
Last Updated: July 13, 2026
Introduction to the Niemann-Pick Diseases
The Niemann-Pick (NP) diseases are a group of autosomal recessive disorders that belong to the family of disorders identified as lysosomal storage disorders.
There are two distinct sub-families of NP diseases. Niemann-Pick type A (NPA) and type B (NPB) diseases are caused by defects in the gene encoding acid sphingomyelinase (ASM). Niemann-Pick type C1 (NPC1) disease is caused by defects in a gene involved in LDL-cholesterol homeostasis identified as the NPC1 gene.
Niemann-Pick Disease, Types A and B
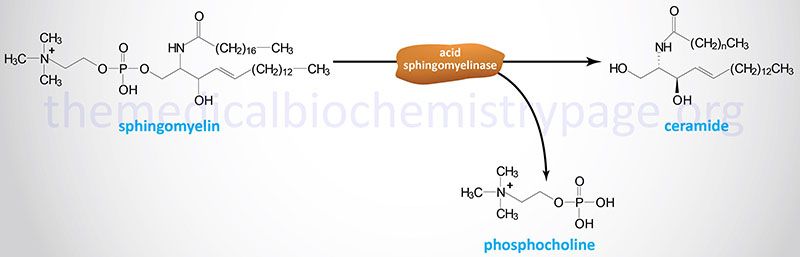
Both Niemann-Pick disease type A and type B are caused by defects in the lysosomal hydrolase, acid sphingomyelinase (ASMase) encoded by the SMPD1 (sphingomyelin phosphodiesterase-1) gene.
Type A Niemann-Pick disease is associated with a rapidly progressing neurodegeneration leading to death by 2 to 3 years of age. In contrast, type B Niemann-Pick disease has a variable phenotype marked primarily by visceral involvement with little to no neurological detriment. Diagnosis of type B Niemann-Pick disease is usually made in early childhood by the presence of hepatosplenomegaly. The most severely affected type B patients exhibit a progressive pulmonary involvement. Both type A and type B Niemann-Pick disease are characterized by the presence of the “Niemann-Pick” cell. This histologically distinct cell type is of the monocyte-macrophage lineage and is a characteristic lipid-laden foam cell.
The course of type A Niemann-Pick disease is rapid. Infants are born following a typically normal pregnancy and delivery. Within 4–6 months the abdomen protrudes and hepatosplenomegaly will be diagnosed. The early neurological manifestations include hypotonia, muscular weakness and difficulty feeding. As a consequence of the feeding difficulties and the swollen spleen, infants will exhibit a decrease in growth and body weight. By the time afflicted infants reach 6 months of age the signs of psycho-motor deterioration become evident. Affected infants becomes weaker and progressively hypotonic. Previous developmental milestones such as sitting alone begin to be lost. Ophthalmic examination reveals a cherry-red spot typical of patients with Tay-Sachs disease (another lysosomal storage disorder; lysosomal storage disease) in about 50% of type A Niemann-Pick disease infants. As the disease progresses spasticity and rigidity increase and infant experience complete loss of contact with their environment. As indicated above, type B Niemann-Pick disease has a much more variable phenotype and most patients do not have neurological involvement and are intellectually normal. There is currently no therapy for either type A or type B Niemann-Pick disease.
Molecular Biology of Niemann-Pick Types A and B
Acid sphingomyelinase is encoded by the SMPD1 gene. The SMPD1 gene is located on chromosome 11p15.4 spanning 5 kb and composed of 6 exons that generate five alternatively spliced mRNAs, each of which encode distinct protein isoforms.
As of 2025 there have been at least 200 pathogenic variants identified in the SMPD1 gene resulting in types A and B Niemann-Pick disease.
There are three disease alleles that represent greater than 90% of the cases of type A Niemann-Pick disease in Ashkenazi Jewish populations. These disease alleles include a single nucleotide deletion resulting in a frame-shift at proline 330 (fsP330) and two different missense variants, one leading to the substitution of leucine for proline at amino acid 302 (L302P) and the other leading to substitution of arginine for leucine at amino acid 496 (R496L). The carrier allele frequency for these type A variants is as high as 1 in 80 in Ashkenazi Jewish populations.
A single pathogenic variant, a 3-base deletion leading to loss of arginine at what would be amino acid position 608 (ΔR608), is commonly associated with type B Niemann-Pick disease. This latter pathogenic variant leaves the ASMase protein with sufficient residual activity to afford protection from the severe neurological symptoms associated with type A NP disease.
Niemann-Pick Disease, Type C1
Niemann-Pick disease type C1 (NPC1) results from an error in the trafficking of exogenous cholesterol, thus it is more commonly referred to as a lipid trafficking disorder even though it belongs to the family of lysosomal storage disorders. The principal biochemical defect in patients with NPC is an accumulation of cholesterol, sphingolipids, and other lipids in the late endosomes/lysosomes (LE/L) of all cells. NPC is a disease characterized by fatal progressive neurodegeneration.
The prevalence of NPC1 disease is more common than NPA and NPB disease combined, however, as indicated above for NPA, certain ethnic groups have significant disease allele carrier frequency. There can be significant clinical heterogeneity associated with NPC disease. Most afflicted individuals have progressive neurological disease with early lethality. The characteristic phenotypes associated with “classic” NPC1 disease are variable hepatosplenomegaly, progressive ataxia, dystonia, dementia and vertical supranuclear gaze palsy (VSGP). These individuals will present in childhood and death will ensue by the second or third decade.
Because of the variable clinical phenotypes of NPC1 disease it has been sub-divided into five presentation classifications: perinatal, early infantile, late infantile, juvenile and adult. VSGP is a characteristic neurological manifestation in NPC1 disease being found in virtually all juvenile and adult cases of the disease. Like NPA and NPB disease, NPC1 pathology is characterized by the presence of lipid-laden foam cells in the visceral organs and the nervous system. There is currently no specific treatment for NPC1 disease.
Molecular Biology of NPC1
The gene, in which pathogenic variants lead to NPC1, is identified as NPC1 (NPC intracellular cholesterol transporter 1) and is located on chromosome 18q11.2 spanning 47 kb and composed of 28 exons that encode a 1278 amino acid precursor protein.
As of 2025 a total of 341 pathogenic variants have identified in the NPC1 gene in Niemann-Pick disease type C afflicted individuals. The most prevalent pathogenic variants are single nucleotide variants and many afflicted individuals are genotypic compound heterozygotes harboring at least two of the variants. The eight most common single nucleotide variants identified are at the following nucleotides:
- 530 [G to A (G>A) variant resulting in cysteine (C) 177 change to tyrosine (Y) (C177Y)]
- 2324 (A>C resulting in QA775P)
- 2974 (G>T resulting in G992W)
- 2974 (G>C resulting in G992R)
- 2974 (G>A resulting in G992R)
- 3019 (C>G resulting in P1007A)
- 3160 (G>A resulting in A1054T)
- 3182 (T>C resulting in I1061T)
Several additional single nucleotide variants have been identified in the NPC1 gene but they do not manifest with disease.
The NPC1 gene encodes a 1278 amino acid precursor protein that contains regions of homology to mediators of cholesterol homeostasis suggesting why LDL-cholesterol accumulates in lysosomes of afflicted individuals. Within the protein are regions of homology to the transmembrane domain of the morphogen receptor patched (of Drosophila melanogaster) and the sterol-sensing domains (SSD) of SCAP (SREBP cleavage-activating protein; SREBP=sterol regulated element binding protein) and HMG-CoA reductase (HMGR).
The NPC1 encoded protein is an integral membrane protein containing 13 putative transmembrane domains. The protein is primarily associated with intraluminal vesicles and multi-vesicular late endosomes. The transmembrane regions of NPC1 are separated by three luminal loops that contain sites of glycosylation. NPC1 also transiently cycles through the trans-Golgi network.
The SSD of NPC1 is composed of 5 of the 13 transmembrane domains. Unlike the SSD in SCAP and HMGR, the NPC1 SSD does not bind the Insig proteins but does bind cholesterol esters and oxysterols. Pathogenic variants in the SSD of NPC1 are commonly found in NPC1 patients. The major function of NPC1 is to facilitate the movement of cholesterol out of LE/L.
Molecular Biology of NPC2
At least 95% of Niemann-Pick type C patients contain pathogenic variants in the NPC1 gene with the remainder harboring pathogenic variants in a second gene identified as NPC2 (NPC intracellular cholesterol transporter 2). The NPC2 gene is located on chromosome 14q24.3 spanning 13.5 kb and composed of 4 exons that generate three alternatively spliced mRNAs, each of which encode a distinct protein isoform.
As of 2025 a total of 30 pathogenic variants in the NPC2 gene have been identified in individuals with Niemann-Pick disease type C disease.
The NPC2 protein was originally identified as epididymal secretory protein E1 (HE1). Like the protein encoded by the NPC1 locus, the NPC2 gene encoded protein also binds cholesteryl esters. However, unlike NPC1, NPC2 does not bind oxysterols. The NPC2 protein is a soluble protein found in the lumen of lysosomes. The NPC2 protein is targeted to the lysosomes by binding the mannose-6-phosphate receptor. The NPC2 protein is involved in cholesterol movement out of LE/L in conjunction with NPC1.
One model proposed for the combined actions of NPC1 and NPC2 is that membrane bound NPC1 interacts with cholesterol that has accumulated in intraluminal vesicle membranes and then transfers the cholesterol to soluble NPC2. The action of NPC2 is then to transfer the cholesterol to the limiting membranes of LE/L which allows for cholesterol distribution to other cellular membranes. An alternative model proposes that NPC2 removes cholesterol from intraluminal vesicles of LE/L and then transfers the cholesterol to NPC1 in the limiting membranes of the LE/L.
Niemann-Pick type C1-like 1
A gene related to the NPC1 gene is called Niemann-Pick type C1-like 1 (NPC1L1). This gene is expressed in the brush border cells of the small intestine and is involved in intestinal absorption of cholesterol. The cholesterol lowering action of the drug ezemitibe (Zetia®) stems from the fact that the drug binds to and interferes with the cholesterol absorption functions of NPC1L1. NPC1L1 is also highly expressed in human liver.
Niemann-Pick type D
A genetic isolate of Niemann-Pick disease, first identified in patients in Colorado and Nova Scotia, Canada, was originally called Niemann-Pick type D (NPD) disease. However, these patients are now known to have harbored specific alleles of the NPC1 locus.