
Last Updated: July 31, 2026
Introduction to Nitrogen Homeostasis and the Urea Cycle
The processes of nitrogen metabolism, which includes the urea cycle to remove waste nitrogen, are critical to the survival of humans given that excess nitrogen, in the form of ammonium ion (NH4+) is exceedingly toxic.
Humans are totally dependent on other organisms for converting atmospheric nitrogen into forms available to the body. Nitrogen fixation is carried out by bacterial nitrogenases forming reduced nitrogen, NH4+, which can then be used by all organisms to form amino acids.
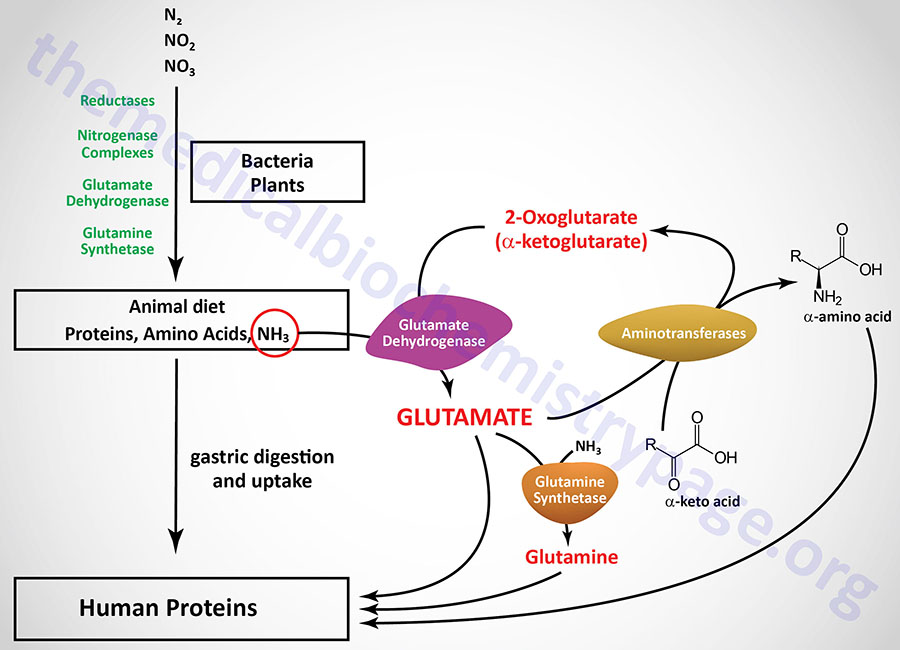
Reduced nitrogen enters the human body as dietary free amino acids, protein, and the ammonia produced by intestinal tract bacteria. A pair of principal enzymes, glutamate dehydrogenase and glutamine synthetase, are found in all organisms and effect the conversion of ammonia into the amino acids glutamate and glutamine, respectively. Amino and amide groups from these two substances are freely transferred to other carbon skeletons by transamination and transamidation reactions.
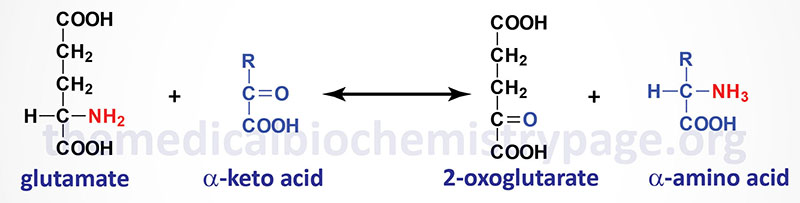
Aminotransferases (transaminases) catalyze the general reaction shown above. The aminotransferases function in both Amino Acid Biosynthesis and Amino Acid Catabolism which allows the amino nitrogen from nearly all amino acids to end up in the nitrogen disposal pathways detailed in this page.
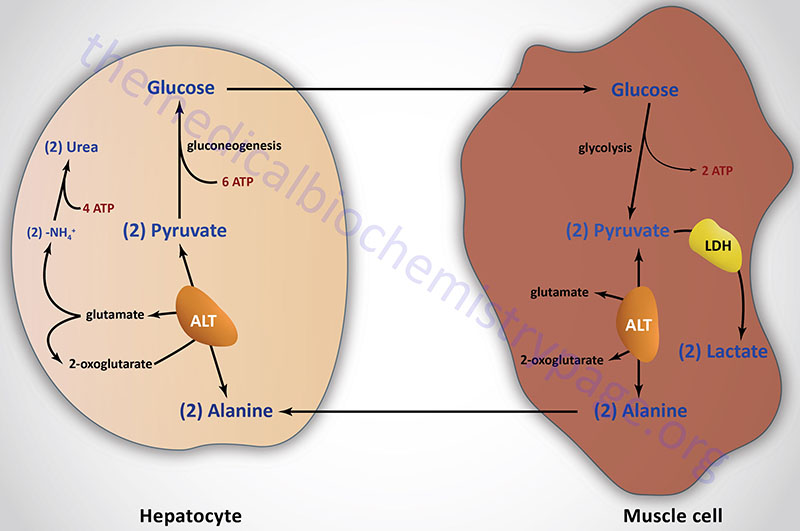
The most common compounds involved as a donor/acceptor pairs in transamination reactions are glutamate and 2-oxoglutarate (α-ketoglutarate). Alanine transaminase has an important function in the delivery of skeletal muscle carbon and nitrogen (in the form of alanine) to the liver. In skeletal muscle, pyruvate is transaminated to alanine, thus affording an additional route of nitrogen transport from muscle to liver. In the liver, alanine transaminase transfers the ammonia to 2-oxoglutarate and regenerates pyruvate. The pyruvate can then be diverted into gluconeogenesis forming new glucose for release to the blood. This process is referred to as the glucose-alanine cycle.
The Glutamate Dehydrogenase Reaction
Humans express two distinct glutamate dehydrogenase genes identified as GLUD1 and GLUD2. The GLUD1 gene encoded enzyme is the primary glutamate dehydrogenase (GDH1) activity in most tissues. This enzyme is localized to the mitochondrial matrix and functions as a homohexameric complex.
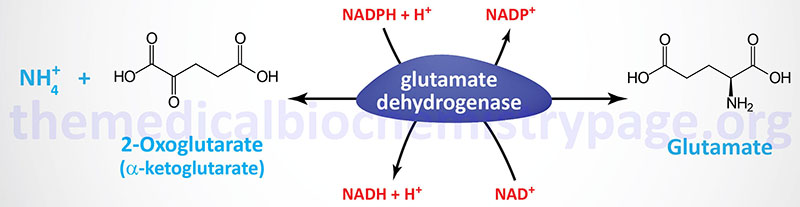
The reaction catalyzed by glutamate dehydrogenase is:
The GLUD1 gene is located on chromosome 10q23.2 and is composed of 18 exons that generate seven alternatively spliced mRNAs that collectively encode three distinct protein isoforms.
The GLUD2 gene is thought to have arisen as a result of a retrotranspostitional event to the X chromosome. The GLUD2 gene is located at Xq24 and is an intronless gene encoding a precursor protein of 558 amino acids. The GLUD2 encoded protein (GDH2) also forms a homohexameric complex in the mitochondrial matrix. Expression of the GLUD2 gene is highest in neural tissues and the regulatory controls over this form of the enzyme are distinct from the GLUD1 encoded enzyme. The GDH2 complex is not inhibited by GTP which allows astrocyte GDH2 to continue to function under conditions of intense excitatory neurotransmission allowing these cells to handle the increased loads of the neurotransmitter glutamate.
Glutamate dehydrogenase (GDH) utilizes both nicotinamide nucleotide cofactors; NAD+ in the direction of nitrogen liberation and NADPH for nitrogen incorporation. In the forward reaction (as shown above) glutamate dehydrogenase is important in converting free ammonia (as ammonium ion, NH4+) and 2-oxoglutarate (α-ketoglutarate) to glutamate, forming one of the 20 amino acids required for protein synthesis while simultaneously reducing the cellular load of potentially toxic ammonium ion. However, it should be recognized that the reverse reaction is a key anaplerotic process linking amino acid metabolism with TCA cycle activity. In the reverse reaction, glutamate dehydrogenase provides an oxidizable carbon source used for the production of energy as well as a reduced electron carrier, NADH.
Regulation of Glutamate Dehydrogenase
As expected for a branch point enzyme with an important link to energy metabolism, glutamate dehydrogenase is regulated by the cell energy charge. ATP and GTP are positive allosteric effectors of the formation of glutamate, and conversely, ATP and GTP exert potent negative allosteric effects on the formation of 2-oxoglutarate.
GTP is unique in its inhibitory effects on GDH, compared to ATP, in that the inhibition by GTP affects all of the subunits of the homohexameric complex regardless of the subunit to which GTP is bound. This effect of GTP makes this nucleotide the more potent regulator of GDH activity.
NADH is also an allosteric inhibitor of the 2-oxoglutarate liberating direction of the GDH reaction. Given that low energy charge would be expected to increase the conversion of glutamate to 2-oxoglutarate, it is not surprising that ADP is a positive allosteric effector of this reaction direction. Thus, when the level of ATP is high, conversion of glutamate to 2-oxoglutarate and other TCA cycle intermediates is limited; when the cellular energy charge is low, glutamate is converted to ammonia and oxidizable TCA cycle intermediates.
Glutamate is also a principal amino donor to other amino acids in subsequent transamination reactions. The multiple roles of glutamate in nitrogen balance make it a gateway between free ammonia and the amino groups of most amino acids.
Glutamine Synthetase
The reaction catalyzed by glutamine synthetase is:
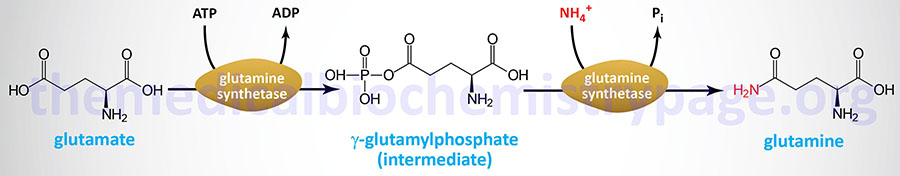
The glutamine synthetase reaction is also important in several respects. First it produces glutamine, one of the 20 major amino acids. Second, in animals, glutamine is the major amino acid found in the circulatory system. Its role there is to carry ammonia to and from various tissues but principally from peripheral tissues to the kidney, where the amide nitrogen is hydrolyzed by the enzyme glutaminase that regenerates glutamate and free ammonium ion. The renal ammonium ion is excreted in the urine.
Glutaminase
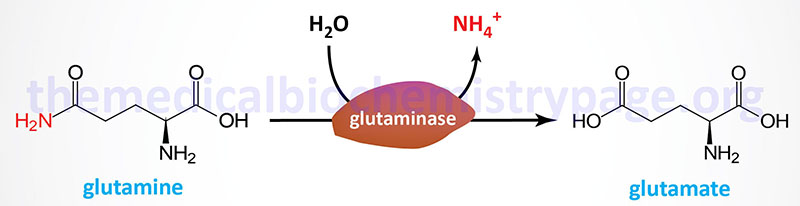
The primary tissues that express glutaminase are the brain and kidney. In the brain the role of glutaminase is in the synthesis of the neurotransmitter glutamate. In the kidneys the role of glutaminase is in acid-base balance as discussed in the section on proximal tubule ammoniagenesis in the Renal Transporters page.
However, the liver also expresses both glutamine synthetase and glutaminase but the enzymes are localized in different subsets of hepatocytes. This ensures that the liver is neither a net producer nor consumer of glutamine. The differences in cell location of these two enzymes allows the liver to scavenge ammonia that has not been incorporated into urea. The enzymes of the urea cycle are located in the same cells as those that contain glutaminase. The result of the differential distribution of these two hepatic enzymes makes it possible to control ammonia incorporation into either urea or glutamine, the latter leads to excretion of ammonia by the kidney.
When acidosis occurs the body will divert more glutamine from the liver to the kidney. This allows for the conservation of bicarbonate ion since the incorporation of ammonia into urea requires bicarbonate (see below). When glutamine enters the kidney, glutaminase releases one mole of ammonia generating glutamate and then glutamate dehydrogenase releases another mole of ammonia generating 2-oxoglutarate (α-ketoglutarate). The ammonia will ionize to ammonium ion (NH4+) which is excreted. The net effect is a reduction in the concentration of hydrogen ion, [H+], and thus an increase in the pH (see also Overview of Renal Acid-Base Balance section of the Renal Transporters: Biochemistry, Physiology, Pharmacology, Pathology page. This process is also covered in the Proximal Tubular Ammoniagenesis section of the same page
The glutamine synthetase enzyme is encoded by the glutamate-ammonia ligase gene, GLUL. The GLUL gene is located on chromosome 1q25.3 and is composed of 9 exons that generate three alternatively spliced mRNAs, all of which encode the same 373 amino acid protein.
There are two distinct glutaminase genes in humans identified as GLS (encoding the GLS1 enzyme) and GLS2 (encoding the GLS2 enzyme). The GLS gene is located on chromosome 2q32.2 and is composed of 22 exons that undergo alternative splicing to yield two mRNAs generating two isoforms of the enzyme. These two GLS-derived isoforms are often referred to as glutaminase C (GAC) and kidney-type glutaminase (KGA) but are collectively the glutaminase 1 (GLS1) enzymes. The GLS encoded isoforms of glutaminase are primarily expressed in the kidneys. GLS encoded kidney-type glutaminase is a protein of 669 amino acids and GLS encoded glutaminase C is a protein of 598 amino acids.
The GLS2 gene encoded glutaminase was originally thought to be liver specific but is in fact expressed in numerous tissues and is important in the glutamate-glutamine cycle in the brain. The GLS2 encoded glutaminase was originally characterized as being dependent on inorganic phosphate (Pi) for activity and is, therefore, also referred to as phosphate-activated glutaminase, PAG. However, both the GLS gene encoded enzymes and the GLS2 encoded enzymes require phosphate for activity, with the GLS encoded enzymes being more sensitive to phosphate than the GLS2 encoded enzymes.
The GLS2 gene is located on chromosome 12q13.3 and is composed of 19 exons that undergo alternative splicing to yield four mRNAs that encode three different isoforms of the enzyme.
The GLS encoded enzymes are inhibited by glutamate but the GLS2 encoded enzyme is not. The GLS2 encoded enzyme is activated by ammonia but the GLS encoded enzymes are not.
Digestive Tract Nitrogen
While glutamine, glutamate, and the remaining nonessential amino acids can be made by animals, the majority of the amino acids found in human tissues necessarily come from dietary sources (about 400g of protein per day). Protein digestion begins in the stomach, where a proenzyme called pepsinogen is secreted, autocatalytically converted to Pepsin A, and used for the first step of proteolysis. However, most proteolysis takes place in the duodenum as a consequence of enzyme activities secreted by the pancreas. All of the serine proteases and the zinc peptidases of pancreatic secretions are produced in the form of their respective proenzymes. These proteases are both endopeptidase and exopeptidase, and their combined action in the intestine leads to the production of amino acids, dipeptides, and tripeptides, all of which are taken up by enterocytes of the mucosal wall.
A circuitous regulatory pathway leading to the secretion of proenzymes into the intestine is triggered by the appearance of food in the intestinal lumen. Special mucosal endocrine cells secret the peptide hormones cholecystokinin (CCK) and secretin into the circulatory system. Together, CCK and secretin cause contraction of the gall bladder and the exocrine secretion of a bicarbonate-rich, alkaline fluid, containing protease proenzymes from the pancreas into the intestine. A second, paracrine role of CCK is to stimulate adjacent intestinal cells to secrete enteropeptidase, a protease that cleaves trypsinogen to produce trypsin. Trypsin also activates trypsinogen as well as all the other proenzymes in the pancreatic secretion, producing the active proteases and peptidases that hydrolyze dietary polypeptides.
Subsequent to luminal hydrolysis, small peptides and amino acids are transferred through enterocytes to the portal circulation by diffusion, facilitated diffusion, or active transport. A number of Na+-dependent amino acid transport systems with overlapping amino acid specificity have been described. In these transport systems, Na+ and amino acids at high luminal concentrations are co-transported down their concentration gradient to the interior of the cell. The ATP-dependent Na+/K+ pump exchanges the accumulated Na+ for extracellular K+, reducing intracellular Na+ levels and maintaining the high extracellular Na+ concentration (high in the intestinal lumen, low in enterocytes) required to drive this transport process. Transport mechanisms of this nature are ubiquitous in the body. Small peptides are accumulated by a proton (H+) driven transport process and hydrolyzed by intracellular peptidases. Amino acids in the circulatory system and in extracellular fluids are transported into cells of the body by at least seven different ATP-requiring active transport systems with overlapping amino acid specificities.
Hartnup disorder is an autosomal recessive impairment of neutral amino acid transport affecting the kidney tubules and small intestine. The disorder results from defects in the specific transport system responsible for neutral amino acid transport across the brush-border membrane of renal and intestinal epithelium. Deficiencies in the solute carrier family 6 (neurotransmitter transporter), member 19 gene (symbol SLC6A19) are associated with Hartnup disorder. The encoded protein is also referred to as the system B(0) neutral amino acid transporter 1 [B(0)AT1] protein. The characteristic diagnostic feature of Hartnup disorder is a dramatic neutral hyperaminoaciduria. Additionally, individuals excrete indolic compounds that originate from the bacterial degradation of unabsorbed tryptophan. The reduced intestinal absorption and increased renal loss of tryptophan lead to a reduced availability of tryptophan for nicotinamide adenine dinucleotide (NAD+ and NADP+) biosynthesis. As a consequence affected individuals frequently exhibit pellagra-like rashes
Many other nitrogenous compounds are found in the intestine as a result of the bacterial protein degradation. Some of the gut microbiota-derived nitrogenous compounds have important physiological effects.
Table of Several Nitrogenous Compounds Produced by Gut Microbiota
| Products of Intestinal Bacterial Activity | ||
| Substrates | Products | |
| Vasopressor Amines | Other | |
| Lysine | Cadaverene | |
| Arginine | Agmatine | |
| Tyrosine | Tyramine | |
| Ornithine | Putrescine | |
| Histidine | Histamine | |
| Tryptophan | Indole and skatole | |
| All amino acids | NH4+ | |
Prokaryotes can make the carbon skeletons of all 20 amino acids and transaminate those carbon skeletons with nitrogen from glutamine or glutamate to complete the amino acid structures. Humans cannot synthesize the branched carbon chains found in branched chain amino acids or the ring systems found in phenylalanine and the aromatic amino acids; nor can we incorporate sulfur into covalently bonded structures. Therefore, the 10 so-called essential amino acids (see Table below) must be supplied from the diet. Nevertheless, it should be recognized that, depending on the composition of the diet and physiological state of an individual, one or another of the non-essential amino acids may also become a required dietary component. For example, arginine is only normally considered to be essential amino acid during early childhood development because enough for adult needs is made by the urea cycle.
To take a different type of example, cysteine and tyrosine are considered non-essential but are formed from the essential amino acids methionine and phenylalanine, respectively. If sufficient cysteine and tyrosine are present in the diet, the requirements for methionine and phenylalanine are markedly reduced; conversely, if methionine and phenylalanine are present in only limited quantities, cysteine and tyrosine can become essential dietary components. Finally, it should be recognized that if the α-keto acids corresponding to the carbon skeletons of the essential amino acids are supplied in the diet, aminotransferases in the body will convert the keto acids to their respective amino acids, largely supplying the basic needs.
Unlike fats and carbohydrates, nitrogen has no designated storage depots in the body. Since the half-life of many proteins is short (on the order of hours), insufficient dietary quantities of even one amino acid can quickly limit the synthesis and lower the body levels of many essential proteins. The result of limited synthesis and normal rates of protein degradation is that the balance of nitrogen intake and nitrogen excretion is rapidly and significantly altered. Normal, healthy adults are generally in nitrogen balance, with intake and excretion being very well matched. Young growing children, adults recovering from major illness, and pregnant women are often in positive nitrogen balance. Their intake of nitrogen exceeds their loss as net protein synthesis proceeds. When more nitrogen is excreted than is incorporated into the body, an individual is in negative nitrogen balance. Insufficient quantities of even one essential amino acid is adequate to turn an otherwise normal individual into one with a negative nitrogen balance.
The biological value of dietary proteins is related to the extent to which they provide all the necessary amino acids. Proteins of animal origin generally have a high biological value; plant proteins have a wide range of values from almost none to quite high. In general, plant proteins are deficient in lysine, methionine, and tryptophan and are much less concentrated and less digestible than animal proteins. The absence of lysine in low-grade cereal proteins, used as a dietary mainstay in many underdeveloped countries, leads to an inability to synthesize protein (because of missing essential amino acids) and ultimately to a syndrome known as kwashiorkor, common among children in these countries.
Essential vs. Nonessential Amino Acids
| Nonessential | Alanine, Asparagine, Aspartate, Cysteine, Glutamate, Glutamine, Glycine, Proline, Serine, Tyrosine |
| Essential | Arginine*, Histidine, Isoleucine, Leucine, Lysine, Methionine*, Phenylalanine*, Threonine, Tryptophan, Valine |
*The amino acids arginine, methionine and phenylalanine are considered essential for reasons not directly related to lack of synthesis. Arginine is synthesized by mammalian cells but at a rate that is insufficient to meet the growth needs of the body and the majority that is synthesized is cleaved to form urea. Methionine is required in large amounts to produce cysteine if the latter amino acid is not adequately supplied in the diet. Similarly, phenylalanine is needed in large amounts to form tyrosine if the latter is not adequately supplied in the diet. For these reasons these three amino acids are more correctly defined as being conditionally essential.
Removal of Nitrogen from Amino Acids
The dominant reactions involved in removing amino acid nitrogen from the body are known as transaminations (see general reaction above). This class of reactions funnels nitrogen from all free amino acids into a small number of compounds; then, either they are oxidatively deaminated, producing ammonia, or their amine groups are converted to urea by the urea cycle. Transaminations involve moving an α-amino group from a donor α-amino acid to the keto carbon of an acceptor α-keto acid. These reversible reactions are catalyzed by a group of intracellular enzymes known as aminotransferases (also called transaminases), most all of which require covalently bound pyridoxal phosphate (vitamin B6) as a cofactor. However, some aminotransferases employ pyruvate as a cofactor.
Aminotransferases (transaminases) exist for all amino acids except threonine and lysine. The most common compounds involved as a donor/acceptor pairs in transamination reactions are glutamate and 2-oxoglutarate (α-ketoglutarate), which participate in reactions with many different aminotransferases. Serum aminotransferases such as aspartate aminotransferase, AST (also called serum glutamate-oxaloacetate transaminase, SGOT) and alanine transaminase, ALT (also called serum glutamate-pyruvate transaminase (SGPT) have been used as clinical markers of tissue damage, with increasing serum levels indicating an increased extent of damage (see the Enzyme Kinetics page for description of the use of enzyme levels in diagnosis).
As indicated earlier, ALT has an important function in the delivery of skeletal muscle carbon and nitrogen (in the form of alanine) to the liver in a series of reactions referred to as the glucose-alanine cycle (see Figure above in Introduction section). In skeletal muscle, pyruvate is transaminated to alanine, thus affording an additional route of nitrogen transport from muscle to liver. In the liver, alanine transaminase transfers the ammonia to 2-oxoglutarate and regenerates pyruvate. The pyruvate can then be diverted into gluconeogenesis allowing the liver to generate endogenous glucose which it can then release to the blood where skeletal muscle can use it for anaerobic energy production.
Humans express two genes encoding alanine transaminase. These genes are identified as GPT (also designated GPT1) and GPT2. The enzyme encoded by the GPT gene is localized to the cytosol, whereas the GPT2 encoded enzyme is localized to the mitochondria.
The GPT gene is located on chromosome 8q24.3 and is composed of 12 exons that generate three alternatively spliced mRNAs, all of which encode the same 496 amino acid protein.
The GPT2 gene is located on chromosome 16q11.2 and is composed of 13 exons that generate two alternatively spliced mRNAs that encode proteins of 523 amino acids (isoform 1) and 423 amino acids (isoform 2)..
Humans express two different AST enzymes, both of which function as homodimeric enzymes. One AST enzyme is a cytosolic enzyme and the other is a mitochondrial enzyme.
The cytosolic AST enzyme is synthesized by the GOT1 (glutamate-oxalate transaminase 1) gene. The GOT1 gene is located on chromosome 10q24.2 and is composed of 9 exons that encode a 413 amino acid protein.
The mitochondrial AST enzyme is synthesized from the GOT2 gene. The GOT2 gene is located on chromosome 16q21 and is composed of 10 exons that generate two alternatively spliced mRNAs that encode two different isoforms: isoform 1 (430 amino acids) and isoform 2 (387 amino acids).
Because of the participation of 2-oxoglutarate in numerous transaminations, glutamate is a prominent intermediate in nitrogen elimination as well as in anabolic pathways. Glutamate, formed in the course of nitrogen elimination, is either oxidatively deaminated by liver glutamate dehydrogenase forming ammonia which is then incorporated into urea, or converted to glutamine by glutamine synthetase and transported to proximal tubule cells in the kidney. There the glutamine is sequentially deamidated by glutaminase and deaminated by kidney glutamate dehydrogenase releasing NH3 which ionizes with H+ forming ammonium ion (NH4+). The NH4+ is excreted in the urine, where it helps increase serum pH in conditions of metabolic acidosis as well as being involved in the maintenance of urine pH in the normal range of pH 4 to pH 8. The extensive production of ammonia by peripheral tissue or hepatic glutamate dehydrogenase is not feasible because of the highly toxic effects of circulating ammonia. Normal serum ammonium concentrations are in the range of 20–40μM, and an increase in circulating ammonia to about 400μM causes alkalosis and neurotoxicity.
A final, therapeutically useful amino acid-related reaction is the amidation of aspartic acid to produce asparagine. The enzyme, asparagine synthetase (gene symbol: ASNS), catalyzes the ATP-requiring transamidation reaction shown below. The ASNS gene is located on chromosome 7q21.3 and is composed of 15 exons that generate seven alternatively spliced mRNAs that collectively encode three distinct protein isoforms.
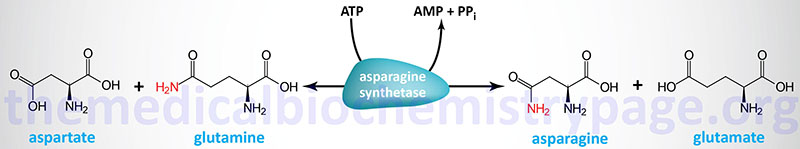
Most cells perform this reaction well enough to produce all the asparagine they need. However, some leukemia cells require exogenous asparagine, which they obtain from the plasma. Chemotherapy using the enzyme asparaginase takes advantage of this property of leukemic cells by hydrolyzing serum asparagine to ammonia and aspartic acid, thus depriving the neoplastic cells of the asparagine that is essential for their characteristic rapid growth.
The Urea Cycle
About 80% of the excreted waste nitrogen is in the form of urea which is produced exclusively in the liver, in a series of reactions that are distributed between the mitochondrial matrix and the cytosol. The series of reactions that form urea is known as the Urea Cycle or the Krebs-Henseleit Cycle.
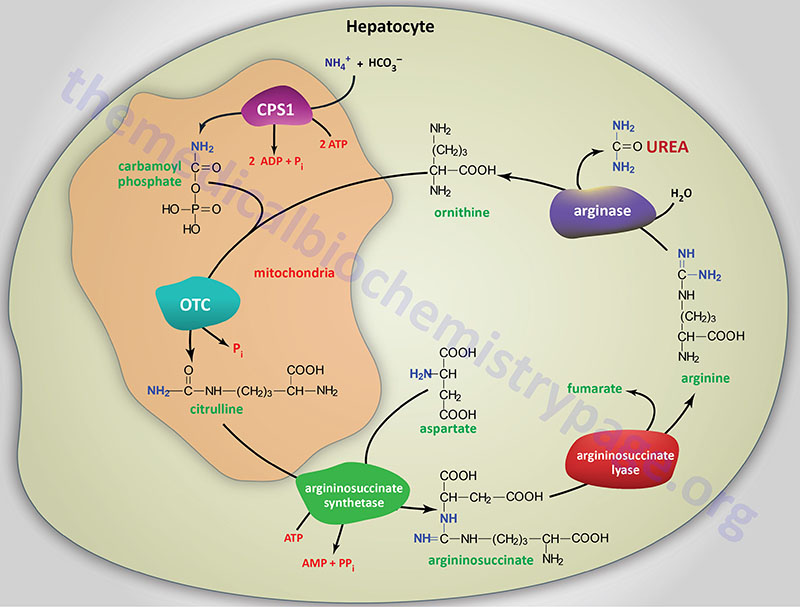
The essential features of the urea cycle reactions are that free ammonium ion, generated predominantly from the glutaminase and glutamate dehydrogenase reactions, is condensed with bicarbonate and eventually converted to urea for excretion. In addition to the arginine produced in the urea cycle, arginine from the diet or from protein breakdown can be cleaved by the cytosolic enzyme arginase, generating urea and ornithine.
Ornithine, arising in the cytosol, is transported to the mitochondrial matrix via the action of ornithine translocase 1, ORNT1. The ORNT1 transporter is a member of the solute carrier family of transporters and as such is encoded by the SLC25A15 gene. Concomitant with ornithine transport into the mitochondria is the export of citrulline to the cytosol, by SLC25A15, where the remaining reactions of the cycle take place. In subsequent reactions of the urea cycle a new urea residue is built on the ornithine from additional ammonium ions, regenerating arginine and perpetuating the cycle.
Pathogenic variants in the SLC25A15 gene result in the disorder termed HHH syndrome, where HHH refers to hyperornithinemia, hyperammonemia, and homocitrullinuria.
Also important in the function of the urea cycle is the mitochondrial transporter called citrin. Citrin is also called aspartate-glutamate carrier 2 (AGC2). Citrin is involved in the mitochondrial uptake of glutamate and export of aspartate and as such functions, in part, in the malate-aspartate shuttle. Citrin is a Ca2+-dependent mitochondrial solute transporter that is also a member of the solute carrier family of transporters and is encoded by the SLC25A13 gene.
Carbamoyl Phosphate Synthetase 1 (CPS1)
The first reaction of the urea cycle involves the activation of carbon and nitrogen forming the high-energy anhydride of carbamoyl phosphate (CP). This activation step requires two equivalents of ATP and the mitochondrial matrix enzyme carbamoyl phosphate synthetase 1 (CPS1 or CPS-I). The reaction catalyzed by CPS1 is the rate-limiting reaction of the urea cycle.
The CPS1 enzyme is encoded by the CPS1 gene which is located on chromosome 2q34 and is composed of 43 exons that generate four alternatively spliced mRNAs. These four mRNAs generate three isoforms of CPS1: isoform a is a protein of 1506 amino acids, isoform b is a protein of 1500 amino acids, and isoform c is a protein of 1049 amino acids.
The catalytic activity of CPS1 is positively regulated by N-acetylglutamate which is produced by N-acetylglutamate synthetase (NAGS). In the absence of N-acetylglutamate there is little, if any, CPS1 activity such that this molecule is often referred to as an obligate activator as opposed to a typical allosteric activator.
There are two carbamoyl phosphate synthetase activities in human cells: the mitochondrial CPS1 which forms CP destined for inclusion in the urea cycle, and a cytosolic carbamoyl phosphate synthetase activity (the CPS2 activity of a tri-functional enzyme complex), which is involved in pyrimidine nucleotide biosynthesis. The cytosolic enzyme (encoded by the CAD gene) that possesses the CPS2 (also designated CPS-II) activity is not affected by N-acetylglutamate.
Ornithine Transcarbamylase (OTC)
In the mitochondria ornithine transcabamylase (OTC; also called ornithine carbamoyltransferase) catalyzes the condensation of ornithine with carbamoyl phosphate, producing citrulline. The energy for the OTC reaction is provided by the high-energy anhydride of carbamoyl phosphate.
The OTC gene is an X-linked gene (Xp11.4) composed of 10 exons that encode a precursor protein of 354 amino acids.
Following the synthesis of citrulline it is transported out of the mitochondria via the action of the solute carrier (SLC) family transporter encoded by the SLC25A15 gene as indicated above. Concomitant with citrulline export to the cytosol is transport of ornithine into the mitochondria.
Argininosuccinate Synthetase
In a 2-step reaction, catalyzed by cytosolic argininosuccinate synthetase (also called argininosuccinate synthase), citrulline and aspartate are condensed to form argininosuccinate. The reaction involves the addition of AMP (from ATP) to the amido carbonyl of citrulline, forming an activated intermediate on the enzyme surface (AMP-citrulline), and the subsequent addition of aspartate to form argininosuccinate.
Argininosuccinate synthetase is encoded by the ASS1 gene. The ASS1 gene is located on chromosome 9q34.11 and is composed of 18 exons that generate two alternatively spliced mRNAs that generate the same 412 amino acid protein.
The human genome contains at least 14 copies of the ASS1 gene all of which are pseudogenes except the one on chromosome 9 which encodes the functional enzyme.
Argininosuccinate Lyase
Arginine and fumarate are produced from argininosuccinate by the cytosolic enzyme argininosuccinate lyase (also called argininosuccinase). Argininosuccinate lyase is functional as a homotetrameric complex. The argininosuccinate lyase protein is encoded by the ASL gene.
The ASL gene is located on chromosome 7q11.21 and is composed of 16 exons that generate four alternatively spliced mRNAs that encode three distinct protein isoforms.
The fumarate, generated via the action of arginiosuccinate lyase, can be reconverted to aspartate for use in the argininosuccinate synthetase reaction. This occurs through the actions of cytosolic versions of the TCA cycle enzymes, fumarate hydratase (fumarase), which yields malate, and malate dehydrogenase, which yields oxaloacetate. The oxaloacetate is then transaminated to aspartate by AST.
Arginase
In the final step of the urea cycle arginase cleaves urea from arginine, regenerating cytosolic ornithine, which can be transported to the mitochondrial matrix for another round of urea synthesis. There are two arginase genes in humans identified as the ARG1 and ARG2 genes. The ARG1 encoded isoform of arginase is a cytosolic enzyme primarily expressed in the liver and functions as the urea cycle enzyme.
The ARG1 gene is located on chromosome 6q23.2 and is composed of 8 exons that generate two alternatively spliced mRNAs encoding arginase-1 isoform 1 (330 amino acids) and arginase-1 isoform 2 (322 amino acids).
The ARG2 encoded arginase (arginase-2) is localized to the mitochondria in non-hepatic tissues, primarily the kidney. The arginase-2 isoform is thought to play a role in arginine metabolism, nitric oxide production, and polyamine metabolism. Recent (2024) work in mice and human liver organoids has found that the ARG2 encoded enzyme functions in the coordination of hepatic urea cycle activity with oxidative metabolism. In these studies, the loss of ARG2, specifically in the liver, resulted in steatohepatitis as a consequence of the enhanced expression of several genes involved in lipogenesis.
The ARG2 gene is located on chromosome 14q24.1 and is composed of 8 exons that encode a precursor protein of 354 amino acids.
Energy Driving the Urea Cycle
Beginning and ending with ornithine, the reactions of the cycle consume three equivalents of ATP and a total of four high-energy nucleotide phosphates. Urea is the only new compound generated by the cycle, all other intermediates and reactants are recycled. The energy consumed in the production of urea is more than recovered by the release of energy formed during the synthesis of the urea cycle intermediates. Ammonia released during the glutamate dehydrogenase reaction is coupled to the formation of NADH. In addition, when fumarate is converted back to aspartate, the malate dehydrogenase reaction used to convert malate to oxaloacetate generates a mole of NADH. These two moles of NADH are subsequently oxidized in the mitochondria yielding six moles of ATP.
Regulation of the Urea Cycle
The urea cycle operates only to eliminate excess nitrogen. On high-protein diets the carbon skeletons of the amino acids are oxidized for energy or stored as fat and glycogen, but the amino nitrogen must be excreted. To facilitate this process, enzymes of the urea cycle are controlled at the gene level. With long-term changes in the quantity of dietary protein, changes of 20-fold or greater in the concentration of cycle enzymes are observed. When dietary proteins increase significantly, enzyme concentrations rise. On return to a balanced diet, enzyme levels decline. Under conditions of starvation, enzyme levels rise as proteins are degraded and amino acid carbon skeletons are used to provide energy, thus increasing the quantity of nitrogen that must be excreted.
Role of N-Acetylglutamate in Urea Cycle Regulation
Short-term regulation of the cycle occurs principally at the CPS1 reaction which is the rate-limiting reaction of the urea cycle. CPS1 is essentially inactive in the absence of the activator N-acetylglutamate such that this molecule is sometimes referred to as an obligate activator as opposed to an allosteric activator. The steady-state concentration of N-acetylglutamate is set by the cellular concentrations of acetyl-CoA and glutamate which are used by the enzyme N-acetylglutamate synthase (NAGS) to form N-acetylglutamate.
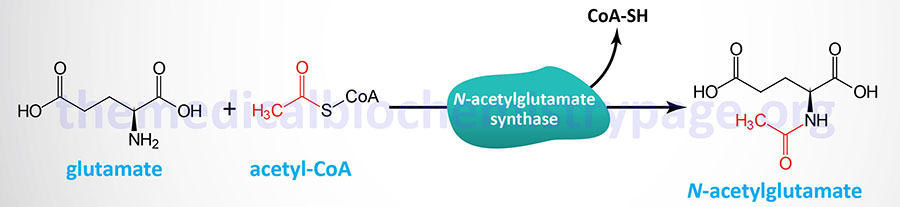
The NAGS gene is located on chromosome 17q21.31 and is composed of 7 exons that encode a mitochondrial protein of 534 amino acids.
The activity of NAGS is allosterically activated by the amino acid and urea cycle intermediate, arginine. Indeed, the allosteric activation of NAGS by arginine explains the therapeutic benefit of adding arginine to the diet of patients with urea cycle disorders. The increased NAGS activity will result in enhanced CPS1 activity resulting in enhanced incorporation of ammonia into the less toxic intermediate, carbamoyl phosphate.
Role of Lysine Succinylation in Urea Cycle Regulation
Post-translational modification of non-histone and histone proteins by acylation of lysine residues is a means to modify protein activity. One of the many types of lysine acylation is succinylation. Like all protein lysine acylations, succinyl-CoA represents the substrate for lysine succinylation (Ksucc) of histone proteins as well as non-histone proteins.
Succinyl-CoA can be derived from several sources and pathways with the most prevalent being from the TCA cycle. Succinyl-CoA is also produced from propionyl-CoA which is an intermediate in the catabolism of the amino acids isoleucine, valine, methionine, and threonine, and from the catabolism of fatty acids with an odd number of carbon atoms, and from the peroxisomal oxidation of dicarboxylic acids. The predominant site of protein succinylation is within the mitochondria and then the nucleus. However, there is ample evidence of cytoplasmic protein succinylation.
Cytosol 2-oxoglutarate (α-ketoglutarate) is transported into the nucleus where nuclear-localized 2-oxoglutarate dehydrogenase complex (OGDHc; also known as α-ketoglutarate dehydrogenase) oxidizes it to succinyl-CoA. Cytoplasmic succinyl-carnitine and succinate are converted to succinyl-CoA, most likely via the action of one or more members of the acyl-CoA synthetase family of enzymes. The succinyl-CoA is then transported into the nucleus.
Succinyl-CoA is a sufficiently energetic compound that non-enzymatic succinylation can occur. Despite this, enzymatic succinylation has been described. The “writer” for enzymatic lysine succinylation has been shown to be the GCN5/PCAF family member GCN5 (KAT2A). The nuclear OGDHc interacts with GCN5 allowing the succinyl-CoA that is generated to be directly accessible by the acetyltransferase. De-succinylation of mitochondrial and nuclear succinylated proteins has been shown to be catalyzed by two members of the sirtuin family, SIRT5 and SIRT7. SIRT5 activity is the major mitochondrial de-succinylase but also functions within the nucleus.
Recent evidence (2024) has identified Ksucc as a mechanism for the regulation of the urea cycle. In studies in mice where the SIRT5 gene was knocked-out in the liver, the level of Ksucc increases in several proteins with the level in the ASS1 encoded enzyme being one the highest. When SIRT5 knock-out mice are challenged with ammonium acetate, their ability to reduce serum ammonia levels are significantly impaired. This suggests that Ksucc of ASS1 leads to impaired enzymatic activity.
There are two lysine residues in human ASS1 that have been identified as sites for succinylation, K112 and K121. When these two residues are both succinylated the activity of the enzyme is significantly impaired. Therefore, Ksucc of ASS1 is a mechanism to regulate the rate of the urea cycle and impairment in the de-succinylation of ASS1 can lead to potentially toxic hyperammonemia.
Urea Cycle Disorders (UCDs)
A complete lack of any one of the enzymes of the urea cycle will result in death shortly after birth. However, deficiencies in each of the enzymes of the urea cycle have been identified in what are referred to as the urea cycle disorders, UCDs. In addition to defects in the five enzymes of the urea cycle proper, pathogenic variants in NAGS are the cause of a form of urea cycle disorder. In addition, pathogenic variants in the gene (SLC25A13) encoding the transporter citrin result in a form of citrullinemia similar to that seen with deficiencies in argininosuccinate synthetase. More information on the individual UCDs can be found in the Introduction to the UCD page.
A common thread to most UCDs is hyperammonemia leading to ammonia intoxication with the consequences described below. Blood chemistry will also show elevations in glutamine. In addition to hyperammonemia, UCDs all present with encephalopathy and respiratory alkalosis. The most dramatic presentation of UCD symptoms occurs in neonates between 24 and 48 hours after birth. Afflicted infants exhibit progressively deteriorating symptoms due to the elevated ammonium levels. Deficiencies in arginase do not lead to symptomatic hyperammonemia as severe or as commonly as in the other UCDs. Deficiencies in carbamoylphosphate synthetase 1 (CPS1), ornithine transcarbamylase (OTC), argininosuccinate synthetase, and argininosuccinate lyase comprise the common neonatal UCDs.
When making a diagnosis of neonatal UCD based upon presenting symptoms and observed hyperammonemia, it is possible to make a differential diagnosis as to which of the four enzyme deficiencies is the cause as shown in the Figure below.
Differential Diagnosis of Urea Cycle Disorders
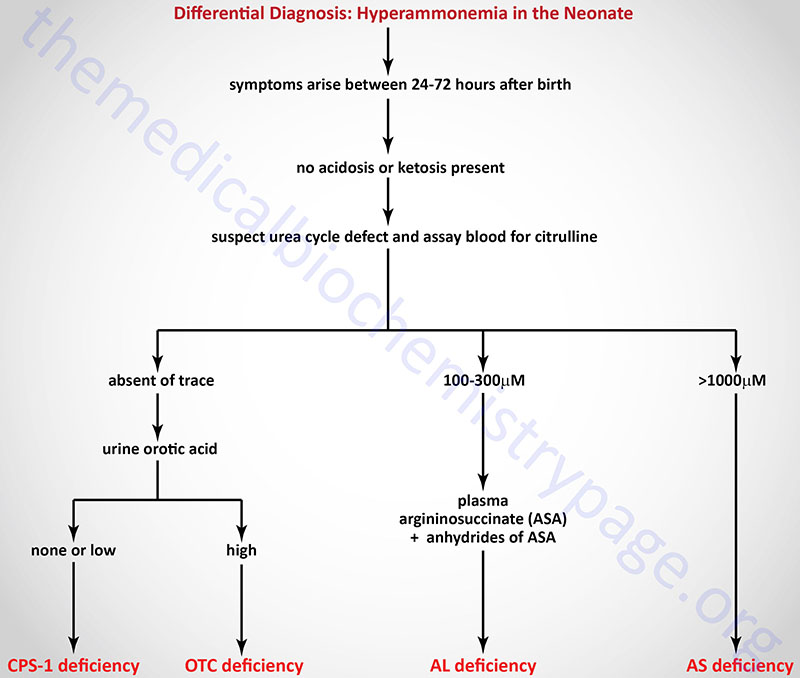
Clinical symptoms are most severe when the UCD is at the level of carbamoyl phosphate synthetase 1 (CPS-1). Symptoms of UCDs usually arise at birth and encompass, ataxia, convulsions, lethargy, poor feeding and eventually coma and death if not recognized and treated properly. In fact, the mortality rate is 100% for UCDs that are left undiagnosed. Several UCDs manifest with late-onset such as in adulthood. In these cases the symptoms are hyperactivity, hepatomegaly and an avoidance of high protein foods.
Treatment of Urea Cycle Disorders
In general, the treatment of UCDs has as common elements the reduction of protein in the diet, removal of excess ammonia and replacement of intermediates missing from the urea cycle. Administration of lactulose (a nonabsorbable disaccharide) results in reduced ammonia production in the gut and, therefore, less ammonium ion is delivered to the portal circulation from the intestines. Bacteria metabolize lactulose to acidic byproducts which then promotes excretion of ammonia in the feces as ammonium ion, NH4+ and interferes with the production of ammonia through reduced catabolism of other nitrogenous compounds.
Antibiotics can be administered to kill intestinal ammonia producing bacteria.
Sodium benzoate and sodium phenylacetate can be administered to covalently bind glycine (forming hippurate) and glutamine (forming phenylacetylglutamine), respectively. These latter compounds, which contain the ammonia nitrogen, are excreted in the feces. Ammunol® is an FDA-approved intravenous solution of 10% sodium benzoate and 10% sodium phenylacetate used in the treatment of the acute hyperammonemia in UCD patients. However, hemodialysis is the only effective means to rapidly reduce the level of circulating ammonia in UCD patients. Buphenyl® is an FDA-approved oral medication for chronic adjunctive therapy of hyperammonemia in UCD patients. Dietary supplementation with arginine or citrulline can increase the rate of urea production in certain UCDs.
Table of Urea Cycle Disorders
| UCD | Enzyme Deficiency | Symptoms/Comments |
| Type I Hyperammonemia, CPSD | Carbamoylphosphate synthetase 1 | with 24h–72h after birth infant becomes lethargic, needs stimulation to feed, vomiting, increasing lethargy, hypothermia and hyperventilation; without measurement of serum ammonia levels and appropriate intervention infant will die: treatment with arginine which activates N-acetylglutamate synthetase |
| N-acetylglutamate synthase deficiency | N-acetylglutamate synthase | severe hyperammonemia, mild hyperammonemia associated with deep coma, acidosis, recurrent diarrhea, ataxia, hypoglycemia, hyperornithinemia: treatment includes administration of the N-acetylglutamate (NAG) analog, N-carbamoylglutamate, which activates CPS-1 similarly to NAG |
| Type 2 Hyperammonemia, OTCD | Ornithine transcarbamylase | most commonly occurring UCD, only X-linked UCD, ammonia and amino acids elevated in serum, increased serum orotic acid due to mitochondrial carbamoyl phosphate entering cytosol and being incorporated into pyrimidine nucleotides which leads to excess production and consequently excess catabolic products: treat with high carbohydrate, low protein diet, ammonia detoxification with sodium phenylacetate or sodium benzoate |
| Classic (type I) Citrullinemia (CTLN1), ASD | Argininosuccinate synthetase | episodic hyperammonemia, vomiting, lethargy, ataxia, seizures, eventual coma: treat with arginine administration to enhance citrulline excretion, also with sodium benzoate for ammonia detoxification |
| Citrullinemia type II (CTLN2) | Citrin (SLC25A13) | recurrent hyperammonemia and citrullinemia with neuropsychiatric symptoms including nocturnal delirium, aggression, irritability, hyperactivity, delusions, disorientation, restlessness, drowsiness, loss of memory, flapping tremor, convulsive seizures, and coma; symptoms often provoked by alcohol and sugar intake, as well as by certain medications, and/or surgery; clinical course in adults with citrullinemia type II is milder than that of CTLN1; citrin deficiency can manifest in newborns as neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) and in older children as failure to thrive and dyslipidemia caused by citrin deficiency (FTTDCD) |
| Argininosuccinic aciduria, ALD | Argininosuccinate lyase (argininosuccinase) | episodic symptoms similar to classic citrullinemia, elevated plasma and cerebral spinal fluid argininosuccinate: treat with arginine and sodium benzoate |
| Hyperargininemia, AD | Arginase | rare UCD, progressive spastic quadriplegia and intellectual impairment, ammonia and arginine high in cerebral spinal fluid and serum, arginine, lysine and ornithine high in urine: treatment includes diet of essential amino acids excluding arginine, low protein diet |
Glutamate and Glutamine: Nitrogen Homeostasis in the Brain
Ammonia is a serious neurotoxin and as such disorders that lead to elevated levels of circulating ammonia, such as UCDs, as well as severe liver dysfunction, can result in serious consequences to the central nervous system (CNS) and even death. Hyperammonemia exerts it primary negative effects on the CNS via actions that disrupt the metabolism and function of the protective glial cells called astrocytes. Ammonia entering the brain from the circulation is initially incorporated into glutamate forming glutamine via the action of glutamine synthetase. This process can affect the levels of glutamate in synaptic neurons.
Within the CNS glutamate is the main excitatory neurotransmitter. Neurons that respond to glutamate are referred to as glutamatergic neurons. Postsynaptic glutamatergic neurons possess three distinct types of receptors that bind glutamate released from presynaptic neurons. These receptors have been identified on the basis of their binding affinities for certain substrates and are, thus referred to as the kainate, 2-amino-3-hydroxy-5-methyl-4-isoxalone propionic acid (AMPA) and N-methyl-D-aspartate (NMDA) receptors. glutamatergic neurons are responsible for the mediation of many vital processes such as the encoding of information, the formation and retrieval of memories, spatial recognition and the maintenance of consciousness. Excessive excitation of glutamate receptors has been associated with the pathophysiology of hypoxic injury, hypoglycemia, stroke and epilepsy.
Within the CNS there is an interaction between the cerebral blood flow, neurons, and the protective astrocytes that regulates the metabolism of glutamate, glutamine, and ammonia. This process is referred to as the glutamate-glutamine cycle and it is a critical metabolic process central to overall brain glutamate metabolism. Using presynaptic neurons as the starting point the cycle begins with the release of glutamate from presynaptic secretory vesicles in response to the propagation of a nerve impulse along the axon. The release of glutamate is a Ca2+-dependent process that involves fusion of glutamate containing presynaptic vesicles with the neuronal membrane.
Following release of the glutamate into the synapse it must be rapidly removed to prevent over excitation of the postsynaptic neurons. Synaptic glutamate is removed by three distinct processes. It can be taken up into the postsynaptic cell, it can undergo reuptake into the presynaptic cell from which it was released or it can be taken up by a third non-neuronal cell, namely astrocytes. Postsynaptic neurons remove little glutamate from the synapse and although there is active reuptake into presynaptic neurons the latter process is less important than transport into astrocytes.
The membrane potential of astrocytes is much lower than that of neuronal membranes and this favors the uptake of glutamate by the astrocyte. Glutamate uptake by astrocytes is mediated by Na+-independent and Na+-dependent systems. The Na+-dependent systems have high affinity for glutamate and are the predominant glutamate uptake mechanism in the central nervous system. There are two distinct astrocytic Na+-dependent glutamate transporters identified as EAAT1 (for Excitatory Amino Acid Transporter 1; also called GLAST) and EAAT2 (also called GLT-1)
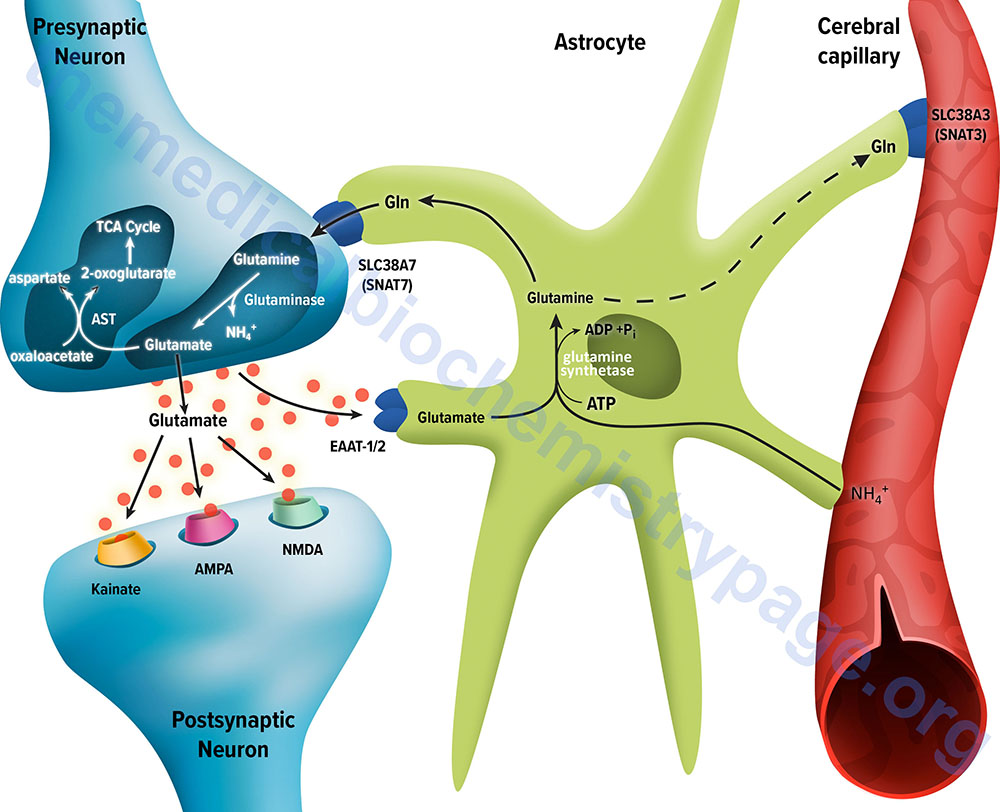
Following uptake of glutamate, astrocytes have the ability to dispose of the amino acid via export into the blood though capillaries that abut the foot processes of the astrocytes. The problem with glutamate disposal via this mechanism is that it would eventually result in a net loss of carbon and nitrogen from the CNS. In fact, the outcome of astrocytic glutamate uptake is its conversion to glutamine. Glutamine thus serves as a “reservoir” for glutamate but in the form of a non-neuroactive compound.
Release of glutamine from astrocytes allows neurons to derive glutamate from this parent compound. Astrocytes readily convert glutamate to glutamine via the glutamine synthetase catalyzed reaction as this microsomal enzyme is abundant in these cells. Indeed, histochemical data demonstrate that the glia are essentially the only cells of the CNS that carry out the glutamine synthetase reaction.
The ammonia that is used to generate glutamine is derived from either the blood or from metabolic processes occurring in the brain. As pointed out below, during periods of hyperammonemia astrocytic glutamine levels may increase so abruptly that the glia may swell resulting in cell damage and death.
Like the uptake of glutamate by astrocytes, neuronal glutamine uptake proceeds via both Na+-dependent and Na+-independent mechanisms. The major glutamine transporter in both excitatory and inhibitory neurons is the system N neutral amino acid transporter identified as sodium-coupled neutral amino acid transporter 7 (SNAT7). SNAT7 is encoded by the SCL38A7 gene.
The predominant metabolic fate of the glutamine taken up by neurons is hydrolysis to glutamate and ammonia via the action of the mitochondrial form of glutaminase encoded by the GLS2 gene. As indicated above, this enzyme is referred to as phosphate-activated glutaminase, PAG. The inorganic phosphate (Pi) necessary for this reaction is primarily derived from the hydrolysis of ATP and its function is to lower the Km of the enzyme for glutamine. During depolarization there is a sudden increase in energy consumption. The hydrolysis of ATP to ADP and Pi thus favors the concomitant hydrolysis of glutamine to glutamate via the resulting increased Pi. Because there is a need to replenish the ATP lost during neuronal depolarization, metabolic reactions that generate ATP must increase.
It has been found that not all neuronal glutamate, that is derived from glutamine, is utilized to replenish the neurotransmitter pool. A portion of the glutamate can be oxidized within the nerve cells following transamination. The principal transamination reaction involves aspartate aminotransferase (AST) and yields 2-oxoglutarate (α-ketoglutarate) which is a substrate in the TCA cycle. Glutamine, therefore, is not simply a precursor to neuronal glutamate but a potential fuel, which, like glucose, supports neuronal energy requirements.
Glutamate, released as a neurotransmitter, is taken up by astrocytes, converted to glutamine, released back to neurons where it is then converted back to glutamate. This process represents the complete glutamate-glutamine cycle. The significance of this cycle to brain glutamate handling is that it promotes several critical processes of CNS function. Glutamate is rapidly removed from the synapse by astrocytic uptake thereby preventing over-excitation of the postsynaptic neuron. Within the astrocyte glutamate is converted to glutamine which is, in effect, a non-neuroactive compound that can be transported back to the neurons. The uptake of glutamine by neurons provides a mechanism for the regeneration of glutamate which is augmented by the generation of Pi as a result of ATP consumption during depolarization. Since the neurons also need to regenerate the lost ATP, the glutamate can serve as a carbon skeleton for oxidation in the TCA cycle. Lastly, but significantly, the incorporation of ammonia into glutamate in the astrocyte serves as a mechanism to buffer brain ammonia.
The glutamate-glutamine cycle does not represent the sole fate of glutamate in the CNS. The brain actively oxidizes glutamate. In fact, data demonstrate that the rate of glutamate oxidation can be so high that the amino acid could theoretically substitute for glucose as an energy source. The primary mechanism of glutamate oxidation appears to be the AST reaction as indicated above. Within the CNS both neurons and astrocytes can also oxidize glutamate via the glutamate dehydrogenase catalyzed reaction. Although this latter reaction could represent a major route for glutamate carbon atoms to be oxidized via the TCA cycle, studies have shown that quantitatively the more significant reaction for glutamate oxidation is the one catalyzed by AST.
During periods of basal metabolism glucose serves as the major metabolic fuel of the brain. During starvation, ketoacidosis results due to increased fatty acid oxidation in the liver. The brain extracts the ketone bodies, β-hydroxybutyrate and acetoacetate, from the blood to use as major fuels during periods when glucose is scarce. Although, as indicated, the brain oxidizes glutamate, there is little passage of either glutamate or glutamine across the blood-brain barrier. Therefore, neither amino acid can serve as a conventional metabolic substrate. Nevertheless, both compounds are important to overall brain energy production. During periods of hypoglycemia, the consumption of glutamate and glutamine increases. Similarly, during periods of acidosis, when glycolytic flux is restricted, astrocytes increase consumption of both glutamate and glutamine. Although the glutamate-glutamine cycle implies a net release of glutamine by astrocytes, these cells also can oxidize glutamine. Just as described above for neurons, astrocytes can convert glutamine back to glutamate via the PAG (glutaminase) catalyzed reaction. Oxidation of glutamate by astrocytes increases as the levels of this amino acid rise.
Experimental evidence indicates that if nerve cell transamination reactions are inhibited there is a concomitant inhibition in the oxidation of glutamate along with significant increases in the intracellular levels of this amino acid. Therefore, since the brain can oxidize glutamate (and similarly glutamine) there is a requirement for a source of nitrogen to compensate for glutamate and glutamine consumed during the process of oxidation. Glutamine can also be transported out of the CNS, a mechanism that may reflect the maintenance of overall metabolic balance.
The major source of carbon atom for glutamate and glutamine synthesis is glucose. Glucose is the ideal substrate for this process since it passes readily from blood to brain via GLUT3-mediated transport and within the brain is oxidized via the TCA cycle to 2-oxoglutarate, which is transaminated via AST to yield glutamate. The major source of the amino groups in glutamate is likely to be another amino acid (or amino acids) since little if any glutamate crosses the blood-brain barrier. The obvious reason for a block to glutamate transport into the brain is that relatively large amounts of glutamate in the extracellular space of the brain would cause inappropriate depolarization of susceptible neurons. Thus, an alternate source of amino groups to support glutamic acid synthesis needs to be made available to the system.
The branched-chain amino acids (BCCA: leucine, isoleucine, and valine) are ideal candidates as the amino donors for brain glutamate synthesis. All three amino acids are readily transported across the blood-brain barrier with leucine crossing more efficiently than any other amino acid. Within many peripheral tissues the BCAA are known to be major sources of the amino group in glutamate.
The brain has abundant BCAA aminotransferase activity and astrocytes are a major cell type involved in the metabolism of BCAA and corresponding α-ketoacids. Experimental evidence has demonstrated that up to 25–30% of the nitrogen present in brain glutamate and glutamine is, in fact, derived from leucine alone. The branched-chain aminotransferase reaction is freely reversible and the parent amino acid is readily regenerated from the corresponding α-ketoacid.
The interrelationship between brain leucine and glutamate suggests that there is likely a leucine-glutamate cycle that functions in concert with the glutamate-glutamine cycle. The interaction of these cycles reflects the fact that leucine entering brain from the periphery is transaminated to yield an α-ketoacid and glutamate. The glutamate can be converted to glutamine, which is released to neurons and there reconverted to glutamate.
Additionally, the α-ketoacid formed from leucine, in the course of the BCAA aminotransferase reaction, is also released from astrocytes and taken up by neurons. Within neurons the α-ketoacid can be transaminated with glutamate to regenerate leucine. The leucine then can be transported back to astrocytes, which completes the putative leucine-glutamate cycle. The metabolic benefits of this proposed cycle are related to the fact that it presents an efficient mechanism for the uptake of amino nitrogen from the periphery and the subsequent synthesis of glutamate and glutamine. Additionally, since the BCAA aminotransferase reaction is freely reversible, the reamination of a branched chain α-ketoacid to a parent amino acid results in the net consumption of glutamate which serves as a type of glutamate buffering.
A final consideration in the context of CNS nitrogen homeostasis is the means by which the brain disposes of waste nitrogen since the brain cannot synthesize urea. The brain generates ammonia and its level of generation rises sharply during neuronal depolarization. The source of the ammonia during this process is the enzymatic hydrolysis of glutamine (derived from astrocytes) within neurons via the PAG catalyzed reaction. A smaller fraction of brain ammonia generation is the result of the oxidative deamination of glutamate via the glutamate dehydrogenase catalyzed reaction. Therefore, the synthesis of glutamine by astrocytes and its transport to the blood provides an important mechanism for the removal of excess nitrogen from the brain.
Branched-Chain Amino Acid Metabolism: Neuronal Nitrogen Homeostasis
Metabolism of the branched-chain amino acids (BCAAs), isoleucine, leucine, and valine, is important not solely for the ability to generate ATP via the oxidation of their carbon skeletons. Metabolism of the BCAAs is an important regulator of overall energy consumption in skeletal muscle, functions to modulate feeding behaviors via altered energy homeostasis in the hypothalamus, serves to regulate excitatory neurotransmitter homeostasis, and also serves to regulate overall nitrogen homeostasis within the brain. The key enzyme in BCAA metabolism that is the major contributor to all the aforementioned functions is BCAA aminotransferase (BCAT). Humans express two genes that encode BCAT activity. These two genes are identified as BCAT1 and BCAT2.
The primary protein encoded by the BCAT1 gene is a cytosolic version of the enzyme and the protein is identified as BCATc. The primary protein encoded by the BCAT2 gene is a mitochondrial version of the enzyme and this protein is designated BCATm.
The BCAT1 gene is located on chromosome 12p12.1 and is composed of 14 exons that generate five alternatively spliced mRNAs, each of which encode a distinct protein isoform.
The BCAT2 gene is located on chromosome 19q13.33 and is composed of 13 exons that generate three alternatively spliced mRNAs, each of which encode a distinct protein isoform.
The BCAT1 gene represents the primary BCAT expressing gene in the brain. Expression of BCAT2 is widely distributed among numerous tissues. Although detectable in the fetal liver, the adult liver does not express either BCAT gene. Within the brain, different cell types predominantly express either the BCAT1 gene or the BCAT2 gene. This differential localization of the two forms of BCAT allows for modulation of nitrogen and neurotransmitter homeostasis. Astroglial cells (astrocytes) express the mitochondrial enzyme (BCATm) encoded by the BCAT2 gene, whereas, neurons express the cytosolic enzyme (BCATc) encoded by the BCAT1 gene.
When branched-chain amino acids enter the vasculature of the brain they are taken up by astroglial cells (astrocytes) which are in direct contact with the blood via cerebral capillaries. Upon entry into the astrocyte the BCAA are deaminated by BCATm. The amino acceptor in these reactions is 2-oxoglutarate (α-ketoglutarate) and the byproducts are glutamate and the corresponding branched-chain α-keto acids (BCKA).
Oxidation of BCKA is very inefficient in astroglial cells and so they are transported out of the cell where they are taken up by the neurons. Within the neuron, the BCKA can be transaminated back to the corresponding BCAA with concomitant production of 2-oxoglutarate from the amino donor, glutamate. The neuronal transamination reaction is catalyzed by BCATc. The resulting BCAA are transported out of the neuron and returned to the astrocyte.
Dependent upon the energy charge in the neuron, the level of glutamate, and on the overall level of neuronal ammonium ion (NH4+), the resulting 2-oxoglutarate can be reductively aminated by glutamate dehydrogenase to yield glutamate, or it can enter the TCA cycle for oxidation to contribute to ATP production. This series of astrocyte BCATm and neuronal BCATc reactions provides a mechanism for efficient nitrogen transfer between astrocytes and neurons and synthesis of glutamate from astrocyte 2-oxoglutarate. This pathway can be pharmacologically disrupted by the use of the drug gabapentin (marketed primarily under the name Neurontin) which inhibits neuronal BCATc resulting in reduced production of the excitatory neurotransmitter, glutamate.
Neurotoxicity Associated with Ammonia
Earlier it was noted that ammonia was neurotoxic. Marked brain damage is seen in cases of failure to make urea via the urea cycle or to eliminate urea through the kidneys. The result of either of these events is a buildup of circulating levels of ammonium ion. Aside from its effect on blood pH, ammonia readily traverses the brain blood barrier and in the brain is converted to glutamate via glutamate dehydrogenase, depleting the brain of 2-oxoglutarate (α-ketoglutarate). As the 2-oxoglutarate is depleted, oxaloacetate falls correspondingly, and ultimately TCA cycle activity comes to a halt. In the absence of aerobic oxidative phosphorylation and TCA cycle activity, irreparable cell damage and neural cell death ensue.
In addition, the increased glutamate leads to glutamine formation. This depletes glutamate stores which are needed in neural tissue since glutamate is both a neurotransmitter and a precursor for the synthesis of γ-aminobutyrate: GABA, another neurotransmitter. Therefore, reductions in brain glutamate affect energy production as well as neurotransmission. For detailed information on the role of the glutamate-glutamine cycle in the brain and its role in regulating neural cell ammonia levels was discussed above.
Additional untoward consequences of hyperammonemia have been attributed to an increase in neural glutamine concentration. Astrocyte cell volume is controlled by intracellular organic osmolyte metabolism. One important organic osmolyte in these cells is glutamine. As glutamine levels rise in the brain concomitant with increased ammonia uptake it was hypothesized that the volume of fluid within glial cells would increase resulting in the cerebral edema seen in infants with hyperammonemia caused by urea cycle defects.
However, there are some problems with this particular model. First, glutamine accounts for no more than 1.5% of the sum of all the osmolytes within the in the brain. Second, a significant proportion of the newly synthesized glutamine can rapidly exit astrocytes due to both diffusion and through the action of specific glutamine transporters. The primary astrocyte glutamine transporter is the sodium-coupled neutral amino acid (system N/A) transporter 3 (SNAT3: a member of the solute carrier family of transporters also identified as SLC38A3). The result of diffusion and transport is a reduction in the glutamine overload within astrocytes. Additionally, an increase in brain glutamine during other causes of hyperammonemia, such as hepatic encephalopathy, has been shown to be accompanied by loss of different low MW osmolytes, including myo-inositol, taurine, and betaine.
A current concept of the role of brain glutamine concentration in the neurotoxicity associated with hyperammonemia relates to its adverse effects on mitochondrial function. In laboratory animals it has been shown that inhibition of glutamine synthesis by methionine sulfoximine (MSO) results in suppression of ammonia-induced formation of reactive oxygen species (ROS). In addition, when glutamine is added directly to astrocyte cell cultures there is a resultant induction of mitochondrial permeability transition (mPT) and swelling of the mitochondria. This effect of glutamine can be blocked by addition of cyclosporine A (CsA), which is a known inhibitor of mPT, as well as by inhibition of mitochondrial glutamine uptake by histidine. Of note is that fact that histidine administration can prevent brain edema during drug-induced liver failure in rodents.
Additionally, studies have demonstrated that when glutamine is administered in the absence of excess ammonia there is a dose-dependent increase in ROS production by astrocytes. This latter effect can be blocked by administration of CsA. Conversely, ammonia itself does not induce mPT or mitochondrial swelling indicating that its direct entry into the mitochondria may not be efficient enough to exert a deleterious effect on mitochondrial function. However, elevation in intracellular ammonia does lead to increased mitochondrial uptake of glutamine.
Within the mitochondria the glutamine is degraded to glutamate and ammonia by a glutaminase encoded by the GLS2 gene. The GLS2 gene encoded glutaminase was originally thought to be liver specific but is in fact expressed in numerous tissues and is important in the glutamate-glutamine cycle in the brain. The GLS2 encoded glutaminase is dependent on inorganic phosphate (Pi) for activity and is, as pointed out earlier, also referred to as phosphate-activated glutaminase, PAG. The action of mitochondrial glutaminase in the astrocytes leads to further increases in intramitochondrial ammonia levels. That the PAG action is significant to glutamine-induced astrocyte swelling is demonstrated by the fact that inhibition of PAG prevents swelling of mitochondria under hyperammonemic conditions.
These results suggest a model whereby, under conditions of hyperammonemia, the accumulation of ammonia in brain cells (in particular astrocytes) potentiates glutamine toxicity by facilitating its uptake into mitochondria. The increased mitochondrial uptake of glutamine leads to increased mitochondrial ammonia production triggering mitochondrial impairment, and an ensuing chain of deleterious events leading to astrocytic dysfunction, swelling and brain edema.