
Last Updated: April 2, 2026
Introduction to Muscle Cell Types
Current understanding of the molecular events underlying muscle contraction is embodied in the sliding filament model of muscle contraction. The model is applicable to smooth, skeletal, cardiac, and other contractile activity, including mechanochemical events such as single cell locomotion and receptor endocytosis. Since the biochemistry of these activities are best understood for skeletal, cardiac, and smooth muscle, this page focuses on these muscle types (noting, where appropriate, differences in other cell types). The biochemical characteristics that differentiate fast-reacting and slow-reacting cells in muscle tissue and the biochemical basis of some common pathophysiological states of muscle, including tetany, fatigue, and rigor mortis are reviewed as well. Cardiac and skeletal muscle cells contain sarcomeres (described in detail below) which gives the cells a “striated” appearance when viewed under a microscope. For this reason these two types of cells are referred to as striated muscle. Smooth muscle cells do not contain sarcomeres and are, therefore, non-striated muscle cells. Skeletal muscle cells form long multinucleated fibers while cardiac muscle cells typically have only one nucleus per cell.
Skeletal muscles comprise about 40% of the mass of the average human body and are formed of long multinucleate, cylindrical cells called muscle fibers. Skeletal muscle fibers are grossly divided into two types; slow twitch (type I) and fast twitch (type II). Type II fibers are further divided into type IIa and type IIb fibers. Type IIa fibers are intermediate fast twitch fibers and can utilize both aerobic and anaerobic metabolism for ATP production. Type IIb fibers are the classic fast twitch fibers. Slow twitch muscle fibers primarily utilize fatty acid oxidation and contain a high concentration of mitochondria and store appreciable amounts of oxygen as oxymyoglobin. These two facts are the reason that slow twitch fibers are red in color. Fast twitch fibers primarily utilize glucose oxidation to pyruvate for ATP production, contain less mitochondria and myoglobin than slow twitch fibers, and thus, are white muscle fibers. Because slow twitch fibers prefer to oxidize fatty acids they are also referred to as oxidative fibers, whereas fast twitch fibers that utilize glucose are referred to as glycolytic fibers. Slow twitch fibers are capable of continuous extended contractions and therefore, do not fatigue quickly. Fast twitch fibers are used for short rapid bursts of energy and as such fatigue more quickly than slow twitch fibers.
The plasma membrane of muscle cells is known as the sarcolemma. Each muscle is made up of bundles of these cells (forming muscle fibers), embedded in a matrix of connective tissue known as the endomysium. The bundle of fibers with its endomysium is surrounded by a more fibrous connective tissue sheath known as the perimysium. The composite of the perimysium and its contents is known as a fasciculus. A complete muscle consists of numerous fasciculi surrounded by a thick outer layer of connective tissue known as the perimysial septa. The translation of contractile activity of individual muscle fibers to anatomical motion take place through this continuous system of connective tissues and sheaths, which ultimately meld into the tendons.
Within the sarcolemma is the sarcoplasm (cytoplasm), containing all the usual subcellular elements plus long prominent myofibrils. Each myofibril is composed of bundles of filamentous contractile proteins, some extending from end to end in the cell. Myofibrils are the most conspicuous elements in skeletal myofibers making up about 60% of myofiber protein. A single myofibril is composed of many short structural units, known as sarcomeres, which are arranged end to end. The proteins at the junctions between sarcomeres form the Z line, and thus a sarcomere extends along a myofibril from one Z line to the next Z line. Sarcomeres are composed mostly of actin thin filaments and myosin thick filaments. Sarcomeres represent the minimal contractile unit of a muscle. It is the coordinated contraction and elongation of millions of sarcomeres in a muscle that gives rise to mechanical skeletal activity. The relationship between muscle proteins and muscles is summarized in the table below:
Table of Muscle Protein and Muscle Structures
| Organization of Contractile Proteins in Muscle | |
| Thick Filament | Composed of hundreds of long, contractile myosin molecules arranged in a staggered side by side complex |
| Thin Filament | Composed of a linear array of hundreds of globular, actin monomers in a double helical arrangement |
| Sarcomere | The unit of contractile activity composed mainly of actin and myosin and extending from Z line (Z band) to Z line in a myofibril |
| Myofibril | End-to-end arrays of identical sarcomeres |
| Myofiber | A single multinucleate muscle cell containing all the usual cell organelles plus many myofibrils |
| Muscle | Organized arrays of muscle fibers |
Organization of the Sarcomere
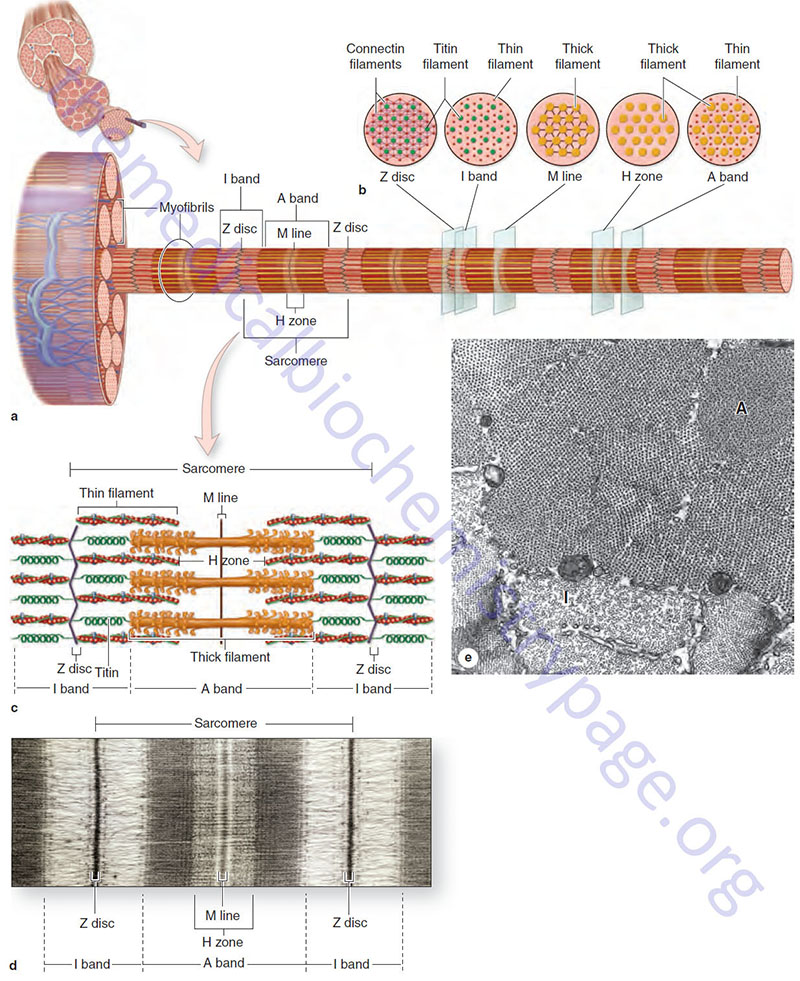
The organization of individual contractile proteins making up a sarcomere is a key feature of the sliding filament model. Each sarcomere is composed of hundreds of filamentous protein aggregates, each known as a myofilament. Two kinds of myofilaments are identifiable on the basis of their diameter and protein composition (see image above). Thick myofilaments are composed of several hundred molecules of one of several different fibrous proteins known as myosin. Thin myofilaments are composed of two helically interwound, linear polymers of globular proteins known as actins. Thin and thick filaments also contain accessory proteins as described below. Proteins of the Z disc, primarily the α-actinins (see next section), serve as an embedding matrix or anchor for one end of the thin filaments, which extend toward the center of sarcomeres on either side of the Z disc. The Z disc proteins often appear continuous across the width of a muscle fiber and seem to act to keep the myofibrils within a myofiber in register.
Actinins
Humans express four α-actinin genes identified as ACTN1, ACTN2, ACTN3, and ACTN4. The distal end of each thin filament is free in the sarcoplasm and the barbed end of the actin filaments are capped with proteins known as actin capping proteins. The actin capping proteins involved in muscle filament formation and stability are members of the F-actin capping protein family. These proteins are heterodimeric complexes formed from an α-subunit and a β-subunit. There are three actin capping α-subunit genes in humans identified as CAPZA1, CAPZA2, and CAPZA3. The single β-subunit gene is identified CAPZB. The CAPZB encoded protein was at one time called β-actinin. The nomenclature CAPZ refers to capping protein of muscle Z line (Z disc).
Myomesins
Also depicted in the image above is a second disk-like protein aggregate, the M-line, which is centrally located in sarcomeres. Like proteins of the Z disc, the proteins of the M line (M band) act as an embedding matrix, in this case for the myosin thick filaments. Thick filaments extend from their point of attachment on both sides of the M line toward the two Z discs that define a sarcomere. The principal proteins associated with the M line (M band) are the myomesins. Humans express three myomesin genes identified as MYOM1, MYOM2, and MYOM3.
Titin
The large protein, titin (encoded by the TTN gene), connects a Z disc with an M line (M band) such that a single titin protein spans half the length of a sarcomere. Titin is the largest human protein and many tissue-specific isoforms are generated as a result of alternative mRNA splicing. The canonical human titin protein consists of 34,350 amino acids. In addition to interacting with proteins in the Z disc and M line, titin also interacts with several other proteins essentially serving as an adhesion template for the assembly of a functional contractile complex in muscle cells.
The TTN gene is located on chromosome 2q31.2 and is composed of 364 exons. Due to alternative splicing at least seven different isoforms of the titin protein are generated. Titin isoform N2-A (33423 amino acids) is the predominant titin protein in skeletal muscle and the N2-B isoform (26926 amino acids) is the predominant titin in cardiac muscle.
Within a sarcomere the thick and thin filaments interdigitate so that in cross section they are seen to form a hexagonal lattice, in which six thin filaments are arrayed around each thick filament. The thick filaments are also arranged hexagonally to each other. During contraction and relaxation the distance between the Z discs varies, decreasing with contraction and increasing with relaxation. The M line, with its attached thick filaments, remains centrally located in the sarcomere. The thin and thick filaments retain their extended linear structure except in extreme situations. Changes in sarcomere length are caused by the thin filaments being pulled along the thick filaments in the direction of the M line.
Proteins of the Myofilaments
The biochemical basis of muscle activity is related to the enzymatic and physical properties of actins, myosins, and the accessory proteins that constitute the thin and thick filaments. The following discussion summarizes the key protein components of the myofilaments and their ATP-dependent interactions, which produce contractile activity. The proteins of the thin and thick filaments can be separated into an actin, a myosin (see below), and several accessory proteins or protein complexes. The accessory proteins/complexes include the α-actinins, Cap Z, the tropomyosins (see next section), and the troponin complex (see next section). Solubilized myosin molecules are long thin (fibrous) proteins with a molecular weight of about 500,000 daltons. Both the actins and the myosins (specifically the heavy chain myosins) possess ATPase activity.
Muscle Actins
The actins were initially characterized as being either α (alpha), β (beta), or γ (gamma) actins. The α-actins are the predominant actins of the contractile apparatus of muscle cells. The β- and γ-actins are involved in the regulation of cell motility functions. There are six actin genes in the human genome identified as ACTA1, ACTA2, ACTB, ACTC1, ACTG1, and ACTG2. The ACTA1, ACTA2, and ACTC1 encoded proteins are all α-actins. The ACTB gene encodes a β-actin. The ACTG1 and ACTG2 genes encode the γ-actins. Expression of the ACTA1 gene predominates in skeletal muscle while the ACTA2 gene is expressed in smooth muscle and the aorta. ACTC1 gene expression predominates in cardiac muscle. Expression of the ACTG2 gene predominates in smooth muscle cells.
Myofilament Compositions
Each myosin molecule is made up of six subunits, two very large, heavy chains (HC), and two pairs of non-identical light chains (LC). Within the myosin molecule it is the HC proteins that possess the ATPase-mediated motor function. In a given muscle fiber the two large subunits are identical, although there are different HC isoforms in different types of muscle fibers.
Heavy chains contain a long linear C-terminal α-helical domain (1,300 amino acids) and a prominent globular N-terminal domain of about 800 amino acids. The two HC α-helical domains are helically interwound, giving the molecules a long, rigid superhelical structure with two globular headpieces.
A complete myosin molecule also contains the two pairs of LC which are associated with the globular headpieces. The LC proteins of a myosin molecule possess molecular weights of 16,000–24,000 Da. The LC proteins were originally characterized by their chemical sensitivities and so are also referred to as alkali light chains and DTNB (Ellman’s reagent) light chains. The alkali light chains are not phosphorylated whereas, the other two light chains in a myosin are phosphorylated. The phosphorylated LC proteins are, therefore, the regulatory light chains of the myosin molecule. The LC proteins bind Ca2+ with high affinity. The phosphorylation of the LC proteins is catalyzed by one of four myosin light chain kinases, MLCK (also identified as MYLK).
The myosin light chain kinases are also regulated by Ca2+ binding to their calmodulin subunits. Phosphorylation of myosins by MLCK serves to regulate the ATPase activity of the myosin molecule as well as its assembly into thick filaments. The different myosin LC proteins, along with different myosin HC proteins constitute the varying myosins from cardiac, skeletal, embryonic, and smooth muscle.
Muscle Heavy Myosins
The human genome contains a large family of myosin protein encoding genes. Although not all of these myosin encoding genes encode myosins involved in muscle contraction, all of the proteins interact with actins, exhibit ATPase enzymatic activity, and are involved in force transduction. The large myosin superfamily is divided into twelve different classes identified as myosin I, II, III, V, VI, VII, IX, X, XV, XVI, XVIII, and XIX.
The myosin heavy chain (HC) genes are all members of the myosin II subfamily of myosins. Humans express 15 functional myosin heavy chain genes identified as MYH1, MYH2, MYH3, MYH4, MYH6, MYH7, MYH7B, MYH8, MYH9, MYH10, MYH11, MYH13, MYH14, and MYH15. Several of the myosin heavy chain genes are clustered in the region of chromosome 17p13 (MYH1, MYH2, MYH3, MYH4, MYH8, MYH10, and MYH13). Expression of the MYH1-MYH4, MYH8, and MYH13 genes predominates in skeletal muscle. Expression of the MYH6, MYH7, and MYH7B genes predominates in cardiac muscle. Expression of MYH11 predominates in smooth muscle. Expression of the MYH9, MYH10, and MYH14 genes is found in non-muscle tissues. The non-muscle myosin heavy chain proteins are involved in the processes of cytokinesis, cell motility, and in the maintenance of cell shape.
Muscle Light Myosins
Although the light chain (LC) proteins of a myosin molecule are called myosin light chains they are not “myosins” but are, nonetheless, important components in the formation of a functional myosin complex. Humans express 13 different myosin light chain genes identified as MYL1, MYL2, MYL3, MYL4, MYL5, MYL6, MYL6B, MYL7, MYL9, MYL10, MYLPF (formerly MYL11), MYL12A, and MYL12B. The LC proteins encoded by MYL1, MYL3, MYL4, MYL6, and MYL6B are all alkali LCs and non-phosphorylated. The regulatory LC proteins, those that are phosphorylated, are encoded by the MYL2, MYL5, MYL9, MYLPF, and MYL12A genes. Less detail is known regarding the functions of the other MYL gene products. MYL1 and MYLPF are both expressed in fast-twitch skeletal muscle. MYL3 and MYL6B are expressed in slow-twitch skeletal muscle with MYL6B also being expressed in non-muscle cells. Expression of the MYL2 and MYL4 genes predominates in cardiac muscle. MYL5 is expressed in the retina and in the brain. MYL6 and MYL12A are expressed in smooth muscle and non-muscle cells.
Myosin Structural Characteristics
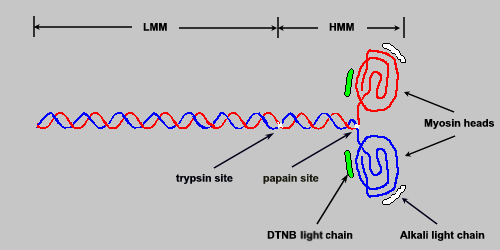
Several functionally important landmarks exist on the myosin molecule. Near the midpoint of the long linear superhelical region is a site defined by its ready susceptibility to proteolytic trypsin digestion. Trypsin cleaves myosin complexes into two portions: one containing both globular headpieces and some superhelical region, and the other consisting of the remaining superhelical portion of the carboxy terminus. The portion containing the headpiece is referred to as heavy meromyosin (HMM; molecular weight 350,000). The C-terminal fragment is referred to as light meromyosin (LMM; molecular weight 125,000).
The significance of the trypsin site is that the susceptibility of myosins to protease action is thought to reflect an interruption in the otherwise rigid superhelix, allowing this site to act as one of a hinge point involved in converting the chemical energy of ATP hydrolysis into the mechanical events of contraction and relaxation. A second proteolytic landmark, susceptible to papain digestion, has also been considered a hinge point. Papain cleaves a site very close to the globular headpieces; these then separate to form two subfragments, each known as a SF-1 (for subfragment 1). The remaining superhelical portion of the molecule is known as SF-2. The ATPase activity of the myosin complex is associated with the SF-1 units.
A thick filament is composed of approximately 400 myosin molecules, 200 arrayed on either side of the M line (M band). These molecules are maintained in bundles by one of several forms of myosin binding protein C, the myomesins of the M line, and the hydrophobic interactions of the myosin molecules themselves. The myosin molecules are most tightly packed in the regions represented by the LMM portion of the molecules. Humans express three myosin binding protein C genes identified as MYBPC1, MYBPC2, and MYBPC3. The MYBPC1 gene is predominantly expressed in slow-twitch muscle, the MYBPC2 gene in fast-twitch muscle, and the MYBPC3 gene in cardiac muscle.
At the trypsin hinge point the heavy meromyosin angles sharply outward from the main axis of the thick filament. This extension of the heavy meromyosin away from the main axis of the thick filament helps bring the headpiece into close proximity to the actin thin filaments lying between the thick filaments. The molecular event underlying muscle contraction is the regulated binding of the myosin headpieces to actin thin filaments, followed by rapid myosin conformational changes about its hinge points with the bound actin being translocated toward the M line.
Organization of Actin Thin Filaments
Thin filaments are composed of many subunits of the globular protein G-actin (G for globular: 42 kD) and several accessory proteins. In thin filaments, G-actin is polymerized into long fibrous (filamentous) arrays known as F-actin (F for filamentous). A pair of linear F-actin arrays is helically wound to form the backbone structure of one complete thin filament.
Each G-actin subunit has one ADP/ATP binding site, presumed to be involved in polymerizing the thin filament. Once polymerized, the actin is capped and the thin filament stabilized by a protein known as β-actinin. In addition to its nucleotide binding site, each G-actin molecule contains a high-affinity myosin head-binding site. In skeletal and cardiac muscle, accessory proteins of the thin filament physically regulate the availability of this site for binding myosin. Thus, the accessory proteins control contractile events.
The main thin filament accessory proteins are the tropomyosins and the troponin complex. Tropomyosins are long, rod-like, αβ helically-interwound heterodimers that span a length of seven G-actin residues. A pair of tropomyosin molecules, in head-to-tail orientation, is associated with every seven pairs of G-actin residues along a thin filament, one tropomyosin molecule in each of the grooves of the F-actin helix. In relaxed muscle, each tropomyosin molecule covers the myosin binding sites of seven G-actin residues, preventing interaction between actin and myosin and thus maintaining the relaxed state. In the absence of calcium, tropomyosins, along with the troponin complex, prevent myosin access to its binding site on actin. Whereas, in the presence of calcium the tropomyosin-troponin complexes allow the myosin-actin interactions to occur. This onset of contractile activity involves the calcium-mediated activation of the troponin complex.
Tropomyosins
Humans express four tropomyosin genes identified as TPM1, TPM2, TPM3, and TPM4. Each of the four human tropomyosin genes contains a basic nine exon structure where there are also several alternative exons that lead to the large number of alternatively spliced mRNAs. For example the TPM1 gene contains two alternative exon 1 (1a and 1B), two alternative exon 2 (2a and 2b), two alternative exon 6 (6a and 6b), and four alternative exon 9 (9a, 9b, 9c, and 9d).
The TPM1 gene is located on chromosome 15q22.2 and is composed of 14 exons that generate at least 40 alternatively spliced mRNAs. The TPM1 encoded proteins are the α-subunits. Expression of TPM1 predominates in striated muscle and as such constitutes the major α-tropomyosin protein in this tissue.
The TPM2 gene is located on chromosome 9p13 and is composed of 11 exons that generate at least 4 alternatively spliced mRNAs. The TPM2 encoded proteins are the β-subunits. Expression of the TPM2 gene predominates in slow-twitch skeletal muscle.
The TPM3 gene is located on chromosome 1q21.3 and is composed of 16 exons that generate at least 15 alternatively spliced mRNAs. The TPM3 encoded proteins are the γ-subunits.
The TPM4 gene gene is located on chromosome 19p13.12-p13.11 and is composed of 13 exons that generate at least 5 alternatively spliced mRNAs. The TPM4 encoded proteins are the δ-subunits.
Given that multiple different tropomyosin mRNAs result from alternative processing of transcripts from the four TPM genes, and that each of these mRNAs encode protein, a common nomenclature has been proposed to simplify and clarify which specific tropomyosin protein is being discussed. For example, proteins encoded by transcripts from the TPM1 gene are designated TPM1.x, where the x relates to the specific transcript isoform. Associated with this nomenclature is an abbreviation for the tissue from which a given tropomyosin protein was historically identified as being most highly associated. These designations are St (striated skeletal or cardiac muscle), Sm (smooth muscle), Br (brain), and Cy (other cytoplasmic). An example of this unified nomenclature is TPM1.1st which was previously identified as α-tropomyosin and skeletal or cardiac tropomyosin. This specific tropomyosin protein is derived from an mRNA generated from the TPM1 gene that contains exons 1a-2b-3-4-5-6b-7-8-9a.
Troponin: a heterotrimeric complex of troponin C (TnC), troponin I (TnI), and troponin T (TnT):
The TnC protein of the complex is the Ca2+-binding subunit, with similarity to calmodulin, whose role is to effect the Ca2+-dependent regulation of muscle contraction. When TnC binds calcium, the whole troponin complex undergoes the conformational changes that move the attached tropomyosin away from the myosin binding sites on actin. This conformational change abolishes the inhibitory action of the TnI protein of the complex. In addition, the conformational change permits nearby myosin heads to interact with myosin binding sites, and contractile activity ensues. Humans express two TnC encoding genes identified as TNNC1 and TNNC2.
The TNNC1 gene is located on chromosome 3p21.1 and is composed of 6 exons that encode a 161 amino acid protein. Expression of the TNNC1 gene is associated with slow-twitch skeletal muscle and cardiac muscle.
The TNNC2 gene is located on chromosome 20q13.12 and is composed of 7 exons that encode a 160 amino acid protein. Expression of the TNNC2 gene is restricted to fast-twitch skeletal muscle fibers.
The TnI protein of the complex is an inhibitory protein that block actin and myosin interactions. The function of the TnI protein is to inhibit the ATPase activity of the actin-myosin complex of the thin filaments that control muscle fiber contraction, thereby, resulting in the relaxation of striated muscle. Humans express three different troponin I genes identified as TNNI1, TNNI2, and TNNI3.
The TNNI1 gene is located on chromosome 1q31.1 and is composed of 9 exons that encode a protein of 187 amino acids. The TNNI1 encoded protein is restricted to slow-twitch skeletal muscle fibers in the adult although the gene is also expressed in cardiac muscle early in development.
The TNNI2 gene is located on chromosome 11p15.5 and is composed of 10 exons that generate three alternatively spliced mRNAs that encode two distinct isoforms of the protein. Expression of TNNI2 is predominantly expressed in fast-twitch skeletal muscle fibers but is also expressed in mammary gland cells, corneal epithelium, and cartilage producing cells.
The TNNI3 gene is located on chromosome 19q13.42 and is composed of 8 exons that encode a 210 amino acid protein. Expression of the TNNI3 gene is restricted to cardiac muscle tissue. Mutations in the TNNI3 gene are associated with certain forms of inherited cardiomyopathy including familial hypertrophic cardiomyopathy type 7 (CMH7) and familial restrictive cardiomyopathy (RCM).
The TnT protein of the complex binds tropomyosin, thereby regulating troponin complex interaction with thin filaments. The entire troponin complex is attached to one end of each tropomyosin molecule and to actin, physically linking these later two proteins. Humans express three troponin T genes identified as TNNT1, TNNT2, and TNNT3.
The TNNT1 gene is located on chromosome 19q13.42 and is composed of 15 exons that generate four alternatively spliced mRNAs which together encode three distinct protein isoforms. Mutations in the TNNT1 gene result in an early lethality (usually before the second year of life) due to respiratory insufficiency. This disorder is commonly known as Amish nemaline myopathy, specifically called nemaline myopathy type 5. The TNNT1 gene is expressed in slow-twitch skeletal muscle fibers.
The TNNT2 gene is located on chromosome 1q32.1 and is composed of 17 exons that generate 13 alternatively spliced mRNAs that encode 8 different protein isoforms, several of which are tissue specific. The primary tissue of TNNT2 expression is cardiac muscle. Mutations in the TNNT2 gene are associated with several inherited cardiomyopathy syndromes.
The TNNT3 gene is located on chromosome 11p15.5 and is composed of 21 exons that generate 17 alternatively spliced mRNAs. Expression of the TNNT3 gene predominates in fast-twitch skeletal muscle.
Thin Filament Activation
Events on the thin filament can be summarized as follows: Prior to the appearance of free calcium in the sarcoplasm, tropomyosin covers the myosin binding sites on actin. The appearance of calcium in the sarcoplasm (cytoplasmic compartment of muscle cells) leads to calcium binding to the TnC subunit of the troponin complex. The resulting conformational changes in the troponin complex move the attached tropomyosin molecule more deeply into the helix groove of F-actin, uncovering the myosin binding sites on G-actin subunits. The exposed sites are then available to interact with myosin headpieces. Removing calcium from the sarcoplasm restores the original conformational states of the troponin complex and tropomyosin, preventing interaction between actin and myosin and leading to the relaxed state.
Excitability of Muscle Cells
Skeletal muscle cells are voluntary cells, meaning that one can make a muscle cell move in response to a voluntary command initiated within the central nervous system. Cardiac and smooth muscle cells are involuntary, meaning that one cannot command these cells to exert an activity. The motor neurons that innervate cardiac and smooth muscle cells are sympathetic and parasympathetic motor neurons of the autonomic nervous system. For more details on the various nerve cell types go to the Biochemistry of Nerve Transmission page.
The details of the biochemistry of skeletal and cardiac muscle cell excitation and contraction are covered in the Skeletal and Cardiac Muscle Excitability page.
Skeletal Muscle Derived Myokines
Like adipose tissue and the liver, skeletal muscle produces and secretes a number of proteins and peptides that exert autocrine, paracrine, and endocrine effects. These proteins are collectively referred to as myokines. Like adipokines and hepatokines, many of the regulatory proteins secreted from skeletal muscle are also secreted by other tissues such as the liver and adipose tissue. For details on the various myokines go the the Secreted Factors: Tissue “Kines” page.
Myosin and the Power Stroke of Contraction
In a rested, non-contracting muscle, myosin binding sites on actin (referred to as cross-bridge sites) are obscured and myosin exists in a high-energy conformational state (M*), poised to carry out a contractile cycle. A contractile cycle is also commonly referred to as the cross-bridge cycle. The energy of ATP hydrolysis is used to drive myosin from a low-energy conformational state (M) to the high-energy state, as depicted in the following equation:
(M-ATP) ↔ (M*-ADP-Pi)
When an action potential reaches the T-tubule system, the associated sarcoplasmic reticulum (SR) is stimulated to release stored calcium (Ca2+). The mechanisms of SR calcium release and re-uptake are described in detail in the Skeletal and Cardiac Muscle Excitability page. When cytosolic calcium increases it binds to TnC in the thin filament causing a conformational change in the troponin-tropomyosin complexes such that myosin binding sites on actin become available. Myosin in the thick filament then binds to actin forming what is referred to as an actomyosin complex.
The formation of an actomyosin complex results in the sequential dissociation of Pi and ADP with conversion of myosin to its low-energy conformational state. These events are accompanied by simultaneous translocation of the attached thin filament toward the M line of the sarcomere, a process that represents chemical energy being transferred to mechanical energy, the actual contraction process. The latter events, summarized in the following two equations, comprise the power stroke of the contractile cycle. Note that the energy of the power stroke is derived from ATP, via ATP-driven conversion of a low-energy myosin conformational state to a high-energy conformational state. A useful analogy is that ATP cocks the myosin trigger and the formation of an actomyosin complex pulls the trigger, releasing the energy stored in cocking the trigger.
(M*-ADP-Pi) + A ↔ (M*-ADP-A) + Pi
(M*-ADP-A) ↔ (M-A) + ADP
At the end of the power stroke the actomyosin complex remains intact until ATP becomes available. ATP binding to myosin is a very exergonic reaction, with the result that ATP displaces actin from the myosin head as indicated by the equation below. Thus, it is often said that ATP is required for muscle relaxation. It is important to note that in relaxed muscle, myosin is in its high-energy conformational state. Note that the final product (M-ATP) is also the first reactant shown in the first equation above, completing the reactions of the contractile cycle. The termination of the contractile cycle in a muscle fiber normally occurs due to the sarcoplasmic (cytoplasmic) Ca2+ being pumped back into the SR (details described below).
(MA) + ATP ↔ (M-ATP) + A
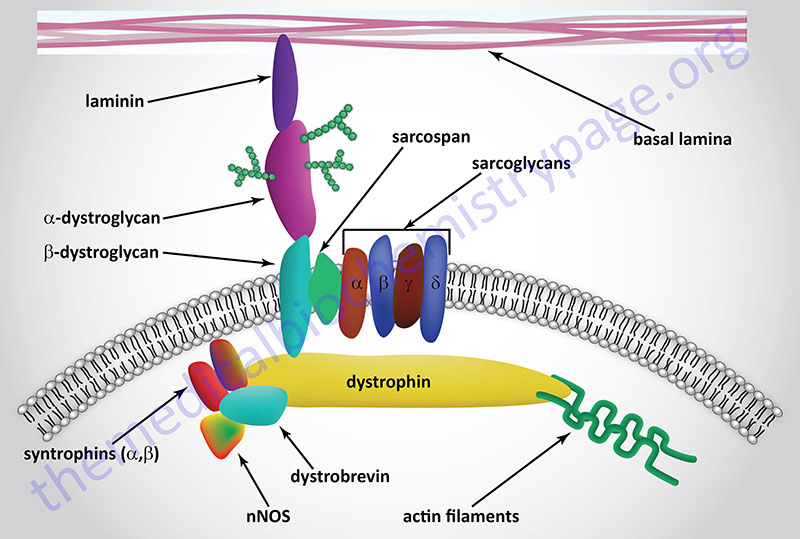
The Dystrophin Complex
The dystrophin-glycoprotein complex (DGC) is a multi-subunit complex within and across the membranes of cardiac and skeletal muscle cells as well as vascular smooth muscle cells. The complex serves both a mechanical stabilizing and a signaling role in these various cell types. The complex mediates interactions between the cytoskeleton, membrane, and the extracellular matrix. The complex is composed of up to 15 different proteins dependent upon its location. These proteins include, extracellular , transmembrane, and intracellular subunits. Within the DGC there is the protein dystrophin, two dystroglycan proteins (α-dystroglycan, α-DG and β-dystroglycan, β-DG), the syntrophins (α and β), the dystrobrevins (α and β), caveolin-3, sarcospan, and a sub-complex composed of members of the sarcoglycan family. There are six known sarcoglycans designated α-, β-, γ-, δ-, ε (epsilon)-, and ζ (zeta)-sarcoglycan. An additional component of the DGC is neuronal nitric oxide synthase (nNOS, NOS-1).
The major intracellular protein is the large dystrophin protein. The dystrophin gene (gene symbol: DMD) is located on the X chromosome (Xp21.2–p21.1) and spans 2.4 million bp (MB) and is composed of 89 exons. In addition to being a large gene composed of numerous exons, the DMD gene contains at least eight independent and tissue-specific promoter elements as well as two different poly(A) addition sites. The results of expression from this complex gene are at least 17 different mRNAs that encode six distinct classifications of dystrophin protein. These different classifications are identified as Dp470, Dp260, Dp140, Dp116, Dp71, and Dp40.
There are four Dp427 mRNAs generated from the DMD gene. The Dp427m mRNA encodes the main dystrophin protein found in muscle. The Dp427c mRNA encodes a neuronal form of dystrophin.
The two Dp260 mRNAs initiate from a promoter and an exon 1 that reside within intron 29 and utilize exons 30-79.
The five Dp140 mRNAs initiate from a promoter and an exon 1 that reside within intron 44. These mRNAs utilize exons 45-79 with alternative splicing of exons 71, 72, 73, 74, and 79 generating the five Dp140 mRNAs..
The single Dp116 mRNA initiates from a promoter and an exon 1 that reside within intron 55 and uses exons 56-79.
The four Dp71 mRNAs use exons 63-79 with a novel 80 to 100 nucleotide exon containing an ATG start site. The different Dp71 mRNAs result from alternative splicing of exons 71 and 78.
The single Dp40 mRNA uses exons 63-70.
The dystrophin proteins serves to link the extracellular portions of the DGC with the actin filaments within the cell. Dystrophin physically interacts with the cytoplasmic tail of β-DG, via a C-terminal domain in dystrophin, and γ-actin in actin filaments (F-actin) via an N-terminal domain. The clinical significance of the dystrophin protein relates to the fact that mutations in the DMD gene are the cause of various muscular dystrophies (see next section): Duchenne muscular dystrophy (DMD), Becker muscular dystrophy (BMD), and X-linked dilated cardiomyopathy (XLDCM).
The Muscular Dystrophies
The muscular dystrophies represent a group of nine characterized disorders, all of which are associated with some level of loss of muscle function along with atrophy of muscle tissue. These diseases are Duchenne muscular dystrophy (DMD), Becker muscular dystrophy (BMD), myotonic dystrophy (DM, for dystrophia myotonica), distal muscular dystrophy, Emery-Dreifuss muscular dystrophy, limb-girdle muscular dystrophy, oculopharyngeal muscular dystrophy, fascioscapulohumeral muscular dystrophy, and congenital muscular dystrophy. Myotonic dystrophy, type 1 results from a trinucleotide repeat expansion (CTG) present in the 3′-untranslated region of the dystrophia myotonica protein kinase gene (DMPK).
The muscular dystrophies that are associated with defects in the gene encoding the intracellular protein, dystrophin (gene symbol, DMD), are known as Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD). Given that the dystrophin gene is found on the X chromosome, both of these diseases are inherited in an X-linked manner. Both DMD and BMD are caused by mutations in the DMD gene and at least 65% of the mutations involve deletion of one or more of the 87 exons that comprise the DMD gene. The vast majority of the deletions found to cause DMD result in frameshift mutations. The primary clinical differences between DMD and BMD are due to the fact that the mutations in the DMD gene that cause Duchene muscular dystrophy result in virtually no function dystrophin protein being made. With Becker muscular dystrophy the mutations result in some functional dystrophin protein ranging from 10%–40% of normal.
Smooth Muscle
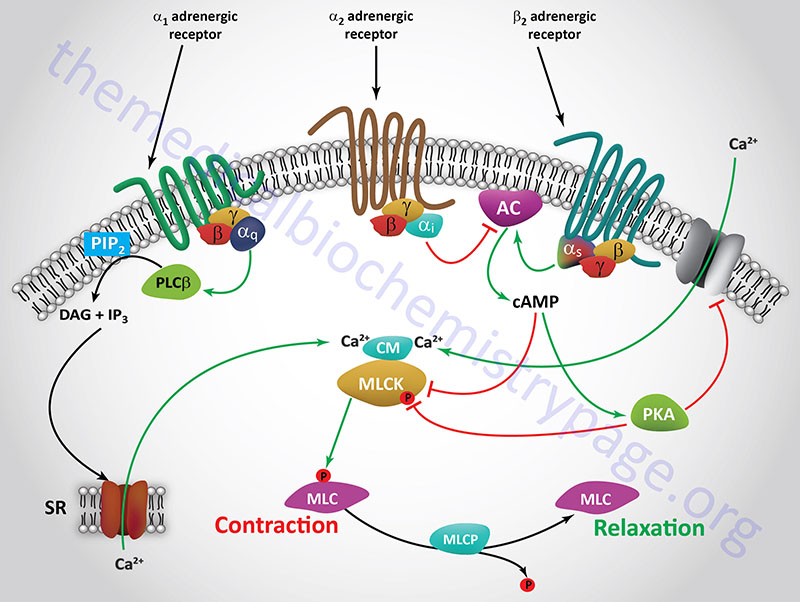
While the sliding filament model adequately describes the basic mechanism of contraction in all muscle types, there are significant differences between striated (skeletal and cardiac) and smooth muscle. An appreciation of these differences stems from the observation that although smooth muscle lacks the troponin complex, its contractile activity is still regulated by cytoplasmic calcium levels. This was explained when it was discovered that a Ca2+/calmodulin (CaCM) binding protein known as caldesmon was involved in regulating the movement of smooth muscle tropomyosin on and off the myosin binding sites of thin filaments. Alterations in smooth muscle cytosolic calcium levels occur via both voltage-dependent activation processes and by receptor-mediated processes.
The voltage-mediated processes involve the activation of plasma membrane voltage-gated calcium channels of the L-type (Cav1, similarly to the processes described above). As described above, the Cav1 channels are composed of five different protein subunits identified as the α1-, α2-, β-, γ, and δ-subunits. In vascular and visceral smooth muscle cells the predominant α1 subunit of the channel is α1C (encoded by the CACNA1C gene) forming the Cav1.2 type calcium channel. However, in some smooth muscle types the α1D isoform (encoded by the CACNA1D gene) is also expressed forming the Cav1.3 type calcium channel. Within smooth muscle the primary β-subunits of the calcium channel are the β2 and β3 isoforms.
The principal receptor-mediated activation of smooth muscle contractile activity occurs in response to α1-adrenergic receptor activation. The α1 adrenergic receptor is coupled to a Gq-type G-protein which activates PLCβ leading to production of inositol-1,4,5-trisphosphate (IP3) and diacylglycerol, DAG. The IP3 binds to receptors on the endoplasmic reticulum leading to release of stored Ca2+, the consequences of which are smooth muscle contraction. In contrast to most of the vasculature, the smooth muscle cells of the vessels in skeletal muscle tissues possess predominantly the β2 adrenergic receptor. The β2 adrenergic receptor is coupled to a Gs type G-protein, the activation of which results in increased cAMP production and activation of PKA. Both PKA and cAMP interfere with smooth muscle contraction leading, instead, to relaxation of the vessels. This allows skeletal muscle cells access to increased nutrients and oxygen in response to stress.
Both the voltage-mediated and the receptor-mediated smooth muscle activation processes lead to increased intracellular calcium levels which lead to increased levels of active CaCM which, in turn, binds caldesmon, removing it from its site on thin filaments. Calmodulin is also a component of the myosin light-chain kinases (MLCK or MYLK) and the activation of calmodulin (CM or CaM) in these enzyme complexes results in phosphorylation of myosin light-chains (MLC or MYL) on serine 19 (S19). Concurrently, tropomyosin is observed to change its location in the helical grooves of F-actin, and the ATPase activity of the actin-myosin complex (actomyosin) is stimulated. Each of these events results in contraction of smooth muscle cells.
Myosin Light Chain Kinases
There are four MLCK (MYLK) genes expressed in humans. These genes are identified as MYLK, MYLK2, MYLK3, and MYLK4. The MYLK gene is the smooth muscle kinase whose activity is described in this section although this gene is also expressed in several non-muscle cell types.
The MYLK gene is located on chromosome 3q21.1 and is composed of 39 exons that generate 7 alternatively spliced mRNAs.
The MYLK2 gene is expressed exclusively in adult skeletal muscle. The MYLK2 gene is located on chromosome 20q11.21 and is composed of 13 exons that encode a 596 amino acid protein.
The MYLK3 gene is expressed predominantly in cardiac muscle. The MYLK3 gene is located on chromosome 16q11.2 and is composed of 14 exons that generate 2 alternatively spliced mRNAs, both of which encode distinct protein isoforms.
The MYLK4 gene encodes a myosin light chain kinase family member, however, the encoded protein most likely phosphorylates non-myosin light chain proteins.
When calcium is depleted, the CaCM complex dissociates and caldesmon is released from its complex with calmodulin and re-associates with thin filaments while simultaneously, MLCK (MYLK) activity is reduced. The actomyosin complex ATPase activity is correspondingly inhibited. In essence, caldesmon in smooth muscle cells replaces the troponin complex as a calcium (CaCM)-dependent regulator of the location of the tropomyosin complex within the context of the thin filaments.
Smooth muscle relaxation is effected via activation of β2-adrenergic receptors which are coupled to Gs-type G-proteins. Activation of β2-adrenergic receptors leads to increased production of cAMP via activation of adenylate cyclase. The increased cAMP levels activate PKA which in turn phosphorylates MLCK (MYLK) resulting in inhibition of MLCK-mediated phosphorylation of myosin light chains. In addition, cAMP itself will inhibit the activity of MLCK (MYLK).
Another path to smooth muscle relaxation initiated via β2 receptor activation is the PKA-mediated phosphorylation of a membrane potassium channel (KATP) which results in depolarization of the cell and closure of the plasma membrane Ca2+ channels. Closure of the Ca2+ channel results in reduced intracellular Ca2+ and thus, reduced levels of active MLCK (MYLK). Activation of α2-adrenergic receptors can interfere with the effects of β2 receptor activation since α2-adrenergic receptors are coupled to Gi-type G-proteins that inhibit the activity of adenylate cyclase.
Myosin Light Chain Phosphatases
Following removal of the initiating stimulus for smooth muscle contraction, all of the events that participated in the activation of contraction must be reversed. With respect to the myosin proteins this entails dephosphorylation of S19 of the myosin light chains. Phosphate removal from myosin light chains is catalyzed by an enzyme called simply, myosin phosphatase. The catalytic activity of myosin phosphatase is a protein that is a member of the protein phosphatase 1 (PP1) family of phosphatases. For more details on the protein serine/threonine phosphatase (PSP) family of enzymes (of which PP1 belongs) go to the Signal Transduction Pathways: Phosphatases page.
The PP1 family of catalytic subunits (referred to as PP1C or PP1c) is composed of four proteins encoded by three distinct genes. These four subunits are identified as PP1Cα (encoded by the PPP1CA gene), PP1Cβ (encoded by the PPP1CB gene), PP1Cγ1 and PP1Cγ2 (encoded by the PP1CC gene). The PP1Cβ protein is also identified as PP1Cδ. These catalytic subunits associate with a variety of different regulatory subunits to control the substrate specificity of a given PP1C in different cell types and under different cellular conditions. The myosin phosphatase activity is associated with the PP1Cβ (PP1Cδ) catalytic subunit. Functional myosin phosphatase also includes one of several regulatory proteins identified as myosin targeting proteins (MYPT).
The MYPT proteins constitute a family of at least five proteins that have been identified as MYPT1, MYPT2, MYPT3, MBS85 (protein phosphatase 1 myosin binding subunit of 85 kDa), and TIMAP (TGF-β1-inhibited, membrane-associated protein). Each of these five targeting proteins contains a domain for binding to PP1Cβ and other protein-protein interaction domains of the ankyrin repeat type and of the leucine zipper type. The MYPT3 and TIMAP proteins are targeted to membranes by being prenylated. Each of these five myosin phosphatase regulatory proteins belong to the larger family of PP1C regulatory proteins (PPP1R) that are encoded by 181 different genes in humans. The MYPT1 protein is encoded by the protein phosphatase 1 regulatory subunit 12A (PPP1R12A) gene, the MYPT2 protein is encoded by the PPP1R12B gene, the MYPT3 proteins is encoded by the PPP1R16A gene, the MBS85 protein is encoded by the PPP1R12C gene, and the TIMAP protein is encoded by the PPP1R16B gene.
Activation of phosphate removal from myosin light chains via the action of myosin phosphatase involves regulated interactions between PP1Cβ and one of the various myosin targeting proteins. Regulation of the interaction between PP1Cβ and MYPT1 is the best understood of all the myosin targeting proteins. MYPT1 activity is regulated by its state of phosphorylation. Phosphorylation of MYPT1 on two threonine residues (T696 and T853) is effected by the monomeric G-protein (RHO) associated kinase identified as ROCK1. Both of these phosphorylation sites in MYPT1 result in increased smooth muscle contractile activity as a result of reduced myosin phosphatase activity. Phosphorylation of MYPT1 on T696 results in a direct decrease in the activity of the associated PP1Cβ subunit, whereas, phosphorylation on T853 of MYPT1 induces its dissociation from myosin light chains resulting in decreased myosin phosphatase activity at the S19 position of the myosin light chains.
Several other kinases such as zipper-interacting protein kinase (ZIPK: encoded by the death associated protein kinase 3, DAPK3, gene), integrin-linked kinase (ILK), myotonic dystrophy kinase (encoded by the DMPK gene), as well as other monomeric G-protein- (Raf-1 and p21)-activated protein kinases have been shown to phosphorylate T696 of MYPT1 resulting in reduced myosin phosphatase activity and, consequently, prolonged smooth muscle contraction.
MYPT1 is also phosphorylated by a member of the PKG family of kinases, specifically PKGIα. This phosphorylation of MYPT1 results in an increase in the level of myosin phosphatase activity and, consequently, smooth muscle relaxation. The residue phosphorylated by PKGIα is a serine at position 695 (S695) which is right next to the threonine phosphorylated by ROCK1. Phosphorylation of S695 in MYPT1, by PKGIα, indirectly results in an increase in the activity of the myosin phosphatase activity of PP1Cβ. Activation of smooth muscle PKGIα is a direct consequence of the production of nitric oxide (NO) by the overlying endothelial cells.
NO diffuses from the endothelium, and when entering the smooth muscle cell binds to the heme moiety of the soluble form of guanylate cyclase resulting in increased production of cGMP, the direct activator of PKGIα. The activation of myosin phosphatase activity by this NO-soluble guanylate cyclase cGMP-PKGIα signaling pathway is key to the role of Ca2+ in the desensitization of smooth muscle contraction. The mechanism by which PKGIα phosphorylation of MYPT1 leads to increased myosin phosphatase activity is due to the inhibition of subsequent T696 phosphorylation by ROCK1. The net effect of PKGIα phosphorylation of MYPT1 is a decrease in the inhibited state of the myosin phosphatase activity of PP1Cβ leading to smooth muscle relaxation.
Adrenergic Receptors in Muscle Functions
The catecholamines, norepinephrine and epinephrine, exert potent effects on cardiac, skeletal, and smooth muscle cells. These effects are exerted in response to binding to one or more of the different types of adrenergic receptors which are described in the following Table.
Table of Adrenergic Receptors
| Receptor Type | Gene Symbol(s) | Expression Profile | Functions / Comments |
| α1 | ADRA1A ADRA1B ADRA1D | predominates in heart, blood vessels, and kidneys, also expressed in adipose tissue | three subtypes: α1A, α1B, α1D; coupled to Gq-type G-proteins, vasoconstrictor for coronary arteries and veins, decreases GI smooth muscle cell motility, induces contraction of smooth muscle in uterus, urethral sphincter, vas deferens, and ureter, modulates glycolysis and gluconeogenesis |
| α2 | ADRA2A ADRA2B ADRA2C | central nervous system (widely distributed); vessels, adipose tissue, kidneys, and platelets | three subtypes: α2A, α2B, α2C; coupled to Gi-type G-proteins, acts within the CNS to decrease blood pressure and exert bradycardic effects, exerts a hypothermic effect, arterial and venous vasoconstriction, inhibits insulin release and stimulates glucagon secretion, modulates gluconeogenesis and glycolysis, inhibits gastric acid secretion and gastric motility, inhibits release of norepinephrine and acetylcholine, involved in thrombus stabilization by inducing platelet aggregation |
| β1 | ADRB1 | heart, kidney, skeletal muscle, lung, colon, liver, thyroid gland, adipocytes (preadipocytes only in BAT) | coupled to Gs-type G-protein, exerts inotropic (contraction strength) and chronotropic (heart rate) effects on the heart, increases fat mobilization from adipose tissue, increases renin release from kidneys, enhances sensation of hunger through release of ghrelin by the stomach |
| β2 | ADRB2 | adipose tissue but not brown adipocytes, bronchioles, skeletal muscle, smooth muscle, lung, kidney, colon, liver, thyroid gland, heart | coupled to Gs-type G-protein, bronchodilator and vasodepressor, induces relaxation of smooth muscle in bronchus, bronchioles, uterus, and detrusor muscle, inhibits release of insulin, stimulates lipolysis, glycolysis, and gluconeogenesis |
| β3 | ADRB3 | abundant in adipocytes of BAT and omental fat, gallbladder and bladder, is not expressed in the heart, skeletal muscle, liver, kidneys, lung, or thyroid gland | coupled to Gs-type G-protein, regulation of lipolysis, principal norepinephrine receptor in BAT, increase lipolysis in BAT and plays major role in adaptive thermogenesis |
When considering the effects of various adrenergic receptor agonist and antagonist effects within the vasculature it is important to understand that the contractile characteristics and the mechanisms that cause contraction of cardiac myocytes and vascular smooth muscle (VSM) are very distinct. Contraction and relaxation of smooth muscle is also discussed in the above section. The contractile properties of cardiac myocytes are fast and of extremely short duration. In contrast, VSM undergoes slow, sustained, tonic contractions. While both cardiac muscle and VSM contain actin and myosin, VSM do not express the regulatory troponin complex as does the heart. An additional difference between VSM and cardiac myocytes relates to the structural arrangement of actin and myosin. In heart muscle these proteins are organized into distinct bands, whereas, in VSM they are not. Although organized differently, the contractile proteins of VSM are indeed highly organized in order to allow for maintaining tonic contractions and reducing vascular diameter.
Contraction of VSM is initiated by several distinct phenomena including mechanical, electrical, and chemical stimuli. Mechanical contraction refers to the passive stretching of VSM from the cell itself and is therefore termed a myogenic response. Electrical stimulation of VSM contraction involves depolarization of the membrane, most often as a result of the opening of voltage gated calcium channels (L-type calcium channels) leading to increased intracellular calcium concentrations. When discussing chemical stimuli, that initiate contraction in VSM, these signals are hormones and neurotransmitters such as the adrenergic molecules, epinephrine and norepinephrine. Binding of norepinephrine or epinephrine to adrenergic receptors on the VSM cell or to receptors on the endothelial cells adjacent to the VSM results in contraction. Although each of these receptor-mediated VSM contraction processes are different, they converge at the point of increased intracellular calcium concentration.
Increases in free intracellular calcium result from either increased calcium influx into the VSM or via the release of sarcoplasmic reticulum (SR) stored calcium. Within the VSM cell, free calcium binds to the regulatory protein, calmodulin. Calcium-calmodulin (CaCM) activates myosin light chain kinase (MLCK; MYLK) which then phosphorylates myosin light chains. Phosphorylation of myosin light chains induces the formation of cross-bridges between the myosin heads and the actin filaments leading to smooth muscle contraction. Conversely, relaxation of VSM cells occurs in response to reduced levels of myosin light chain phosphorylation.
Adrenergic receptor stimulation by epinephrine or norepinephrine involves G-protein-coupled signal transduction pathways that impinge upon levels of the PKA activating molecule, cAMP. Since α1 receptors are coupled to the activation of Gq type G-proteins there is a resultant increase in release of intracellular calcium via the action of the second messenger IP3 binding to SR membrane receptors. The consequences of the released calcium are, therefore, VSM contraction. Norepinephrine is the major activator of α1 receptors. Norepinephrine also activates α2 receptors which are Gi coupled receptors. The resultant inhibition of cAMP production due to the inhibition of adenylate cyclase also leads to increased MLCK (MYLK) activity. The effects, therefore, of norepinephrine at α1 and α2 receptors are the same but elicited via different signaling pathways.
On the other hand, epinephrine activates β2 receptors which are coupled to Gs proteins which activate adenylate cyclase resulting in increased cAMP concentrations and a consequent increase in the activity of the kinase, PKA. Although it might seem counterintuitive for this pathway to be activated under conditions where VSM relaxation is being triggered, the increased cAMP levels induced by VSM β2 receptor activation result in inhibition of MLCK (MYLK), thereby reducing myosin light chain phosphorylation. In addition, activated PKA phosphorylates a plasma membrane localized inwardly rectifying potassium channel (Kir) in VSM resulting in hyperpolarization of the cell preventing the Ca2+ influx that is required for contraction. The net effect of both of these β2 receptor-mediated events is VSM relaxation.
Activation of the β1 receptor in the heart results in an increase in both the inotropic (heart rate) and the chronotropic (strength of contraction) activity of the heart muscle. Pharmacologic antagonism of the β1 receptor in the heart (go to the Biochemistry of Nerve Transmission page for details of the pharmacology), therefore, results in decreasing heart rate and contractility. The overall effect is a decrease in blood pressure. This is the basis for the use of beta blocker drugs in the treatment of hypertension.
The β2 receptors are prevalent in the smooth muscle cells of the bronchioles of the lungs and arteries of skeletal muscle. Activation of the β2 receptors in bronchioles causes them to dilate which allows more oxygenated air to enter the lungs. Simultaneously, activation of β2 receptors in the arteries of skeletal muscle causes them to dilate to allow increased blood flow into this tissue. Both of these receptor-mediated activities allow for an enhanced response to stress such as is typical of the fight-or-flight response.
It is important to note that norepinephrine also binds weakly to β2 receptors which results in vasodilation as for the case of epinephrine. This phenomenon is most noticeable pharmacologically when α1 blockers are utilized. Under normal physiological conditions this vasodilator effect of norepinephrine is overwhelmed by α1 receptor-mediated vasoconstriction. Equally important is the fact that, although epinephrine binds with highest affinity to β2 receptors on VSM to induce vasodilation, at high concentrations it is able to bind to α1 and α2 receptors which can override β2 receptor effects leading to vasoconstriction.
Acetylcholine and Receptors in Muscle Functions
A different type of nerve transmission occurs when an axon terminates on a skeletal muscle fiber, at a specialized structure called the neuromuscular junction. An action potential occurring at this site is known as neuromuscular transmission. At a neuromuscular junction, the axon subdivides into numerous terminal boutons that reside within depressions formed in the skeletal muscle plasma membrane. At these locations the skeletal muscle membrane is thickened and is referred to as the motor end plate. The space between the terminal boutons and the motor end plate is similar to the synaptic cleft that exists where the pre-synaptic and post-synaptic membranes of neurons are in close proximity. The particular transmitter in use at the neuromuscular junction is acetylcholine, ACh. Acetylcholine is a simple molecule synthesized from choline and acetyl-CoA through the action of choline acetyltransferase. Neurons that synthesize and release ACh are termed cholinergic neurons.
Two main classes of ACh receptors have been identified on the basis of their responsiveness to the toadstool alkaloid muscarine and to nicotine, respectively. The muscarinic receptors (mAChR) and the nicotinic receptors (nAChR). Details of the ACh receptors can be found in the Biochemistry of Nerve Transmission page. The muscarinic receptors are G-protein coupled receptors (GPCR) and are also referred to as metabotropic receptors. The nicotinic receptors are ligand-gated ion channels which are also referred to as ionotropic receptors. Both receptor classes are abundant in the human brain. The are five subtypes of muscarinic receptors, identified as M1–M5, that are classified based upon pharmacological activity. The M1, M3, and M5 muscarinic receptors are coupled to the Gq type G-proteins that activate PLCβ. The M2 and M4 receptors are coupled to Gi type G-proteins that inhibit adenylate cyclase. Muscarinic receptor desensitization occurs in response to phosphorylation of the receptors by kinases that are members of the G-protein coupled receptor kinase (GRK) family. For example the M2 receptor is phosphorylated by GRK2 (originally called β-adrenergic receptor kinase-1, βARK1). More information on the GRK family can be found in the Signal Transduction Pathway: G-Proteins and GPCR page.
Muscarinic receptors are found at the postganglionic parasympathetic neuroeffector junctions in vascular smooth muscle (VSM) cells, other smooth muscle cells such as in the gut and in exocrine glands, and the heart. Classical responses to activation of M2 and M3 receptors are salivation, urination, defecation, pupillary constriction, vasodilation, cardiac slowing, depressed atrioventricular (AV) nodal conduction, and bronchoconstriction. With respect to muscle and ACh actions, the M2 receptors are found on cardiac muscle cells and the M2 and/or M3 receptors are found on different types of smooth muscle cells. Activation of cardiac M2 receptors results in reduced chronotropic (rate) activity via the inhibition of electrical signaling through the sinoatrial (SA; pacemaker node) and AV nodes. Activation of vascular smooth muscle M3 receptors results in vasoconstriction.
When an action potential reaches the terminal bouton of a pre-synaptic motor neuron, a voltage-gated calcium channel is opened. The influx of Ca2+ ions stimulates the exocytosis of pre-synaptic vesicles containing ACh, releasing the neurotransmitter into the synaptic cleft. Once released, ACh binds to nicotinic ACh receptors (nAChR) on the post-synaptic membrane, which in the case of the neuromuscular junction, is the motor end plate membrane of the muscle cell. Activation of nAChR in the motor end plate results in an increase in Na+ and K+ conductance. The resulting influx of Na+ into the skeletal muscle cell produces a depolarizing potential.
As a result of this depolarization, action potentials are conducted in both directions, away from the motor end plate, along the muscle fiber. This action potential results in the initiation of muscle contraction. Once released from the motor neuron, ACh is catabolized through the action of acetylcholinesterase. The autonomic nerves (sympathetic and parasympathetic) that make contacts with cardiac and vascular smooth muscle cells are distinct from that of the neuromuscular junctions of skeletal muscle. These latter interactions are discussed in the section above.
Tetany and Rigor Mortis
Tetany, a condition of hyper-contracted muscle that sometimes follows a prolonged period of repetitive, summed muscle stimulation, is caused by the depletion of ATP and other high-energy phosphates that help maintain normal ATP levels. The latter include other nucleoside triphosphates (NTPs), creatine phosphate (CP), and ADP, as illustrated in the three equations below. The three reactions are carried out by nucleoside diphosphokinase, creatine kinase and adenylate kinase, respectively.
NTP + ADP → NDP + ATP
CP + ADP → Creatine + ATP
ADP + ADP → AMP + ATP
Since tetanic stimulation raises sarcoplasmic calcium and depletes ATP, the end result is a highly contracted muscle with calcium bound to TnC and no ATP available to re-sequester calcium into the cisternae of the SR, nor to break actomyosin cross-bridges. Under these conditions, mitochondria will preferentially pump calcium into the mitochondrial matrix, ultimately removing calcium bound to TnC, obscuring myosin binding sites on thin filaments, and, allowing the muscle to assume a flaccid state. However, the absence of ATP results in myosin remaining in its low-energy conformational state, with the result that new cycles of muscle stimulation will result in only limited ability of the muscle to generate contractile activity. Muscles in this physiological state are said to be fatigued.
In death, all reactions tend toward equilibrium. Among the first of these processes is that of ion equilibration across all compartments of the body as ion pumps loose their energy supplies. In the case of muscle, this results in cisternal and extracellular calcium leaking into the sarcoplasm, raising calcium concentrations to high levels. The calcium induces conformational changes in the troponin-tropomyosin complex, exposing myosin binding sites on thin filaments. The resulting uncontrolled contractile activity hastens the total exhaustion of ATP supplies and ends with all or nearly all myosin molecules in cross-linked actomyosin complexes. The rigid state of muscles that develops shortly after death is due to this highly cross-linked state of thin and thick filaments and is known as rigor mortis.