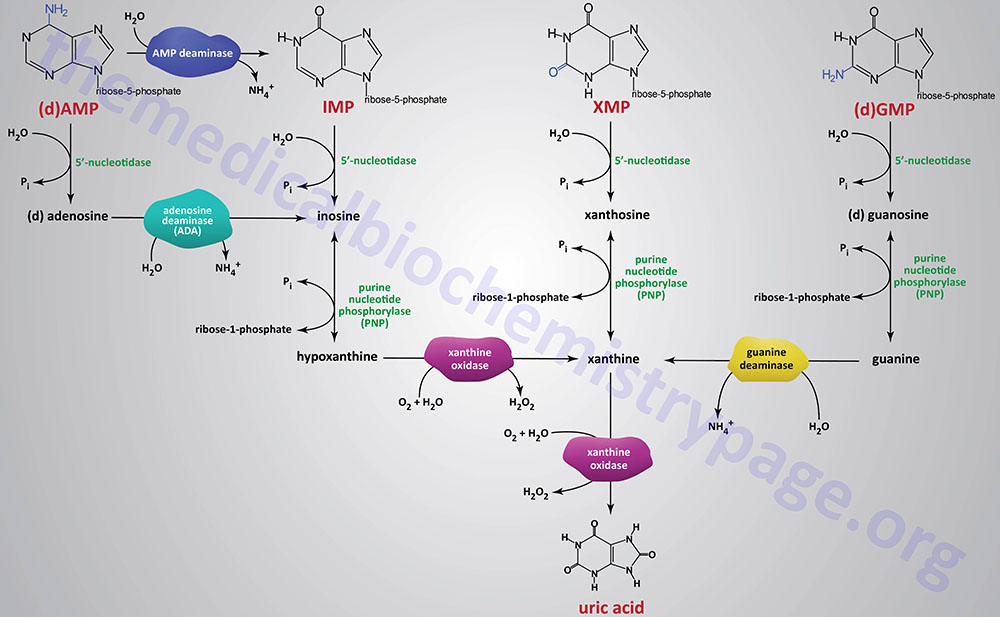
Dephosphorylation of nucleoside monophosphates is catalyzed by 5′-nucleotidases. Humans express seven 5′-nucleotidase genes with five encoding cytosolic enzymes, one encoding a mitochondrially localized enzyme, and one gene encoding an extracellular enzyme that is tethered to the plasma membrane via a GPI linkage.
Catabolism of AMP and dAMP can occur by conversion first to IMP through the action of AMP deaminase or following 5′-nucleotidase action generating adenosine or deoxyadenosine. In this latter pathway the nitrogen is removed from adenosine generating inosine by the critical enzyme, adenosine deaminase, ADA.
Humans express three AMP deaminase genes: AMPD1, AMPD2, and AMPD3.
The AMPD1 gene is located on chromosome 1p13.2 and is composed of 16 exons that generate two alternatively spliced mRNAs encoding isoform 1 (747 amino acids) and isoform 2 (776 amino acids). The AMPD1 encoded enzymes are primarily responsible for skeletal muscle deamination of AMP. Mutations in the AMPD1 gene represent the most common cause of metabolic myopathy.
The AMPD2 gene is located on chromosome 1p13.3 and is composed of 20 exons that generate five alternatively spliced mRNAs that collectively encode four distinct protein isoforms. The AMPD2 encoded enzymes are primarily responsible for liver deamination of AMP.
The AMPD3 gene is located on chromosome 11p15.4 and is composed of 19 exons that generate five alternatively spliced mRNAs that collectively encode four distinct protein isoforms. The AMPD3 encoded enzymes are primarily responsible for erythrocyte deamination of AMP.
The ADA gene is located on chromosome 20q13.12 and is composed of 12 exons that generate three alternatively spliced mRNAs, each of which encode a distinct protein isoform. The highest levels of ADA expression are observed in the duodenum of the small intestines. Loss of ADA activity results in the potentially lethal disorder, ADA-deficient severe combined immunodeficiency, SCID.
The ribose is removed from the nucleotides by purine nucleoside phosphorylase (PNP) yielding the nucleobases, hypoxanthine, xanthine, and guanine. Purine nucleotide phosphorylase is encoded by the PNP gene. The PNP gene is located on chromosome 14q11.2 and is composed of 6 exons that encode a 289 amino acid protein.
The nitrogen is removed from guanine by guanine deaminase yielding xanthine. Guanine deaminase is encoded by the GDA gene. The GDA gene is located on chromosome 9q21.13 and is composed of 27 exons that generate seven alternatively spliced mRNAs that collectively encode five distinct protein isoforms.
Hypoxanthine and xanthine are then converted to the terminal product of purine catabolism, uric acid, by the enzyme xanthine oxidase. The enzymatic activity called xanthine oxidase is the term used for the modified form of the enzyme xanthine dehydrogenase which is a molybdenum-dependent hydroxylase that functions as a homodimer. The conversion xanthine dehydrogenase to xanthine oxidase results from reversible sulfhydryl oxidation as well as from irreversible proteolytic action.
Xanthine dehydrogenase is encoded by the XDH gene. The XDH gene is located on chromosome 2p23.1 and is composed of 37 exons that generate a 1337 amino acid protein.
Salvage of Purine Nucleotides
The re-synthesis of nucleotides from the purine bases and purine nucleosides takes place in a series of steps known as the salvage pathways. The free purine bases, adenine, guanine, and hypoxanthine, can be reconverted to their corresponding nucleotides by phosphoribosylation where PRPP, like in the de novo synthesis pathway, serves as the activated form of ribose-5′-phosphate.
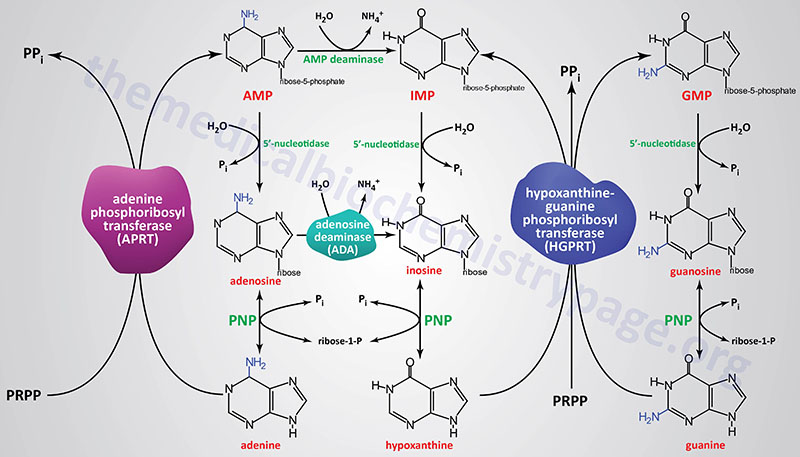
Most all cells carry out purine nucleotide salvage, with the notable exception being lymphocytes in which the activities of the purine salvage pathways are reduced compared to other cells which makes lymphocytes particularly dependent on the pathways of de novo purine synthesis for proliferation. This dependence of lymphocyte proliferation on de novo purine synthesis makes them particularly susceptible to inhibition of enzymes such as IMP dehydrogenase. Mycophenolic acid, an inhibitor of both the IMPDH1 and IMPDH2 encoded enzymes is often used as an immunosuppressant in patients following kidney transplantation.
Two key transferase enzymes are involved in the salvage of purines: adenosine phosphoribosyltransferase (APRT), which catalyzes the following reaction:
adenine + PRPP ↔ AMP + PPi
and hypoxanthine-guanine phosphoribosyltransferase (HGPRT), which catalyzes the following reactions:
hypoxanthine + PRPP ↔ IMP + PPi
guanine + PRPP ↔ GMP + PPi
Adenosine phosphoribosyltransferase is encoded by the APRT gene. The APRT gene is located on chromosome 16q24.3 and is composed of 5 exons that generate two alternatively spliced mRNAs encoding isoform a (180 amino acids) and isoform b (134 amino acids).
Hypoxanthine-guanine phosphoribosyltransferase is encoded by the HPRT1 gene. The HPRT1 gene is located on the X chromosome (Xq26.2–q26.3) and is composed of 9 exons that encode a 218 amino acid protein.
A critically important enzyme of purine salvage in rapidly dividing cells is adenosine deaminase (ADA) which catalyzes the deamination of adenosine to inosine. Deficiency in ADA results in a form of severe combined immunodeficiency, SCID (and briefly outlined below).
Additional enzymes are utilized in the salvage of the deoxynucleotides. These additional enzymes have overlapping specificities and their activities control the rate limiting phosphorylation of deoxynucleosides. These additional enzymes are deoxycytidine kinase, deoxyguanosine kinase, and the two forms of thymidine kinase, one that is cytosolic (TK1) and one that is mitochondrial (TK2).
Deoxycytidine kinase is encoded by the DCK gene which is located on chromosome 4q13.3 and is composed of 8 exons that generate seven alternatively spliced mRNAs that collectively encode five distinct protein isoforms. Deoxycytidine kinase can phosphorylate deoxyadenine, deoxycytidine, and deoxyguanosine.
Deoxyguanosine kinase is a mitochondrial enzyme that is encoded by the DGUOK gene. The DGUOK gene is located on chromosome 2p13.1 and is composed of 8 exons that generate seven alternatively spliced mRNAs, that collectively encode five distinct protein isoforms. Deoxyguanosine kinase can phosphorylate deoxyadenine, deoxycytidine, and deoxyguanosine in the mitochondria.
The TK1 gene is located on chromosome 17q25.3 and is composed of 6 exons that generate three alternatively spliced mRNAs, each of which encode a distinct protein isoform. The TK1 encoded proteins phosphorylate thymidine generating dTMP. Expression of the TK1 gene is regulated in a cyclic manner with peak expression during S-phase (DNA synthesis). The product of the TK1 reaction, dTMP, is ultimately converted to dTTP through the actions of thymidylate kinase (encoded by the DTYMK gene) and then nucleotide diphosphate kinases. The resultant dTTP serves as a feed-back inhibitor of the thymidine kinase, thus regulating the cellular levels of dTTP.
The TK2 gene is located on chromosome 16q21 and is composed of 11 exons that generate seven alternatively spliced mRNAs, each of which encode a distinct protein isoform. The TK2 encoded proteins are responsible for mitochondrial DNA replication by phosphorylation of thymidine, deoxycytidine, and deoxyuridine.
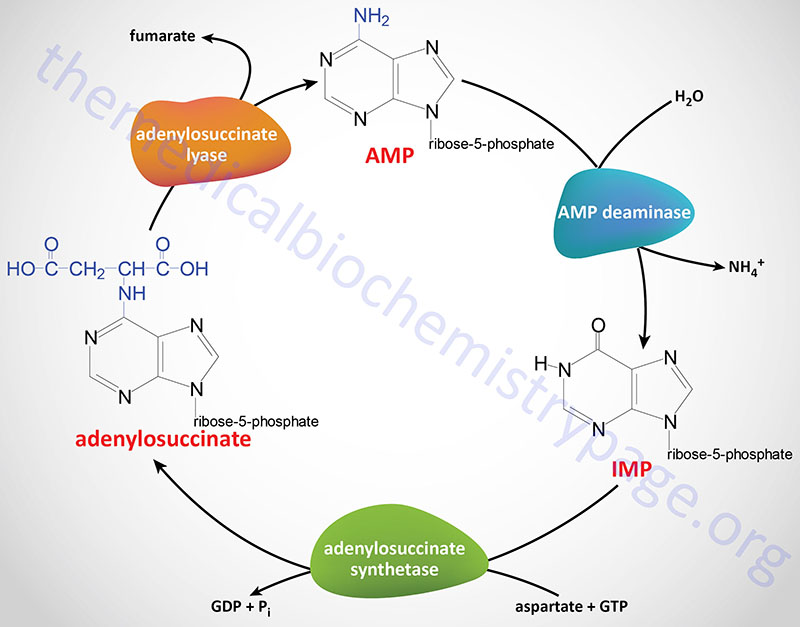
Purine Nucleotide Cycle
The synthesis of AMP from IMP and the salvage of IMP via AMP catabolism have the net effect of deaminating aspartate to fumarate. This process has been termed the purine nucleotide cycle (see diagram below). This cycle is a critical metabolic process in skeletal muscle cells. During intense muscle activity the demand for ATP increases with a concomitant demand to increase the level of TCA cycle activity, in order to generate more NADH for the production of ATP. However, skeletal muscle cells lack many of the enzymes of the major anaplerotic reactions that provide substrates for the TCA cycle. In these cells the fumarate, produced via the purine nucleotide cycle, as well as succinate, and malate represent the primary anaplerotic intermediates for enhanced muscle TCA cycle activity.
In the kidney the purine nucleotide cycle represents a means to release ammonia without increases in blood levels.
Clinical Significances of Purine Metabolism
Clinical problems associated with nucleotide metabolism in humans are predominantly the result of abnormal catabolism of the purines. The clinical consequences of abnormal purine metabolism range from mild to severe and even fatal disorders. Clinical manifestations of abnormal purine catabolism arise from the insolubility of the degradation byproduct, uric acid.
Metabolic Disturbances Resulting from Hyperuricemia
Elevated levels of uric acid have been found to be directly correlated to the development of metabolic dysfunction-associated fatty liver disease (MAFLD), diabetes, the metabolic syndrome, and obesity. In addition, hyperuricemia is associated with atherosclerosis, hypertension, dyslipidemia, and increased likelihood for stroke. MAFLD is also referred to as metabolic dysfunction-associated steatotic liver disease, MASLD. MAFLD was formerly referred to as non-alcoholic fatty liver disease, NAFLD.
Within the liver, as well as other tissues such as the kidneys, adipose tissue, and the vasculature, high levels of uric acid are associated with increased mitochondrial stress resulting in elevation in reactive oxygen species (ROS) production. The increased ROS production by uric acid is, in part, the result of the translocation of the catalytic (NOX4) subunit of NADPH oxidase to the mitochondria. Additionally, high uric acid levels contribute to the activation of endoplasmic reticulum (ER) stress.
The increase in mitochondrial ROS production by uric acid results in inhibition of aconitase of the TCA cycle leading to accumulation of citrate. The citrate is then transported out to the cytosol where the action of ATP-citrate lyase generates acetyl-CoA and oxaloacetate. The increased production of acetyl-CoA is a direct contributor to the increased production of fatty acids and ultimately triglycerides and the consequent infiltration of fat in tissues, particularly the liver.
Reactive oxygen species are known to activate kinases of the MAP kinase family such as JNK. The activation of JNK results in the phosphorylation and activation of the JUN transcription factor. JUN is known to activate transcription of the ACC1 and FAS genes contributing to increased fatty acid synthesis. ER stress results in the increased cleavage and release of SREBP-1c contributing to the increased expression of lipogenic genes with the consequences being increased fat deposition in tissues such as the liver.
Gout
The details of gout are covered in the Gout: Hyperuricemia page. Briefly, gout is a condition that results from the precipitation of urate as monosodium urate (MSU) or calcium pyrophosphate dihydrate (CPPD) crystals in the synovial fluid of the joints, leading to severe inflammation and arthritis. The inflammatory response is due to the crystals engaging the caspase-1-activating inflammasome resulting in the production of interleukin-1β (IL-1β) and IL-18. Most forms of gout are the result of excess purine production and consequent catabolism or to a partial deficiency in the salvage enzyme, HGPRT.
Most forms of gout can be treated by administering the antimetabolite: allopurinol. Allopurinol is specifically a pro-drug that is metabolized to oxypurinol which is a structural analog of hypoxanthine that strongly inhibits xanthine oxidase.
Lesch-Nyhan Syndrome and SCID
Two severe disorders, both quite well described, are associated with defects in purine metabolism: Lesch-Nyhan syndrome and the ADA-deficient form of severe combined immunodeficiency disease (SCID).
Lesch-Nyhan syndrome is an X-linked recessive disorder that results from the loss of a functional hypoxanthine-guanine phosphoribosyltransferase which is encoded by the HPRT1 gene (also often called HGPRT). Patients who inherit loss of function mutations in the HPRT1 gene exhibit not only severe symptoms of gout but also a severe malfunction of the nervous system. In the most serious cases, patients resort to self-mutilation. Common intervention in the self mutilation behavior is to surgically remove the patients teeth following acquisition of the adult teeth. Death usually occurs before patients reach their 20th year.
SCID refers to a group of potentially fatal disorders due to a combined loss of function of both T- and B-lymphocytes. There are at least 13 known and characterized genetic causes of SCID. The most common (45%) cause of SCID is the X-linked disorder resulting from loss of function of the common gamma (γ) chain of the T-cell receptor and other interleukin (IL) receptors. The second most common (15%) form of SCID is caused by defects in the enzyme adenosine deaminase, ADA. This is the enzyme responsible for converting adenosine to inosine in the catabolism of the purines. This deficiency selectively leads to a destruction of B and T lymphocytes, the cells that mount immune responses. In the absence of ADA, deoxyadenosine is phosphorylated to yield levels of dATP that are 50-fold higher than normal. The levels are especially high in lymphocytes, which have abundant amounts of the salvage enzymes, including nucleoside kinases. High concentrations of dATP inhibit ribonucleotide reductase (see below), thereby preventing other dNTPs from being produced. The net effect is to inhibit DNA synthesis.
Since lymphocytes must be able to proliferate dramatically in response to antigenic challenge, the inability to synthesize DNA seriously impairs the immune responses, and the disease The accumulating dATP also induces DNA strand breakage in non-dividing lymphocytes via a direct activation of a major protease (caspase 9) involved in apoptosis (programmed cell death). In addition, S-adenosylhomocysteine hydrolase activity is markedly inhibited by 2′-deoxyadenosine resulting in accumulation of S-adenosylhomocysteine which in turn results in reduced synthesis of S-adenosylmethionine (AdoMet), a critical substrate in transmethylation reactions. ADA deficient SCID is usually fatal in infancy unless special protective measures are taken. A less severe immunodeficiency results when there is a lack of purine nucleoside phosphorylase (PNP) activity, another purine degrading enzyme.
One of the many glycogen storage diseases von Gierke disease also leads to excessive uric acid production. This disorder results from a deficiency in glucose 6-phosphatase activity. The increased availability of glucose-6-phosphate increases the rate of flux through the pentose phosphate pathway, yielding an elevation in the level of ribose-5-phosphate and consequently PRPP. The increases in PRPP then result in excess purine biosynthesis followed by catabolism to uric acid.
Table of Disorders of Purine Metabolism
| Disorder | Affected Gene | Nature of Defect | Comments |
| Gout | PRPS1 | increased enzyme activity | increased PRPP synthetase activity results in excess purine synthesis and consequent catabolism to uric acid resulting in hyperuricemia |
| Gout | HPRT1 | enzyme deficiency | hyperuricemia |
| Gout | G6PC1 | enzyme deficiency | hyperuricemia; enzyme is deficient in von Gierke disease |
| Lesch-Nyhan syndrome | HPRT1 | lack of enzyme | associated with bizarre self-mutilating behavior in addition to hyperuricemia and gout |
| ADA-deficient SCID | ADA | lack of enzyme | ADA (adenosine deaminase) deficient SCID represents 15% of all cases of SCID |
| Immunodeficiency | PNP | lack of enzyme | decreased T-cells; increased risk for autoimmune disorders; also associated with spasticity, developmental delay, intellectual disability, and ataxia |
| Renal lithiasis | APRT | lack of enzyme | associated with excess production and renal excretion of 2,8-dihydroxyadenine (DHA) resulting in DHA crystal nephropathy |
| Xanthinuria | XDH | lack of enzyme | very low uric acid (hypouricemia) associated with high levels of xanthine in blood and urine; can lead to renal xanthine stones and kidney failure |
| von Gierke disease | G6PC1 | enzyme deficiency |
Pyrimidine Nucleotide Biosynthesis
Synthesis of the pyrimidines is less complex than that of the purines, since the base is much simpler. The first completed base is derived from one mole of glutamine, one mole of ATP and one mole of CO2 (which form carbamoyl phosphate) and one mole of aspartate. An additional mole of glutamine and ATP are required in the conversion of UTP to CTP. The pathway of pyrimidine biosynthesis is diagrammed below.
The synthesis of pyrimidines differs in two significant ways from that of purines. First, the ring structure is assembled as a free base, not built upon PRPP. PRPP is added to the first fully formed pyrimidine base (orotic acid), forming orotate monophosphate (OMP), which is subsequently decarboxylated to UMP. Second, there is no branch in the pyrimidine synthesis pathway.
The carbamoyl phosphate used for pyrimidine nucleotide synthesis is derived from glutamine and bicarbonate, within the cytosol, as opposed to the urea cycle carbamoyl phosphate derived from ammonia and bicarbonate in the mitochondrion. The urea cycle reaction is catalyzed by carbamoyl phosphate synthetase 1 (CPS1, CPS-1, CPS-I) whereas the pyrimidine nucleotide precursor is synthesized by the CPS2 (CPS-2, CPS-II) activity of the tri-functional rate-limiting enzyme of pyrimidine nucleotide biosynthesis.
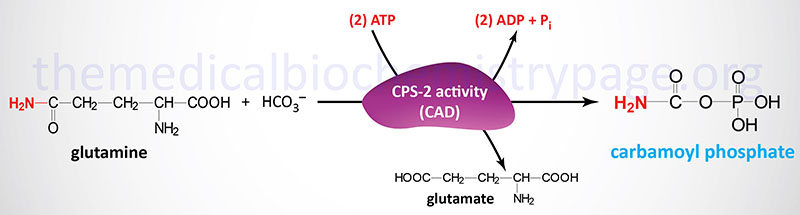
The carbamoyl phosphate that is produced by this enzyme is then condensed with aspartate in the second step of the reaction catalyzed by the aspartate transcarbamylase (ATCase) activity of the enzyme.
The third step of pyrimidine nucleotide biosynthesis is catalyzed by the dihydroorotase activity (previously referred to as carbamoyl aspartate dehydratase) of the tri-functional enzyme. The official name for this tri-functional enzyme is carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase.
Carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase are encoded by the CAD gene. The CAD gene is located on chromosome 2p23.3 which is composed of 45 exons that generate two alternatively spliced mRNAs, one encoding a protein of 2225 amino acids (isoform 1) and the other a protein of 2162 amino acids (isoform 2).
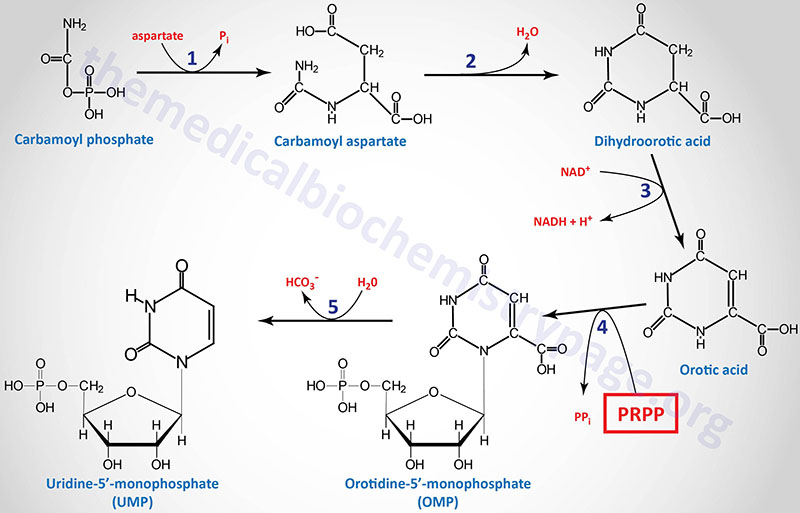
1. aspartate transcarbamylase (ATCase) activity of the CAD gene encoded enzyme
2. dihydroorotase (previously called carbamoyl aspartate dehydratase) activity of the CAD gene encoded enzyme
3. dihydroorotate dehydrogenase (DHODH gene activity)
4. orotate phosphoribosyltransferase activity of the UMPS gene
5. orotidine-5′-phosphate decarboxylase (OMP decarboxylase) activity of the UMPS gene
Following the synthesis of dihydroorotic acid by the tri-functional CAD enzyme the compound is oxidized to orotic acid by dihydroorotate dehydrogenase (reaction 3). Dihydroorotate dehydrogenase is a mitochondrial enzyme tethered to the outer face of the inner mitochondrial membrane.
Dihydroorotate dehydrogenase is encoded by the DHODH gene. The DHODH gene is located on chromosome 16q22.2 and is composed of 10 exons that encode a protein of 395 amino acids.
The last two reactions (4 and 5), which generate the first fully formed pyrimidine nucleotide (UMP), are catalyzed by the bi-functional homodimeric enzyme identified as UMP synthetase. This enzyme possesses orotate phosphoribosyltransferase activity in the N-terminal domain and OMP decarboxylase activity in the C-terminal domain.
UMP synthetase is encoded by the UMPS gene. The UMPS gene is located on chromosome 3q21.2 and is composed of 7 exons that encode a protein of 480 amino acids.
Following the completion of UMP synthesis this nucleotide is phosphorylated twice to yield UTP (ATP is the phosphate donor). The first phosphorylation is catalyzed by uridylate kinase and the second by ubiquitous nucleoside diphosphate kinase. The synthesis of CTP occurs through the amination of UTP by the action of CTP synthase.
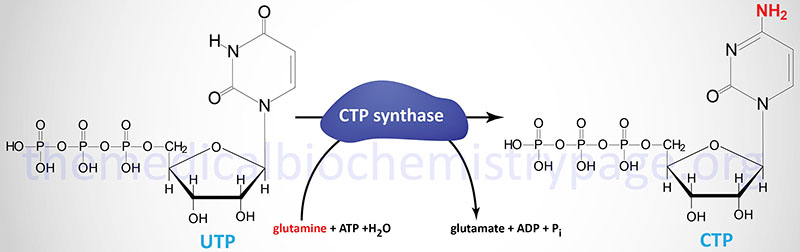
Humans possess two distinct CTP synthase genes, CTPS1 and CTPS2. The CTPS1 gene is located on chromosome 1p34.2 and is composed of 22 exons that generate two alternatively spliced mRNAs, both of which encode distinct protein isoforms.
The CTPS2 gene is located on chromosome Xp22.2 and is composed of 25 exons that generate three alternatively spliced mRNAs, each of which encode the same 586 amino acid protein.
Roles of Pyrimidine Nucleotides Beyond DNA and RNA Synthesis
In addition to their roles in the synthesis of DNA and RNA, the pyrimidines CTP and UTP are important in numerous other metabolic processes.
CTP serves an important function in phospholipid synthesis through activation of choline (as UDP-choline) and ethanolamine (as UDP-ethanolamine) for the synthesis of phosphatidylcholines, phosphatidylethanolamines, and phosphatidylserines. CTP is also used to activate diacylglycerol (as UDP-diacylglycerol) for the synthesis of cardiolipins and other diphosphatidylglycerols.
UTP serves an important function in the activation of various carbohydrates (referred to as nucleotide sugars) for use in various glycosylation reactions that include protein glycosylation. UTP is used to activate glucose (UDP-glucose), galactose (UDP-galactose), and N-acetylgalactosamine (UDP-GalNAc).
When energy levels are reduced the actions of uridine salvage enzymes, the uridine phosphorylases, predominate in the direction of the release of ribose-1-phosphate (R1P) and uracil from uridine. The ribose-1-phosphate can then contribute to glucose homeostasis by first being converted to fructose-6-phosphate (F6P) and glyceraldehyde-3-phosphate by the non-oxidative branch of the pentose phosphate pathway (PPP). In this pathway the uridine-derived R1P is converted to ribose-5-phosphate (R5P) via the activity of phosphoglucomutase (primarily the PGM2 encoded enzyme). Through the actions of enzymes of the PPP the R5P, and other intermediates of the PPP, are ultimately converted to F6P and glyceraldehyde-3-phosphate. Both F6P and glyceraldehyde-3-phosphate can contribute to ATP production via glycolysis as well as glucose synthesis via gluconeogenesis. The ability of uridine to contribute to overall glucose and energy homeostasis is not limited to any specific tissue. Indeed, the process occurs throughout the body.
Role of Pyrimidine Nucleotides in Lipid Metabolism
Inhibition of pyrimidine biosynthesis, by deletion of the CAD, or DHODH, or UMPS genes, has been shown to correlate with deficiency in pyruvate production. Given that pyruvate is a major source of acetyl-CoA for lipid (fatty acids, cholesterol, etc.) biosynthesis, the role of pyrimidines in pyruvate production is critical for overall lipogenesis.
The mechanism by which pyrimidines contribute to the generation of pyruvate is via the regulation of the activity of the PDH complex (PDHc). The PDHc is a multi-subunit complex that requires several vitamin-derived cofactors including thiamine pyrophosphate (TPP), NAD+, FAD, CoASH, and lipoic acid.
The production of TPP from thiamine requires the enzyme thiamine pyrophosphokinase 1, TPK1. The activity of TPK1 was originally thought to be dependent on ATP as the phosphate donor, however, recent work has shown that the enzyme requires UTP, not ATP as the phosphate donor. This is the reason that pyrimidine depletion, or blocking of pyrimidine biosynthesis, results in severely impaired lipogenesis.
Synthesis of the Thymine Nucleotides
The thymine nucleotides can be derived by de novo synthesis from dUMP or by salvage pathways from deoxyuridine or deoxythymidine.
The de novo pathway to dTTP synthesis first requires the use of dUMP from the metabolism of either UDP or CDP. The dUMP is converted to dTMP (5-methyl-dUMP) by the action of thymidylate synthase (sometimes called thymidylate synthetase as in the case of the gene name). The methyl group (recall that thymine is 5-methyl uracil) is donated by N5,N10-methylene THF. The unique property of the action of thymidylate synthase is that in the course of the reaction the THF is converted to dihydrofolate (DHF), the only such reaction yielding DHF from a THF derivative.
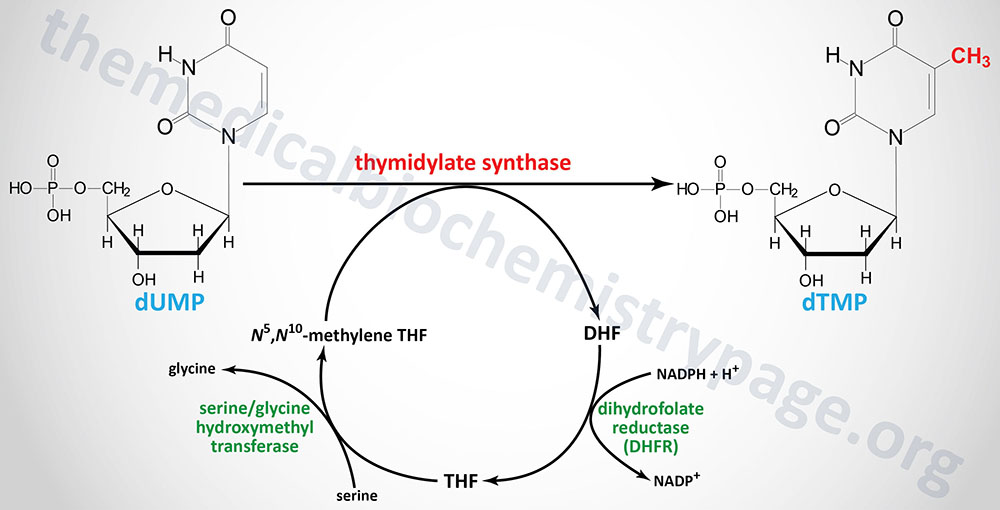
Thymidylate synthase is encoded by the TYMS gene. The TYMS gene is located on chromosome 18p11.32 and is composed of 8 exons that generate three alternatively spliced mRNAs, each of which encode distinct protein isoforms. A naturally occurring antisense RNA (NAT) is derived by transcription in the opposite direction to a portion of the 3′-end of the TYMS gene. This NAT was originally identified as rTSα but the gene is now identified as ENOSF1 (enolase superfamily member 1). Expression of TYMS and the antisense RNA varies inversely during progression through the cell cycle.
In order for the thymidylate synthase reaction to continue, THF must be regenerated from DHF. This is accomplished through the action of dihydrofolate reductase which is encoded by the DHFR gene. The DHFR gene is located on chromosome 5q14.1 and is composed of 6 exons that generate three alternatively spliced mRNAs that encode three distinct isoforms of DHFR.
THF is then converted to N5,N10-methylene THF via the action of serine hydroxymethyl transferase (encoded by the SHMT1 gene). The crucial role of DHFR in thymidine nucleotide biosynthesis makes it an ideal target for chemotherapeutic agents (see below).
Clinical Relevance of Tetrahydrofolate
Tetrahydrofolate (THF) is regenerated from the dihydrofolate (DHF) product of the thymidylate synthetase reaction by the action of dihydrofolate reductase (DHFR), an enzyme that requires NADPH. Cells that are unable to regenerate THF suffer defective DNA synthesis and eventual death. For this reason, as well as the fact that dTTP is utilized only in DNA, it is therapeutically possible to target rapidly proliferating cells over non-proliferating cells through the inhibition of thymidylate synthetase or through inhibition of DHFR.
The class of molecules used to inhibit thymidylate synthetase are fluorinated pyrimidines. Molecules of this class include 5-fluorouracil (fluorouracil: 5-FU), 5-fluorodeoxyuridine (floxuridine; FUdR), and 5-fluoro-2′-deoxyuridine monophosphate (FUdR-MP). Fluorouracil within cells to numerous metabolic intermediates including FUdR-MP (also written FdUMP), 5-fluorodeoxyuridine triphosphate (FdUTP), and 5-fluorouridine triphosphate (FUTP). It is FUdR-MP (FdUMP) that inhibits thymidylate synthetase. The other major metabolites, FdUTP and FUTP, inhibit DNA synthesis and repair and RNA synthesis, respectively. The many different DHFR inhibitors are termed antifolates or antimetabolites and include methotrexate, aminopterin, trimethoprim, and pyrimethamine. Each of these is a structural analog of folic acid, hence the term antifolate.
Regulation of Pyrimidine Biosynthesis
The regulation of pyrimidine synthesis occurs mainly at the first step which is catalyzed by the trifunctional enzyme encoded by the CAD gene. The ATCase activity of the enzyme is inhibited by CTP and activated by ATP. The carbamoyl-phosphate synthetase activity of this complex is termed carbamoyl-phosphate synthetase 2 (CPS2), as opposed to CPS1 which is involved in the urea cycle. The CAD encoded enzyme is localized to the cytoplasm and the CPS2 activity utilizes glutamine as the nitrogen donor for the synthesis of carbamoyl phosphate. CPS1 of the urea cycle is localized in the mitochondria and utilizes ammonia. The CPS2 domain is activated by ATP and inhibited by UDP, UTP, dUTP, and CTP.
The role of glycine in the regulation of the CAD gene tri-functional enzyme is exerted on the ATCase activity of the complex. ATP levels also regulate pyrimidine nucleotide biosynthesis at the level of PRPP formation. An increase in the level of PRPP results in an activation of pyrimidine synthesis.
There is also regulation of OMP decarboxylase activity of the bi-functional OMP synthase enzyme. The decarboxylase activity domain is competitively inhibited by UMP and, to a lesser degree, by CMP. Finally, CTP synthase is feedback-inhibited by CTP and activated by GTP.
Catabolism of Pyrimidine Nucleotides
Catabolism of the pyrimidine nucleotides leads ultimately to β-alanine, when CMP and UMP are degraded, or β-aminoisobutyrate (BAIBA), when dTMP is degraded, and NH4+ and CO2. The β-alanine (3-aminopropanoic acid) and β-aminoisobutyrate serve as –NH2 donors in transamination of 2-oxoglutarate (α-ketoglutarate) to glutamate. A subsequent reaction converts the products to malonyl-CoA (which can be diverted to fatty acid synthesis) or methylmalonyl-CoA (which is converted to succinyl-CoA and can be shunted to the TCA cycle).
During the catabolism of thymine and uracil, these nucleotides are first converted to dihydrothymine and 5,6-dihydrouracil, respectively, through the action of dihydropyrimidine dehydrogenase (encoded by the DPYD gene). Dihydropyrimidine dehydrogenase represents the rate-limiting enzyme in the catabolism of both thymine and uracil.
Dihydrothymine is then converted to N-carbamoyl-β-aminoisobutyrate (N-carbamoyl-BAIBA) via the action of dihydropyrimidase (encoded by the DPYS gene). Finally, N-carbamoyl-BAIBA is converted to D-BAIBA via the action of beta-ureidopropionase 1 (encoded by the UPB1 gene). The UPB1 encoded enzyme is also known as β-alanine synthase.
The DPYS encoded enzyme converts dihydrouracil to β-ureidopropionate. The UPB1 encoded enzyme then converts β-ureidopropionate to β-alanine, CO2, and NH4+.
One important metabolic significance of thymine catabolism is the production of β-aminoisobutyric acid (BAIBA). During exercise, skeletal muscle cells produce and secrete numerous factors referred to as myokines and also as exerkines. One such exerkine, that exerts effects on both fatty acid metabolism and glucose metabolism, is β-aminoisobutyrate (BAIBA). BAIBA is a non-protein amino acid that exists in two enantiomeric forms, D-BAIBA and L-BAIBA.
The DPYD gene is located on chromosome 1p21.3 and is composed of 26 exons that generate two alternatively spliced mRNAs. The major mRNA encodes a protein of 1025 amino acids. The highest levels of expression of the DPYD gene are found in the liver and peripheral blood mononuclear cells (PBMC).
The DPYS gene is located on chromosome 8q22.3 and is composed of 10 exons that generate two alternatively spliced mRNAs. The major mRNA encodes a 519 amino acid protein. The highest levels of expression of the DPYS gene are found in the liver and kidney.
The UPB1 gene is located on chromosome 22q11.23 and is composed of 13 exons that encode a 384 amino acid protein. The highest levels of expression of the UPB1 gene are found in the liver and kidney.
Salvage of Pyrimidine Nucleotides
The salvage of pyrimidine bases has less clinical significance than that of the purines, owing to the solubility of the by-products of pyrimidine catabolism. However, as indicated above, the salvage pathway to thymidine nucleotide synthesis is especially important in the preparation for cell division. Uracil can be salvaged to form UMP through the concerted action of uridine phosphorylase and uridine kinase, as indicated:
uracil + ribose-1-phosphate ↔ uridine + Pi
uridine + ATP ↔ UMP + ADP
Humans express two uridine phosphorylase genes identified as UPP1 and UPP2. When energy levels are reduced the actions of uridine phosphorylase predominate in the direction of ribose-1-phosphate and uracil production. The ribose-1-phosphate can then contribute to glucose homeostasis by first being converted to fructose-6-phosphate and glyceraldehyde-3-phosphate by the non-oxidative branch of the pentose phosphate pathway. The ability of uridine to contribute to overall glucose homeostasis is not limited to any specific tissue. Indeed, the process occurs throughout the body.
Deoxyuridine is also a substrate for uridine phosphorylase.
Formation of dTMP, by salvage of thymidine, requires thymine phosphorylase and the previously discussed cytoplasmic thymidine kinase 1 (TK1):
thymine + deoxyribose-1-phosphate ↔ thymidine + Pi
thymidine + ATP → dTMP + ADP
The salvage of deoxycytidine is catalyzed by deoxycytidine kinase:
deoxycytidine + ATP ↔ dCMP + ADP
Deoxyadenosine and deoxyguanosine are also substrates for deoxycytidine kinase, although the Km for these substrates is much higher than for deoxycytidine.
The major function of the pyrimidine nucleoside kinases is to maintain a cellular balance between the level of pyrimidine nucleosides and pyrimidine nucleoside monophosphates. However, since the overall cellular and plasma concentrations of the pyrimidine nucleosides, as well as those of ribose-1-phosphate, are low, the salvage of pyrimidines by these kinases is relatively inefficient.
Clinical Significances of Pyrimidine Metabolism
Because the products of pyrimidine catabolism are soluble, few disorders result from excess levels of their synthesis or catabolism. Two inherited disorders affecting pyrimidine biosynthesis are the result of deficiencies in the bifunctional enzyme catalyzing the last two steps of UMP synthesis, orotate phosphoribosyltransferase and OMP decarboxylase. These deficiencies result in two types of orotic aciduria (type 1 and type 2) that result in impaired growth, and a severe anemia associated with hypochromic erythrocytes and megaloblastic bone marrow, both of which are the result of the block to DNA synthesis. Leukopenia is also common in orotic acidurias. These disorders can be treated with uridine and/or cytidine, which leads to increased UMP production via the action of nucleoside kinases. The UMP then inhibits the CPS2 activity of the CAD encoded enzyme, thus attenuating orotic acid production.
Table of Disorders of Pyrimidine Metabolism
| Disorder | Defective Enzyme | Comments |
| Orotic aciduria, type 1 | due to defects in both the orotate phosphoribosyltransferase and OMP decarboxylase activities of the bifunctional enzyme encoded by the UMPS gene | see above for details; NO associated hyperammonemia; normal BUN measurements |
| Orotic aciduria, type 2 | due to defects in the OMP decarboxylase activity of the bifunctional enzyme encoded by the UMPS gene | see above for details; NO associated hyperammonemia; normal BUN measurements |
| Orotic aciduria due to OTC deficiency (no hematologic component) | the urea cycle enzyme ornithine transcarbamylase is deficient | increased mitochondrial carbamoyl phosphate exits and augments cytoplasmic pyrimidine biosynthesis; hepatic encephalopathy; associated with hyperammonemia; is NOT associated with megaloblastic anemia since no defect in nucleotide metabolism |
| β-aminoisobutyric aciduria | transaminase, affects urea cycle function during deamination of α-amino acids to α-keto acids | clinically benign; frequent in Orientals |
| drug induced orotic aciduria | OMP decarboxylase activity of the bifunctional enzyme encoded by the UMPS gene | allopurinol and 6-azauridine treatments cause orotic acidurias without a hematologic component; their catabolic by-products inhibit the OMP decarboxylase activity of the UMPS encoded bifunctional enzyme |
Formation of Deoxyribonucleotides
The typical cell contains 5 to10 times as much RNA (mRNAs, rRNAs and tRNAs) as DNA. Therefore, the majority of nucleotide biosynthesis has as its purpose the production of rNTPs. However, because proliferating cells need to replicate their genomes, the production of dNTPs is also necessary. This process begins with the reduction of rNDPs, followed by phosphorylation to yield the dNTPs. The phosphorylation of dNDPs to dNTPs is catalyzed by the same nucleoside diphosphate kinases that phosphorylates rNDPs to rNTPs, using ATP as the phosphate donor.
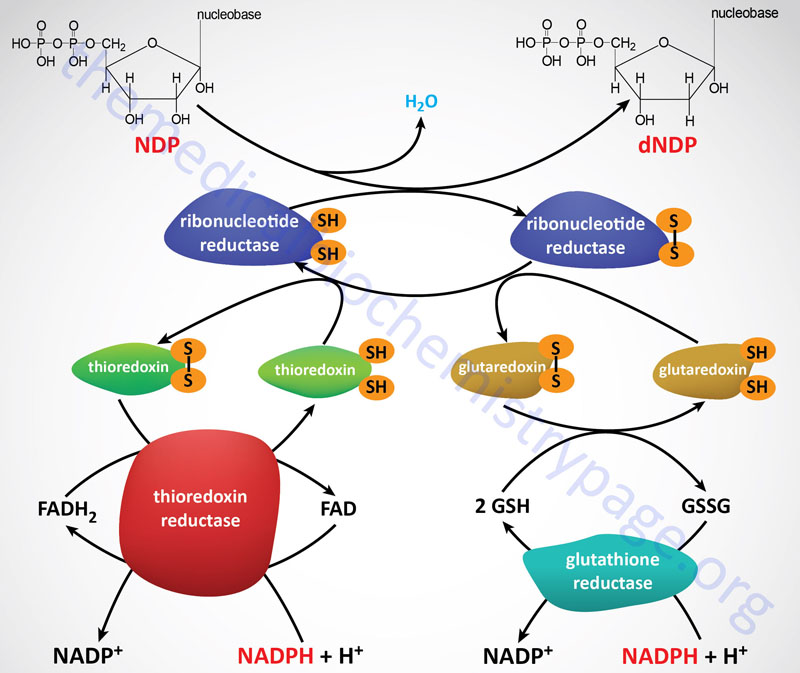
Ribonucleotide reductase (RR) is a heterotetrameric enzyme that contains redox-active thiol groups for the transfer of electrons during the ribonucleotide reduction reactions. Functional RR complexes are composed of two catalytic subunits identified as M1 (large subunit) and M2 (small subunit). Within the functional RR complex there are two copies of the M1 subunit and two copies of the M2 subunit. In addition, there is a regulatory subunit identified as M2B. The gene encoding the M2B subunit (RRM2B) is inducible by the p53 tumor suppressor.
The M1 protein is encoded by the RRM1 gene. The RRM1 gene is located on chromosome 11p15.4 and is composed of 20 exons generate four alternatively spliced mRNAs, each of which encode a distinct protein isoform. The RRM1 gene resides in the region of chromosome 11 that is deleted in Beckwith-Wiedemann syndrome.
The M2 protein is encoded by the RRM2 gene. The RRM2 gene is located on chromosome 2p25.1 and is composed of 14 exons that generate two alternatively spliced mRNAs encoding two distinct protein isoforms (isoform 1: 449 amino acids and isoform 2: 389 amino acids).
The M2B protein is encoded by the RRM2B gene. The RRM2B gene is located on chromosome 8q22.3 and is composed of 9 exons that generate three alternatively spliced mRNAs encoding three distinct protein isoforms.
Following the formation of a deoxynucleotide, the oxidized thiol group in RR must be returned to its reduced state. The reduction the RR thiol groups is carried out by either of two systems, the thioredoxin system and the glutaredoxin system. The ultimate source of the electrons is NADPH. The thioredoxin system involves the protein identified as thioredoxin (abbreviated Trx) and the enzymes identified as thioredoxin reductases (abbreviated TrxR). The TrxR enzymes function as homodimers and contain a flavin (FAD) prosthetic group and possess a binding site for NADPH which is the terminal electron donor in the reduction of RR.
Humans express two genes encoding thioredoxin, TXN and TXN2. The TXN2 encoded enzyme is localized to the mitochondria where it is involved in the regulation of mitochondrial membrane potential. The TXN encoded enzyme is involved in the cytoplasmic thioredoxin system.
The TXN gene is located on chromosome 9q31.3 and is composed of 5 exons that generate two alternatively spliced mRNAs encoding two distinct cytoplasmic protein isoforms.
The TNX2 gene is located on chromosome 22q12.3 and is composed of 4 exon that encode a 166 amino acid protein.
Humans express three thioredoxin reductase enzymes encoded by the TXNRD1, TXNRD2, and TXNRD3 genes. Each of the TXNRD encoded enzymes contain selenocysteine residues that are incorporated during their translation.
The TXNRD1 encoded protein is identified as TrxR1. The TXNRD1 gene located on chromosome 12q23.3 and is composed of 18 exons that generate seven alternatively spliced mRNAs encoding five different isoforms of TrxR1. All of the TXNRD1 encoded enzymes are cytoplasmic and are the principal enzymes involved in deoxynucleotide synthesis.
The TXNRD2 encoded protein is identified as TrxR2. The TXNRD2 gene located on chromosome 22q11.21 and is composed of 22 exons that generate six alternatively spliced mRNAs resulting in six different isoforms of TrxR2. Although all six TXNRD2 encoded enzymes are classified as mitochondrial enzymes the isoform 3 and isoform 4 proteins lack the N-terminal mitochondrial targeting sequence and are, therefore, predicted to be localized to the cytosol. The TXNRD2 encoded proteins, localized to the mitochondria, are primarily involved in scavenging reactive oxygen species in this organelle.
The TXNRD3 encoded protein is identified as TrxR3. The TXNRD3 gene located on chromosome 3q21.3 and is composed of 16 exons that generate two alternatively spliced mRNAs resulting in two different isoforms of TrxR3. The mRNA encoding the TrxR3 isoform 1 protein utilizes a non-AUG codon (CUG) to initiate translation. Like the TXNRD1 encoded proteins, the TXNRD3 encoded proteins are involved in the formation of deoxynucleotides.
The glutaredoxin system involves one of several proteins of the glutaredoxin family, the anti-oxidant peptide, glutathione (abbreviated GSH), and the enzyme glutathione reductase. Like the TrxR enzymes, functional glutathione reductase contains an FAD prosthetic group and an NADPH-binding site.
The glutaredoxins are a family of glutathione-dependent proteins that function in a variety of cellular redox reactions including the formation of deoxynucleotides. Humans express five genes that encode proteins containing the glutaredoxin functional domain. Four of the five proteins are called glutaredoxins, the fifth protein is the enzyme identified as prostaglandin E synthase 2 (encoded by the PTGES2 gene). The four glutaredoxin proteins are encoded by the GLRX, GLRX2, GLRX3, and GLRX5 genes.
The GLRX gene is located on chromosome 5q15 and is composed of 3 exons that generate four alternatively spliced mRNAs, each of which encode the same 106 amino acid cytoplasmic protein. The GLRX encoded protein is the primary glutaredoxin involved in the formation of deoxynucleotides.
The GLRX2 gene is located on 1q31.2 and is composed of 5 exons that generate three alternatively spliced mRNAs that encode three distinct protein isoforms that possess distinct subcellular patterns of localization that include the mitochondria, cytosol, and nucleus.
The GLRX3 gene is located on chromosome 10q26.3 and is composed of 13 exons that generate three alternatively spliced mRNAs encoding two distinct protein isoforms. The primary function of the GLRX3 encoded proteins is in the regulation of the function of a specific PKC isoform (PKCθ).
The GLRX5 gene is located on chromosome 14q32.13 and is composed of 2 exons that encode a 157 amino acid precursor protein. The GLRX5 encoded protein is localized to the mitochondria where it functions in the formation of iron-sulfur centers in complexes of the oxidative phosphorylation pathway. Mutations in the GLRX5 gene are associated with a form of hyperglycinemia referred to as variant nonketotic hyperglycinemia. The most common causes of variant nonketotic hyperglycinemia are mutations in the LIAS gene involved in the synthesis of lipoic acid. Classic nonketotic hyperglycinemia results from mutations in genes encoding subunits of the glycine cleavage complex responsible for the catabolism of glycine.
The glutathione reductase gene (symbol: GSR) is located on chromosome 8p12 and is composed of 13 exons that generate four alternatively spliced mRNAs that encode four distinct mitochondrial protein isoforms.
Salvage of Deoxynucleotides
The salvage pathway to dTTP synthesis involves the thymidine kinase enzymes which can use either thymidine or deoxyuridine as substrate:
thymidine + ATP ↔ TMP + ADP
deoxyuridine + ATP ↔ dUMP + ADP
As indicated above, humans express two thymidine kinase genes, one whose encoded protein is cytosolic (TK1) and the other mitochondrial (TK2). The activity of the TK1 gene is unique in that it fluctuates with the cell cycle, rising to peak activity during DNA synthesis (S-phase). Expression of the TK2 gene does not change during the cell cycle.
Regulation of dNTP Formation
Ribonucleotide reductase is the only enzyme used in the generation of all the deoxyribonucleotides. Therefore, its activity and substrate specificity must be tightly regulated to ensure balanced production of all four of the dNTPs required for DNA replication. Such regulation occurs by binding of nucleoside triphosphate effectors to either the activity sites or the specificity sites of the enzyme complex. The activity sites bind either ATP or dATP with low affinity, whereas the specificity sites bind ATP, dATP, dGTP, or dTTP with high affinity. The binding of ATP at activity sites leads to increased enzyme activity, while the binding of dATP inhibits the enzyme. Indeed, the binding of dATP dramatically decreases the activity of RR towards all four NDPs and explains, in part, the severely reduced deoxynucleotide production in ADA deficient SCID. The binding of nucleotides at specificity sites effectively allows the enzyme to detect the relative abundance of the four dNTPs and to adjust its affinity for the less abundant dNTPs, in order to achieve a balance of deoxynucleotide production.
Interconversion of the Nucleotides
Nucleoside Monophosphate Kinases
During the catabolism of nucleic acids, nucleoside mono- and diphosphates are released. The nucleosides do not accumulate to any significant degree, owing to the action of nucleoside kinases. These include both nucleoside monophosphate (NMPK) kinases and nucleoside diphosphate (NDPK) kinases.
The NMP kinases catalyze ATP-dependent reactions of the type:
(d)NMP + ATP ↔ (d)NDP + ADP
There are four classes of NMP kinases that catalyze, respectively, the phosphorylation of:
- AMP and dAMP; family of kinases known as adenylate kinases; nine human adenylate kinase genes
- GMP and dGMP; family of two enzymes knows as guanylate kinases encoded by GUK1 and GUK2 genes
- CMP, UMP, and dCMP; family of two cytidine and uridine kinases (UCK1 and UCK2) that do not phosphorylate the deoxynucleotides and a family of two kinases known as cytidine/uridine monophosphate kinases 1 (CMPK1) and CMPK2 that phosphorylate the deoxynucleotides
- dTMP; conversion of dTMP to dTDP is catalyzed by thymidylate kinase which is encoded by the DTYMK gene. Mutations in the DTYMK gene are associated with childhood-onset form of neurodegeneration associated with progressive microcephaly.
The adenylate kinase family of enzymes is important for ensuring adequate levels of energy in cells such as liver and muscle. The predominant reaction catalyzed by adenylate kinases is:
2ADP ↔ AMP + ATP
Nucleoside Diphosphate Kinases
The NDP kinases belong to a family of kinases identified as the NME family. This nomenclature stems from the identification of the NM23 (non-metastatic clone 23, also known as NME) gene whose encoded protein was originally identified as being responsible for metastasis suppression in the murine melanoma model system. The NM23 gene was found to encode one of the subunits of NDPK, identified as NDPK A. The human NME/NM23/NDPK family is composed of nine members identified as NME1-NME9.
The various members of the NDPK family catalyze reactions of the type:
N1TP + N2DP ↔ N1DP + N2TP
N1 can represent a purine ribo– or deoxyribonucleotide; N2 a pyrimidine ribo– or deoxyribonucleotide. The activity of the NDP kinases can range from 10 to 100 times higher than that of the NMP kinases. This difference in activity maintains a relatively high intracellular level of (d)NTPs relative to that of (d)NDPs. Unlike the substrate specificity seen for the NMP kinases, the NDP kinases recognize a wide spectrum of (d)NDPs and (d)NTPs.
In addition to the role of NDPK in maintaining the pool of NTP and dNTP for nucleic acid (DNA and RNA) homeostasis, these enzymes provide a pool of CTP utilized in the process of phospholipid synthesis, UTP utilized in the synthesis of glycoconjugates such as glycoproteins, and GTP required for GPCR-mediated signal transduction.