
Last Updated: July 13, 2026
Introduction to the Morquio Syndromes
The Morquio syndromes (mucopolysaccharidosis type IVA and type IVB; MPS IVA and MPS IVB) represent a group of related autosomal recessive disorders that belong to the sub family of lysosomal storage disorders that were historically referred to as the mucopolysaccharidoses. The Morquio syndromes are characterized by the lysosomal accumulation of glycoconjugates (glycolipids, glycoproteins and glycosaminoglycans) due to deficiencies in lysosomal hydrolases responsible for the degradation of of these classes of molecules.
Although Morquio syndrome is the historical name associated with these two related disorders, current nomenclature recommends the use of the mucopolysaccharidosis (MPS) designations, MPS IVA and MPS IVB.
Molecular Biology of Morquio Syndrome Type A
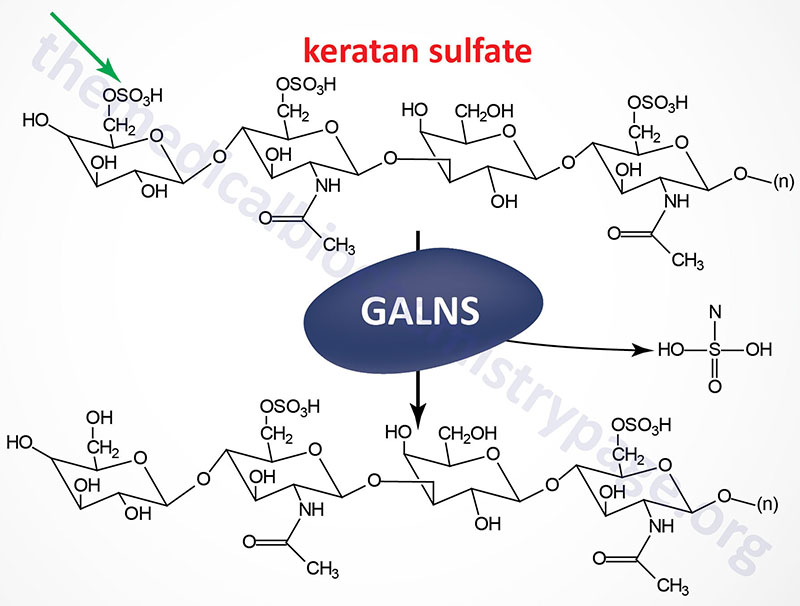
Morquio syndrome type A (MPS IVA) results from the lysosomal accumulation of glycoconjugates derived primarily from keratan sulfates and chondroitin 6-sulfates. These glycoconjugates accumulate in the lysosomes as a consequence of defects in the gene encoding N-acetylgalactosamine 6-sulfatase (also called galactose 6-sulfatase).
The GALNS [galactosamine (N-acetyl)-6-sulfatase] gene is located on chromosome 16q24.3, spanning 50 bp and comprising 16 exons that generate three alternatively spliced mRNAs, each of which encode a distinct protein isoform.
Removal of the 26 amino acid N-terminal signal peptide from the isoform 1 protein results in a mature glycoprotein of 496 amino acid residues. N-acetylgalactosamine 6-sulfatase is a member of a family of sulfatases that includes many of the enzymes involved in glycosaminoglycan degradation.
As of 2025 there have been a total of 168 pathogenic variants identified in the GALNS gene that are associated with Morquio syndrome type A (MPS IVA).
Molecular Biology of Morquio Syndrome Type B
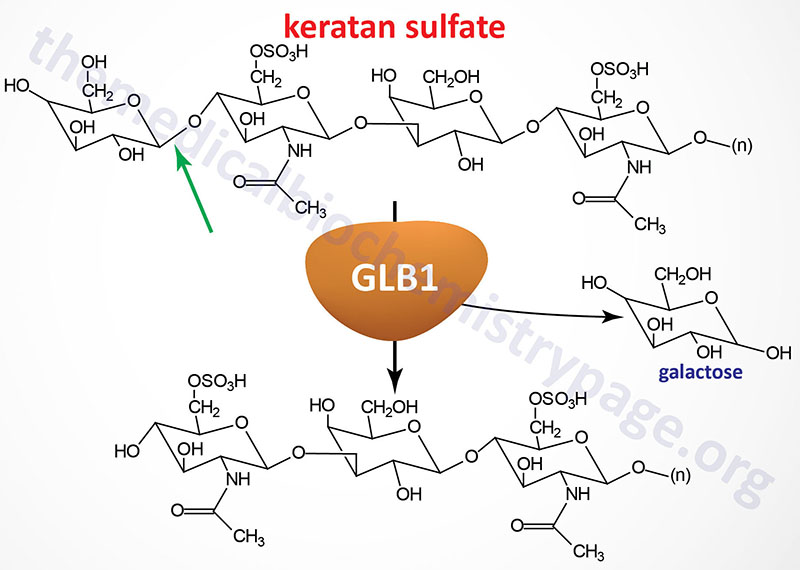
Morquio syndrome type B (MPS IVB) results from the lysosomal accumulation of glycoconjugates with terminal β-galactosyl residues. These glycoconjugates accumulate in the lysosomes as a consequence of defects in the lysosomal hydrolase, β-galactosidase-1. The β-galactosidase-1 enzyme is encoded by the GLB1 (galactosidase, beta 1) gene.
The GLB1 gene is located on chromosome 3p22.3 spanning 62.5 kb and is composed of 19 exons that generate five alternatively spliced mRNAs. The major GLB1 encoded protein is a 677 amino acid precursor protein that is processed to 654 amino acid glycoprotein that functions in a complex with PPCA (protective protein/cathepsin A) and saposin B. See the Sphingolipid Metabolism and the Ceramides page or the Gaucher Disease page for a description of the activities of the saposins. The activity of PPCA is to stabilize β-galactosidase.
The major substrates for β-galactosidase are GM1 gangliosides, GA1, lactosylceramides, asialofetuin, keratan sulfates and galactose-containing oligosaccharides. The primary syndrome resulting from defects in the GLB1 gene is GM1 gangliosidosis. The pathogenic variants in GLB1 that underlie Morquio type B cause the enzyme to lose specificity for keratan sulfates but retains the ability to degrade GM1 gangliosides. Several missense variants in GLB1 have been identified as causing Morquio type B.
As of 2025 a total of 174 pathogenic variants have been identified in the GLB1 gene. Some of these pathogenic variants result in GM1 gangliosidosis, while others are the cause of Morquio type B (MPS IVB). Some of the pathogenic variants are associated with both disorders.
In addition to the action of β-galactosidase-1, on terminal β-linked galactose residues, another β-galactosidase activity, identified as galactosylceramidase (also called galactocerebrosidase), exhibits a similar enzymatic profile but with substrate preference towards galactosylceramides, galactosylsphingosines, lactosylceramides and monogalactosyl diglyceride. Deficiencies in galactosyceramidase result in Krabbe disease (globoid cell leukodystrophy, GLD).
Clinical Features of Morquio Syndromes
The major clinical symptoms associated with the Morquio syndromes are short-trunk dwarfism, spondyloepiphyseal (skeletal) dysplasia and fine corneal implants. Unlike many mucopolysaccharidotic diseases, patients with either form of Morquio syndrome retain normal intelligence.
Clinical Features of Morquio Syndrome Type A (MPS IVA)
The clinical spectrum of Morquio type A is broad but all patients exhibit skeletal and central nervous system deficits. Most patients are normal at birth and then begin to display growth retardation with a short trunk and neck. A waddling gait with a tendency to fall are characteristically seen in these patients.
The typical skeletal abnormalities seen in Morquio type A patients include the short-trunk dwarfism indicated earlier, platyspondylia (flatness of the bodies of the vertebrae), hyperlordosis (exaggerated lumbar curvature of the spine), scoliosis, ovoid deformities of the vertebrae, short phalanges, metacarpal deformities and odontoid hypoplasia (decreased ossification of the odontoid bone which is the anterior process of the second vertebra). This latter symptom results in the most severe consequences for patients with Morquio type A. The instability of the odontoid process can result in life-threatening atlantoaxial subluxation which is a misalignment between the 1st and 2nd cervical vertebrae. In the severe forms of Morquio type A patients have a shortened life span, usually not surviving beyond the third decade.
Treatment of Morquio Type A
Currently there is an FDA approved enzyme replacement therapy for the treatment of Morquio syndrome type A in young children. The drug is called elosulfase alpha (Vimizim) which was developed by BioMarin Pharmaceuticals and approved for use in 2014.
Clinical Features of Morquio Syndrome Type B (MPS IVB)
Morquio syndrome type B was originally identified as a milder form of the type A disease but subsequently shown to be the result of defects in a different gene than those causing the type A phenotype. Morquio type B is characterized by a generalized skeletal dysplasia resulting in short stature, pectus carinatum (protrusion of the sternum), platyspodylia, scoliosis and odontoid hypoplasia. There is no central nervous system involvement in Morquio type B disease and intelligence is normal. As indicated above, this disorder is caused by pathogenic variants in the β-galactosidase gene that are distinct from those resulting in GM1 gangliosidosis.