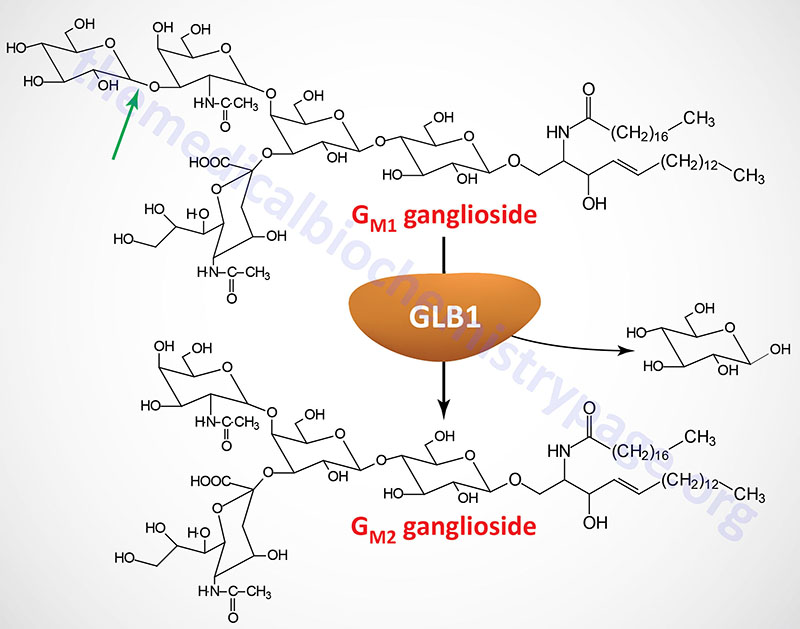
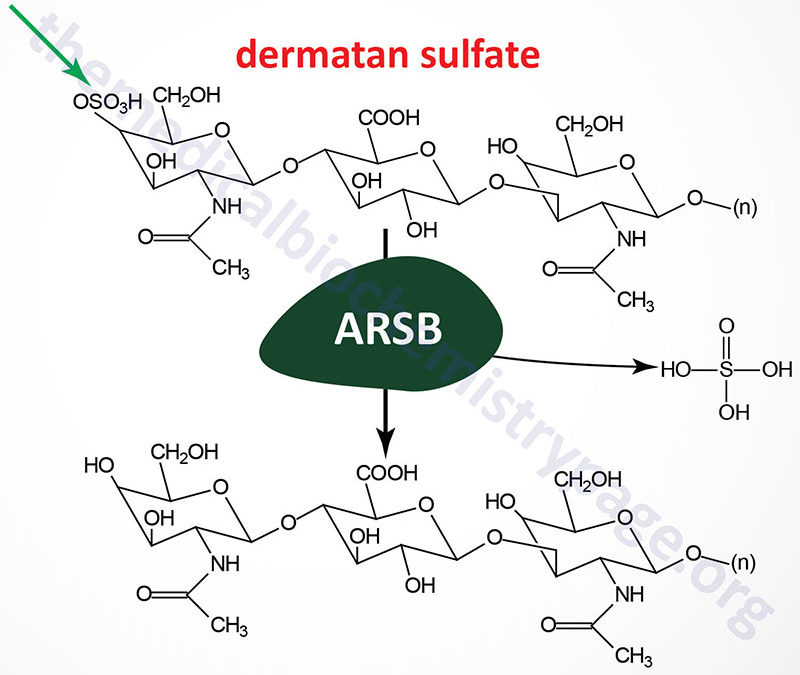
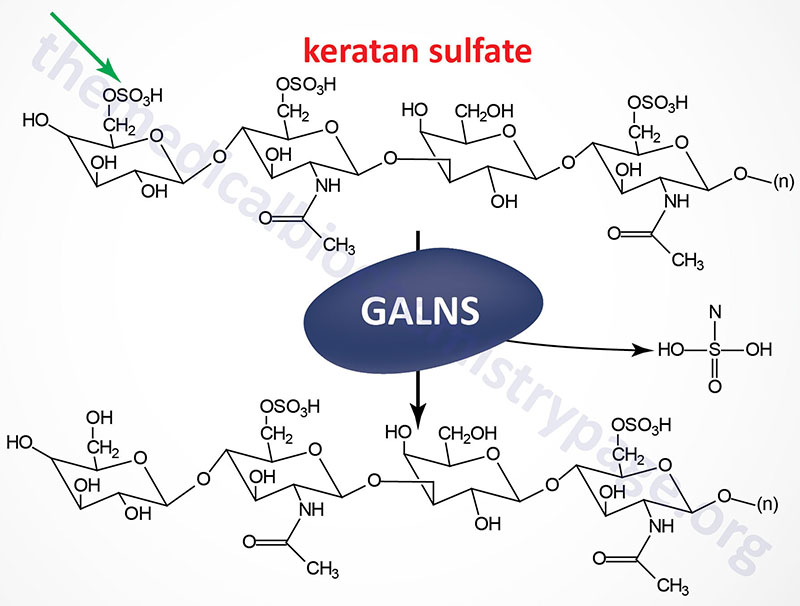
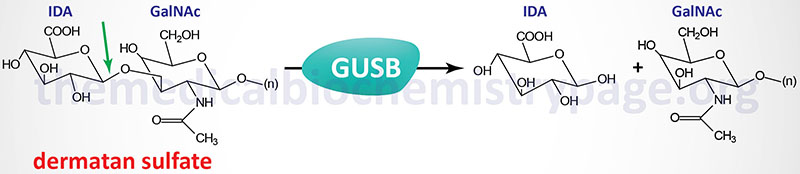
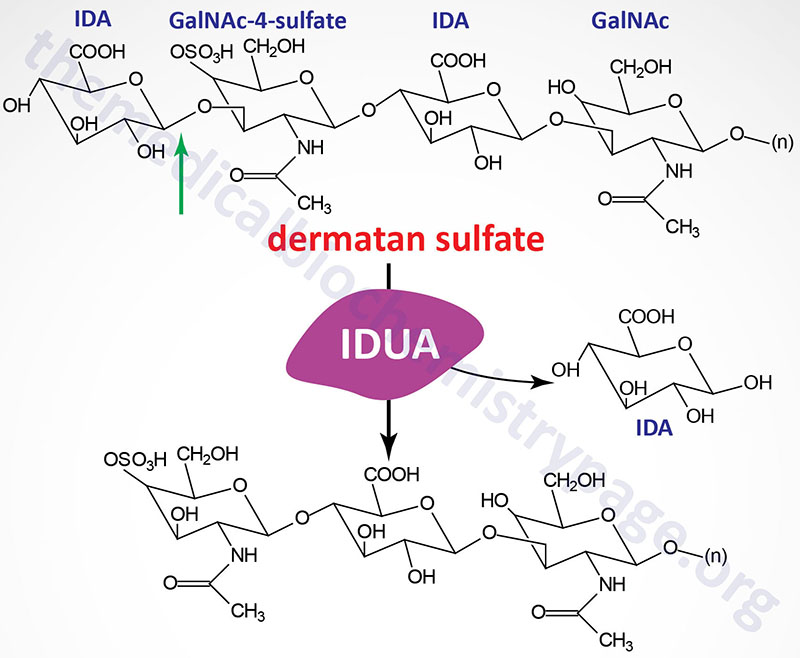
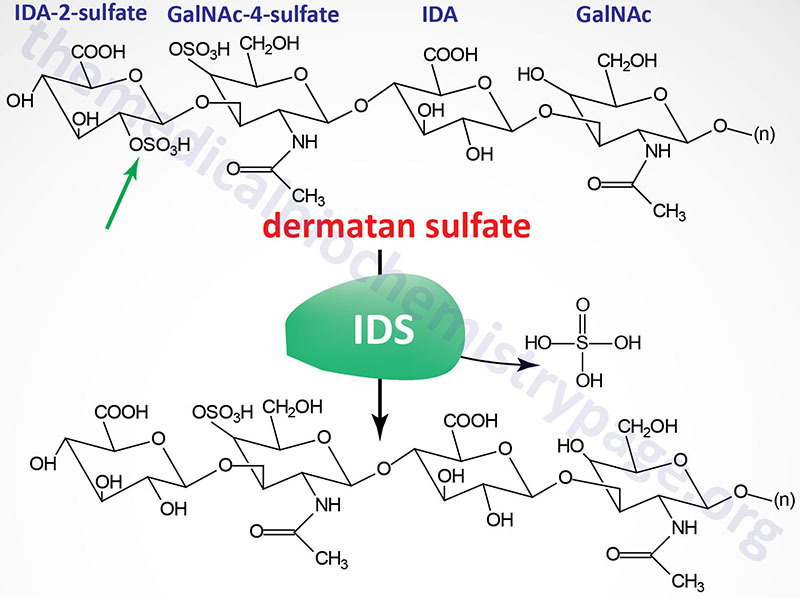
Last Updated: April 2, 2026 Introduction to the Lysosomal Storage Disorders The term "lysosomal storage disorder" (LSD; also referred to as lysosomal storage disease) encompasses a large family of inherited metabolic disorders that are associated with the pathological...