
Last Updated: May 18, 2026
Introduction to Structures and Functions of Biological Membranes
Biological membranes are composed of lipid, protein, and carbohydrate that exist in a fluid state. Biological membranes are the structures that define and control the composition of the space that they enclose. All membranes exist as dynamic structures whose composition changes throughout the life of a cell. In addition to the outer membrane that results in the formation of a typical cell (this membrane is often referred to as the plasma membrane), cells contain intracellular membranes that serve distinct functions in the formation of the various intracellular organelles, e.g. the nucleus, the peroxisomes, the lysosomes, the endoplasmic reticulum (ER), and the mitochondria.
The nucleus and the mitochondria represent unique membrane enclosed organelles given that both are composed of two lipid bilayers. The nuclear membrane is most often referred to as the nucleolemma and is composed of closely associated inner and outer lipid bilayers. The space between the inner and outer nuclear membranes is referred to as the perinuclear space. This space is in contact with the lumen of the endoplasmic reticulum (ER) via connections between the outer nuclear membrane and the ER membranes. The inner and outer nuclear membranes are also connected at thousands of locations via multiprotein complexes that generate pores in the nuclear membrane called nuclear pore complexes. The nuclear pores are through which RNA and proteins are transported. The mitochondrial inner and outer membranes separated by a greater distance than those of the nucleus creating the inner membrane space.
The two mitochondrial membranes are also functionally very distinct and subjected to very different transport controls. Whereas the outer mitochondrial membrane is fairly permeable to most small molecules, the inner mitochondrial membrane is essentially impermeable and transport across the membrane in either direction requires highly specific transporter proteins.
Composition and Structure of Biological Membranes
As indicated above, biological membranes are composed of lipids, proteins, and carbohydrates. The carbohydrates of membranes are attached either to lipid forming glycolipids of various classes, or to proteins forming glycoproteins. The lipid and protein compositions of membranes vary from cell type to cell type as well as within the various intracellular compartments that are defined by intracellular membranes. Protein concentrations can range from around 20% to as much as 70% of the total mass of a particular membrane.
The lipids making up components of membranes are of three major classes that includes glycerophospholipids, sphingolipids, and cholesterol. For information on the structures of these different lipid classes see the Biochemisty of Lipids page, Synthesis of Phospholipids page, Sphingolipid Metabolism and the Ceramides page and the Cholesterol: Synthesis, Metabolism, and Regulation page.
Sphingolipids and glycerophospholipids constitute the largest percentage of the lipid weight of biological membranes. The hydrocarbon tails of these two classes of lipid result in steric limitations to their packing such that they will form disk-like micelles. The structure of these micelles results from the interactions of the hydrophobic tails of the lipids and the exposure of the polar head groups to the aqueous environment. This orientation results in what is referred to as a lipid bilayer and is diagrammed in the figure below. Lipid bilayers are essentially two-dimensional fluids and the lipid components of the bilayer can diffuse laterally and in fact evidence demonstrates that this lateral diffusion occurs readily.
The lipid composition of biological membranes exhibits a distinct inner versus outer polarity. For example the cytoplasmic side of the plasma membrane (also referred to as the inner leaflet) is enriched in the aminophospholipids phosphatidylserine (PS) and phosphatidylethanolamine (PE), whereas the exoplasmic side (also referred to as the outer leaflet) is enriched phosphatidylcholine (PC) and sphingomyelin.
Lipids in the bilayer can also undergo transverse diffusion (also called a flip-flop) where the lipid diffuses from one surface to the other. However, because the flip-flop requires the polar head group to pass through the hydrocarbon core of the bilayer the process is extremely rare.
Enzymes have been identified that facilitate the flip-flop process and these enzymes (transporter proteins) are referred to as flippases, floppases, and scramblases (phospholipid scramblases, PLSCR). Although flippases and floppases belong to the same family of membrane transporters, flippases are distinguished from floppases by the direction of the catalyzed lipid movement. Flippases are ATP-dependent transporters that catalyze the movement of lipids from the exoplasmic side (outer leaflet) of the membrane to the cytoplasmic side (inner leaflet) while floppases are ATP-dependent transporters that catalyze the reverse reaction. Scramblases are ATP-independent transporters that transport negatively charged phospholipids in either direction across the membrane.
Humans express 14 flippase and floppase genes, all of which are members of the ATPase family of membrane transporters, specifically the ATP8, ATP9, ATP10, and ATP11 subfamilies. Humans express five scramblase genes identified as PLSCR1–PLSCR5.
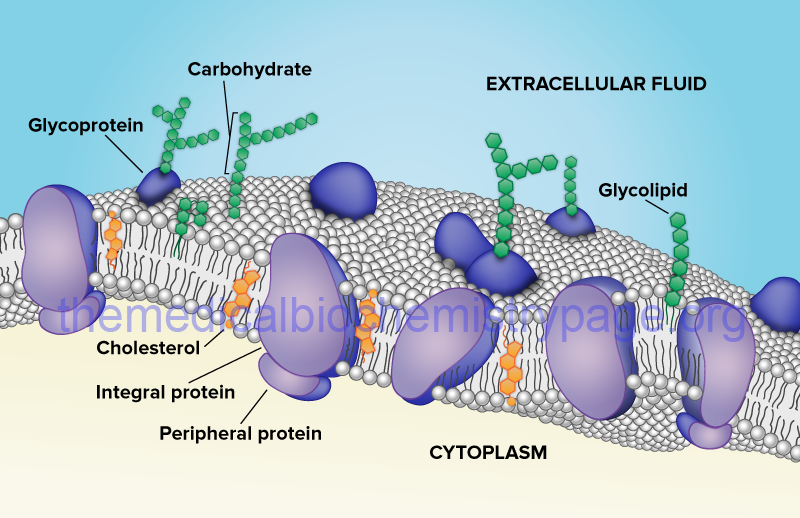
Biological membranes also contain proteins, glycoproteins, and lipoproteins (see the Glycoprotein: Synthesis and Clinical Consequences and Protein Modifications pages). Proteins associated with membranes are of two general types: integral and peripheral. Integral membrane proteins (also called intrinsic proteins) are tightly bound to the membrane through hydrophobic interactions and are inserted into and/or penetrate the lipid bilayer. In contrast, peripheral membrane proteins (also called extrinsic proteins) are only loosely associated with the membrane either through interactions with the polar head groups of the lipids or through interactions with integral membrane proteins. Peripheral membrane proteins are most often, if not exclusively, found on the cytosolic face of the plasma membrane or the luminal surface of subcellular organelle membranes.
Proteins that are found associated with membranes can also be modified by lipid attachment (lipoproteins). The lipid portion of a lipoprotein anchors the protein to the membrane either through interaction with the lipid bilayer directly or through interactions with integral membrane proteins. Lipoproteins associated with membranes contain one of three types of covalent lipid attachment. The lipids are isoprenoids such as farnesyl and geranylgeranyl residues (see the Protein Modifications page for the mechanism of protein prenylation), fatty acids such as myristic and palmitic acid, and glycosylphosphatidylinositol, GPI. Proteins modified with GPI are termed glipiated proteins.
Activities of Biological Membranes
Although biological membranes contain various types of lipids and proteins, their distribution between the two different sides of the bilayer is asymmetric. As a general example the outer surface of the bilayer is enriched in phosphatidylethanolamine, whereas the intracellular surface is enriched in phosphatidylcholine. Carbohydrates, whether attached to lipid or protein, are almost exclusively found on the external surfaces of membranes. The asymmetric distribution of lipids and proteins in membranes results in the generation of highly specialized sub-domains within membranes. In addition, there are highly specialized membrane structures such as the endoplasmic reticulum (ER), the Golgi apparatus and vesicles. The most important vesicles are those that contain secreted factors. Membrane bound proteins (e.g. growth factor receptors) are processed as they transit through the ER to the Golgi apparatus and finally to the plasma membrane. As these proteins transit to the surface of the cell they undergo a series of processing events that includes glycosylation.
The vesicles that pinch off from the Golgi apparatus are termed coated vesicles. The membranes of coated vesicles are surrounded by specialized scaffolding proteins that will interact with the extracellular environment. There are three major types of coated vesicles that are characterized by their protein coats. Clathrin-coated vesicles contain the protein clathrin and are involved in transmembrane protein, GPI-linked protein and secreted protein transit to the plasma membrane. COPI (COP: coat protein complex I) forms the surface of vesicles involved in the transfer of proteins between successive Golgi compartments and in the process of retrograde transport of proteins back to the ER. COPII forms the surface of vesicles that transfer proteins from the ER to the Golgi apparatus. Clathrin-coated vesicles are also involved in the process of endocytosis such as occurs when the LDL receptor binds plasma LDLs for uptake by the liver. The membrane location of these types of receptors is called a clathrin-coated pit.
In addition, certain cells have membrane compositions that are unique to one surface of the cell versus the other. For instance epithelial cells have a membrane surface that interacts with the luminal cavity of the organ and another that interacts with the surrounding cells. The membrane surface of cells that interacts with luminal contents is referred to as the apical surface or domain, the rest of the membrane is referred to as the basolateral surface or domain. The apical and basolateral domains do not intermix and contain different compositions of lipid and protein.
Most eukaryotic cells are in contact with their neighboring cells and these interactions are the basis of the formation of organs. Cells that are touching one another are in metabolic contact which is brought about by specialized tubular particles called junctions. Mammalian cells contain three major types of cell junctions called gap junctions, tight junctions, and adherens junctions.
Gap Junctions
Gap junctions are intercellular channels designed for intercellular communication and their presence allows whole organs to be continuous from within. One major function of gap junctions is to ensure a supply of nutrients to cells of an organ that are not in direct contact with the blood supply. Gap junctions are formed from a type of protein called a connexin (also called gap junction proteins). Connexin proteins form complexes in the membrane (called connexons) that generate the channels. Humans express a total of 22 gap junction protein genes that are divided into five families identified as GJA-GJE. A related nomenclature uses the designation CX to reflect the name connexin. For example the GJA1 (gap junction protein alpha 1) gene is also known as the CX43 gene.
Tight Junctions
Tight junctions (also referred to as occluding junctions) are primarily found in the epithelia and endothelia and are designed for occlusion. Tight junctions act as barriers that regulate the movement of solutes and water between various epithelial and endothelial layers. The tight junction serves as a barrier to the paracellular movement of ions and molecules, as well as to the movement of proteins and lipids between the apical and the basolateral domains of the plasma membrane. In addition to forming barrier complexes in the membrane, tight junction proteins also coordinate numerous other signal transduction proteins and intracellular trafficking proteins. At least 40 proteins have been found to be involved in the formation of the various tight junctions. These proteins are divided into four major categories; scaffolding, regulatory, transmembrane, and signaling. The major transmembrane proteins of tight junctions are the protein occludin (encoded by the OCLN gene), the claudin proteins (23 proteins encoded by genes designated CLDN), and the junctional adhesion molecules [JAM; these proteins belong to immunoglobulin (Ig) superfamily].
Adherens Junctions
Adherens junctions are composed of transmembrane proteins that serve to anchor cells via interactions with the extracellular matrix and intracellular proteins that interact with the actin filaments that control cell movement and shape. The major transmembrane proteins of adherens junctions are members of the cadherin family of Ca2+-dependent adhesion molecules. The intracellular proteins of an adherens junction are the α-catenins, a β-catenin family protein called junction plakoglobin (is also referred to as γ-catenin), and δ-catenins.
Humans express three α-catenin genes identified as CTNNA1, CTNNA2, and CTNNA3 and two δ-catenin genes identified as CTNND1 and CTNND2. Junction plakoglobin is encoded by the JUP gene.
Related to the adherens junctions are the desmosomes (junctions of epithelia and cardiac muscle) and hemidesmosomes (of keratinocytes) that are also involved in membrane anchoring functions.
Given the predominant lipid nature of biological membranes many types of molecules are restricted in their ability to diffuse across a membrane. This is especially true for charged ions, water and hydrophilic compounds. The barrier to membrane translocation is overcome by the presence of specialized channels and transporters. Although channels and transporters are required to move many types of molecules and compounds across membranes, some substances can pass through from one side of a membrane to the other through a process of diffusion. Diffusion of gases such as O2, CO2, NO, and CO occurs at a rate that is solely dependent upon concentration gradients. Lipophilic molecules will also diffuse across membranes at a rate that is directly proportional to the solubility of the compound in the membrane. Although water can diffuse across biological membranes, the physiological need for rapid equilibrium across plasma membranes has led to the evolution of a family of water transporting channels that are called aquaporins (see section below).
Transmembrane Protein Classes
There are numerous classes of protein that span the membrane of cells, be it the plasma membrane or intracellular organellar membranes. The transmembrane proteins include the various ion channels, other types of channel proteins, transporter proteins, growth factor receptors, and cell adhesion molecules. All transmembrane proteins, regardless of function, are classified dependent upon their structure.
There are four main classifications for transmembrane proteins, type I, II, III, and IV. Types I, II, and III are all characterized by passing through the membrane once, referred to as single-pass transmembrane proteins. Type IV transmembrane proteins pass through the membrane several times and, therefore, they are all referred to as multiple-pass transmembrane proteins.
Type I transmembrane proteins are anchored to the membrane via a sequence of hydrophobic amino acids referred to as the stop-transfer sequence and this class all have the C-terminus of the protein inside the cell and the N-terminus outside. A typical example of a type I transmembrane protein is the LDL receptor.
Type II transmembrane proteins are anchored to the membrane via a signal-anchor sequence and have the C-terminus outside the cell and the N-terminus inside. An example of a type II transmembrane protein is the transferrin receptor.
Type III transmembrane proteins do not have a signal sequence and the N-terminus of the protein is outside the cell. An example of a type III transmembrane protein would be any member of the cytochrome P450 family of xenobiotic metabolizing enzymes found in the liver.
Type IV transmembrane proteins are typified by the G-protein coupled receptor (GPCR) superfamily of receptor proteins that span the membrane seven times. This class of receptor is often referred to as the serpentine receptor family because of the multiple membrane spans. Another example of a type IV transmembrane protein is the α-subunit of a typical Na+/K+-ATPase (see below). Type IV transmembrane proteins are divided into type IV-A and type IV-B where the IV-A members have the N-terminus inside the cell and the C-terminus outside and the IV-B members are oriented in the opposite direction. The Na+/K+-ATPase α-subunit proteins are type IV-A multi-pass transmembrane proteins, whereas, all GPCRs are members of the type IV-B family.
Membrane Channels
The definition of a channel (or a pore) is that of a protein structure that facilitates the translocation of molecules or ions across the membrane through the creation of a central aqueous channel in the protein. This central channel facilitates diffusion in both directions dependent upon the direction of the concentration gradient. Channel proteins do not bind or sequester the molecule or ion that is moving through the channel. Specificity of channels for ions or molecules is a function of the size and charge of the substance. The flow of molecules through a channel can be regulated by various mechanisms that result in opening or closing of the passageway. More details on the numerous types of ion channels are discussed in the sections below.
Membrane channels are of three distinct types; α-channels, β-barrel channels, and pore-forming toxin channels.
α-Channels
The α-type channels are homo- or hetero-oligomeric structures that in the latter case consist of several dissimilar proteins. This class of channel protein has between 2 and 22 transmembrane α-helical domains which explains the derivation of their class. Molecules move through α-type channels down their concentration gradients and thus, require no input of metabolic energy. Some channels of this class are highly specific with respect to the molecule translocated across the membrane while others are not.
In addition, there may be differences from tissue to tissue in the channel used to transport the same molecule. As an example, there are 40 different K+-specific voltage-gated channels in humans. The transport of molecules through α-type channels occurs by several different mechanisms. These mechanisms include changes in membrane potential (termed voltage-regulated or voltage-gated), phosphorylation of the channel protein, intracellular Ca2+, G-proteins, and organic modulators.
β-Barrel Channels
The β-barrel channels (also called porins) are so named because they have a transmembrane domain that consists of β-strands forming a β-barrel structure. Porins are found in the outer membranes of mitochondria. The mitochondrial porins are voltage-gated anion channels that are involved in mitochondrial homeostasis and apoptosis.
Pore-Forming Toxin Channels
The pore-forming toxins represent the third class of membrane channels. Although this is a large class of proteins first identified in bacteria, there are a few proteins of this class expressed in mammalian cells. The defensins are a family of small cysteine-rich antibiotic proteins that are pore-forming channels found in epithelial and hematopoietic cells. The defensins are involved in host defense against microbes (hence the derivation of their name) and may be involved in endocrine regulation during infection.
Aquaporins
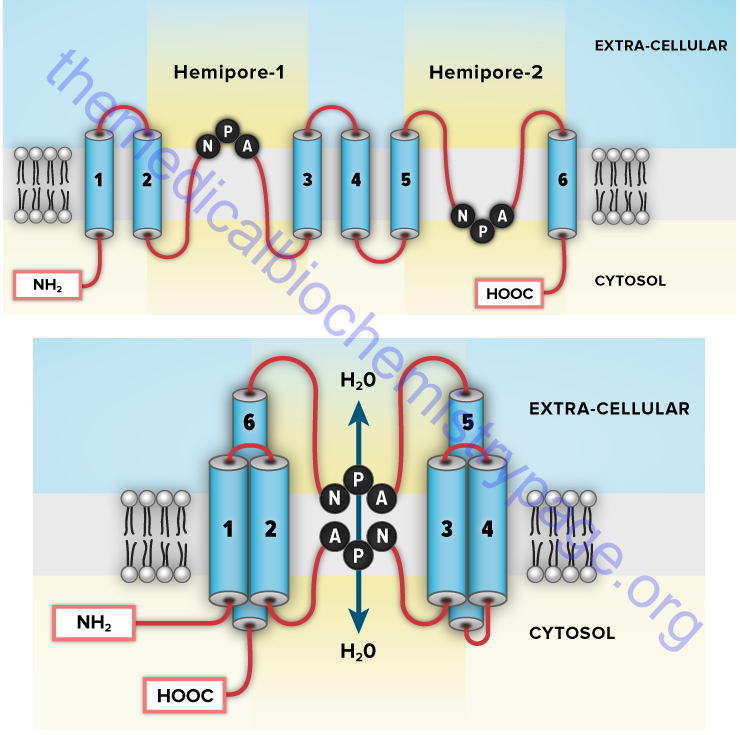
Aquaporins (AQP) are a family of α-type channels responsible for the transport of water across membranes. At least 14 aquaporin protein encoding genes have been identified in humans (termed MIP, AQP1–AQP11, AQP12A, and AQP12B). MIP is major intrinsic protein of lens fiber and has also been called aquaporin 0 (AQP0). Several AQP genes encode proteins that transport water and glycerol and sometimes other small molecules. These latter aquaporins are also referred to as the aquaglyceroporins. The AQP3, AQP7, and AQP9 genes encode the human aquaglyceroporins.
The aquaporins assemble in the membrane as homotetramers with each monomer consisting of six transmembrane α-helical domains forming the distinct water pore. Probably the most significant location of aquaporin expression is in the kidney. The proximal tubule expresses AQP1, AQP7, AQP8, and AQP11, while the collecting ducts express AQP2, AQP3, AQP4, AQP5, AQP6, and AQP8. Loss of function of the renal aquaporins is associated with several disease states. Mutations in the AQP2 gene are associated with nephrogenic diabetes insipidus (NDI), acquired hypokalemia, and hypercalcemia.
Membrane Transporters
Transporters are distinguished from channels because they catalyze (mediate) the movement of ions and molecules by physically binding to and moving the substance across the membrane. Transporter activity can be measured by the same kinetic parameters applied to the study of enzyme kinetics. Transporters exhibit specificity for the molecule being transported as well as show defined kinetics in the transport process. Transporters can also be affected by both competitive and noncompetitive inhibitors. Transporters are also known as carriers, permeases, translocators, translocases, and porters. Mediated transporters are classified based upon the stoichiometry of the transport process. Uniporters transport a single molecule at a time, symporters simultaneously transport two different molecules in the same direction, and antiporters transport two different molecules in opposite directions.
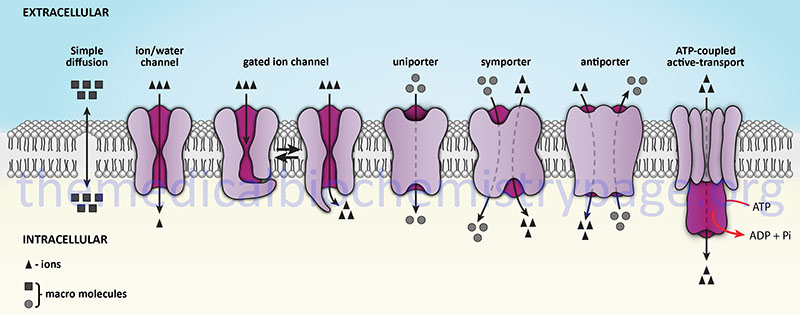
The action of transporters is divided into two classifications: passive-mediated transport (also called facilitated diffusion) and active transport. Facilitated diffusion involves the transport of specific molecules from an area of high concentration to one of low concentration which results in an equilibration of the concentration gradient. Glucose transporters are a good example of passive-mediated (facilitative diffusion) transporters. More information on the different glucose/hexose transporters can be found in the Glycolysis and the Regulation of Blood Glucose page. Another important class of passive-mediated transporters are the K+ channels (see section above). In contrast, active transporters transport specific molecules from an area of low concentration to that of high concentration. Because this process is thermodynamically unfavorable, the process must be coupled to an exergonic process, most often the hydrolysis of ATP.
The ATPase Family of Transporters
There are many different classes of membrane transporters that couple the hydrolysis of ATP to the transport of specific molecules. In general these transporters are referred to as ATPases. These ATPases are so named because the ATP hydrolysis that occurs during the transport process is coupled to the autophosphorylation of the transporter. There are four primary types of ATPase transporters that function in eukaryotes. In addition to the four classes of ATPase described in this section, another important class of transporters that function via the use of ATP hydrolytic energy is the ATP-binding cassette (ABC) transporter family. An additional family of functionally diverse ATPase-related proteins is called the AAA ATPase family. The name of this family is derived from ATPases Associated with diverse cellular Activities. The AAA family is composed of 52 functional genes whose encoded proteins all contain a conserved domain of approximately 230 amino acids. Several members of the AAA family function in the processes of DNA replication, exocytosis and endocytosis, and several are components of the 26S proteasome.
E-type ATPases
E-type ATPases are cell surface transporters that hydrolyze a range of nucleoside triphosphates that includes extracellular ATP. These transporters derive their nomenclature from the fact that they are involved in Extracellular transport. The activity of the E-type ATPases is dependent on Ca2+ or Mg2+ and it is insensitive to specific inhibitors of P-type, F-type, and V-type ATPases. The E-type ATPases can hydrolyze other NTPs besides ATP, and some can utilize NDPs. The most common substrates are ATP, ADP, and UTP. There are at least three classes of E-type ATPases.
F-type ATPases
F-type ATPases function in the translocation of H+ in the mitochondria during the process of oxidative phosphorylation. F-type transporters contain rotary motors. The nomenclature of F-type ATPases derives from phosphorylation Factor. Because these transporters transport H+ they are also referred to as H+-transporting ATPases. Additional common nomenclature for these ATPases is F0F1-ATPase. The F0 subunit is the rotary core of the ATPase that is connected to the F1 catalytic core of the mitochondrial ATP synthase complex.
P-type ATPases
P-type ATPases are mostly found in the plasma membrane and are involved in the transport of H+, K+, Na+, Ca2+, Cd2+, Cu2+, Mg2+, Co2+, Ag2+, and Zn2+. These transporters represent one of the largest families found in both prokaryotes and eukaryotes. The P-type ATPases are grouped into five classes designated P1–P5 with several classes further divided into subclasses designated A, B, C etc. For example the P2 class contains the A, B, C, and D subclasses. The P-type ATPases contain a core cytoplasmic domain structure that includes a phosphorylation domain (P domain), a nucleotide-binding domain (N domain), and an actuator domain (A domain). The P-type ATPases also possess ten transmembrane helixes termed M1–M10 where helixes M1–M6 comprise the core of the membrane transport domain. The P-type ATPases are also referred to as the E1-E2 ATPases.
V-type ATPases
V-type ATPases have homology to the F-type ATPases such as ATP synthase (complex V) of the oxidative phosphorylation process. V-type ATPases also contain rotary motors like F-type ATPases. The V nomenclature is derived from the fact that these transporters are located in Vacuoles. In addition to vacuoles, V-ATPases are located in acidic vesicles and lysosomes as well as the plasma membranes of numerous cell types such as intercalated cells of the nephrons of the kidney and macrophages and neutrophils. The V-type ATPases are involved in the processes of neurotransmitter release, protein trafficking, receptor-mediated endocytosis, and active transport of metabolites.
Similar to the F-type ATPases, the V-ATPases are multi-subunit complexes that have two main structures identified as V1 and V0. The V0 domain is composed of membrane associated subunits while the V1 domain is composed of peripherally associated subunits. The standard nomenclature has all the subunit of the V1 domain possessing upper case letters while the subunits of the V0 domain possess lower case letters.
The vacuolar V-ATPase is a proton (H+) transporter and as such is also designated as the H+-ATPase. The human V-ATPase V1 domain is composed of 13 subunits while the V0 domain is composed of 10 subunits. All of the genes encoding V1 domain subunits are designated ATP6V1 while those encoding V0 subunits are designated ATP6V0 except for the gene encoding the V0 a3 subunit which is designated TCIRG1 (T cell immune regulator 1, ATPase H+ transporting V0 subunit a3). The TCIRG1 gene was also designated ATP6V0A3.
A-type ATPases
A fifth family of ATPase transporters is the A-type family found only in prokaryotes. A-type ATPases are Archaeal bacterial transporters that function like the F-type class of ATPases.
Table of the ATPase Family of Transporters
| ATPase Family/ Type | Function | Member Names / Comments |
| 1 / P-type | Na+/K+ transporters | ATP1A1, ATP1A2, ATP1A3, ATP1A4 the ATP1A genes all encode the catalytic α-subunit of the transporter ATP1B1, ATP1B2, ATP1B3 the ATP1B genes all encode the regulatory glycoprotein β-subunit of the transporter ATP1B4: encoded protein in humans does not function as a Na+/K+-ATPase β-subunit, but instead interacts with the nuclear transcriptional co-regulator SNW domain containing 1, SNW1 (also known as SKI-interacting protein, SKIP) |
| 2 / P-type | Ca2+ transporters | ATP2A1: also known as SERCA1 (sarco/endoplasmic reticulum Ca2+-ATPase); found in cardiac muscle fast twitch fibers ATP2A2: also known as SERCA2; found in cardiac muscle slow twitch fibers ATP2A3: also known as SERCA3; ubiquitously expressed in muscle cells ATP2B1, ATP2B2, ATP2B3, and ATP2B4 are all plasma membrane-associated Ca2+ transporters ATP2B1: also identified as PCMA1b (plasma membrane Ca2+ transporter 1b) ATP2B2: also identified as PCMA2a (plasma membrane Ca2+ transporter 2a) ATP2B3: also identified as PCMA3a (plasma membrane Ca2+ transporter 3a) ATP2B4: also identified as PCMA4b (plasma membrane Ca2+ transporter 4b) ATP2C1, ATP2C2 are secretory pathway Ca2+ transporters |
| 4 / P-type | H+/K+ transporters | ATP4A: encodes the α-subunit of the stomach parietal cell H+ transporter; also identified as the α1 subunit; also expressed in the kidneys; the α2 subunit (encoded by the ATP12A gene) is termed the non-gastric (colonic) transporter which is also expressed in the kidneys ATP4B: encodes the β-subunit of the stomach parietal cell H+ transporter |
| 5 / F-type | H+ transporters, mitochondrial | ATP5A1: encodes the α-subunit of mitochondrial F1 (catalytic core) complex ATP5B: encodes the β-subunit of mitochondrial F1 complex ATP5C1: encodes the γ-subunit of mitochondrial F1 complex ATP5D: encodes the δ-subunit of mitochondrial F1 complex ATP5E: encodes the ε-subunit of mitochondrial F1 complex ATP5F1: encodes the B1-subunit of mitochondrial F0 (proton channel) complex ATP5G1: encodes the C1-subunit of mitochondrial F0 complex ATP5G2: encodes the C2-subunit of mitochondrial F0 complex ATP5G3: encodes the C3-subunit of mitochondrial F0 complex ATP5H: encodes the D-subunit of mitochondrial F0 complex ATP5I: encodes the E-subunit of mitochondrial F0 complex ATP5J: encodes the F6-subunit of mitochondrial F0 complex ATP5J2: encodes the F2-subunit of mitochondrial F0 complex ATP5L: encodes the G-subunit of mitochondrial F0 complex ATP5L2: encodes the G2-subunit of mitochondrial F0 complex ATP5O: encodes the O-subunit (OSCP: oligomycin sensitivity-conferring protein) of mitochondrial F0 complex MT-ATP6: mitochondrially encoded ATP synthase 6 MT-ATP8: mitochondrially encoded ATP synthase 8 |
| 6 / V-type | H+ transporters, lysosomal | ATP6AP1 and ATP6AP2 encode vacuolar (V-type) ATPase accessory proteins ATP6AP2 encoded protein is also known to bind the hormone renin (and prorenin) resulting in the activation of the conversion of angiotensinogen to angiotensin I ATP6V1A, ATP6V1B1, ATP6V1B2, ATP6V1C1, ATP6V1C2, ATP6V1D, ATP6V1E1, ATP6V1E2, ATP6V1F, ATP6V1G1, ATP6V1G2, ATP6V1G3, ATP6V1H all encode components of the cytosolic catalytic (V1) complex of V-type ATPases; the V1 complexes are all composed of eight different subunits, three A subunits, three B subunits, two G subunits and the C, D, E, F, and H subunits (A3B3CDEFG2H); the A3B3 hexamer is the catalytic (ATP hydrolysis) complex ATP6V0A1, ATP6V0A2, ATP6V0A4, ATP6V0B, ATP6V0C, ATP6V0D1, ATP6V0D2, ATP6V0E1, ATP6V0E2; all encode components of the transmembrane (V0) complex of V-type ATPases; the V0 complexes are composed of at least five different subunits identified as a, b, c’, c”, d, e |
| 7 / P-type | Cu2+ transporters | ATP7A: encodes the Cu2+-transporting ATPase α-polypeptide; localized to the Golgi to supply copper-dependent enzymes with copper; migrates to plasma membrane to participate in copper efflux when intracellular copper levels are elevated; defects in gene result in Menkes disease ATP7B: encodes the Cu2+-transporting ATPase β-polypeptide; localized to the Golgi to supply copper-dependent enzymes with copper; migrates to plasma membrane to participate in copper efflux when intracellular copper levels are elevated; particularly important in the export of hepatocyte copper to the bile canaliculi; defects in gene result in Wilson disease |
| 8 / P-type | phospholipid flippases | ATP8A1, ATP8A2 ATP8B1, ATP8B2, and ATP8B3: involved in phosphatidylserine (PS) and phosphatidylethanolamine (PE) transport from one side of a membrane to the other ATP8B4: putative phospholipid transporter |
| 9 / P-type | phospholipid flippases | ATP9A, ATP9B |
| 10 / P-type | phospholipid flippases | ATP10A: involved in phosphatidylserine (PS) and phosphatidylethanolamine (PE) transport; this gene is maternally expressed and is located in the region of chromosome 15 commonly deleted in Angelman syndrome ATP10B, ATP10D |
| 11 / P-type | phospholipid flippases | ATP11A, ATP11B, ATP11C |
| 12 / P-type | H+/K+ transporters, non-gastric | ATP12A: originally identified as the non-gastric (colonic) H+/K+-ATPase; encodes the catalytic subunit of ouabain-sensitive H+/K+-ATPases; also identified as the α2-subunit of H+/K+-ATPases |
| 13 / P-type | inorganic cation transporters | ATP13A1, ATP13A2, ATP13A3, ATP13A4, ATP13A5 ATP13A2 (also known as PARK9): mutations in gene associated with Kufor-Rakeb syndrome, KRS; KRS is also known as Parkinson disease 9, a juvenile-onset, levodopa-responsive form of the disease |
Na+/K+-ATPases
One of the most thoroughly studied classes of ATPases are the Na+/K+-ATPases found in plasma membranes. These transporters, sometimes called Na+/K+-pumps, are involved in the transport of Na+ out of, and K+ into, cells. The extrusion of Na+ allows cells to control their water content. In addition, the fact that three moles of Na+ are transported out and only two moles of K+ are transported into the cell, an electrochemical gradient is established. This is the basis for the electrochemical excitability of nerve cells. In fact, it is this transporter action that is the major requirement for ATP production from glucose oxidation in the central nervous system.
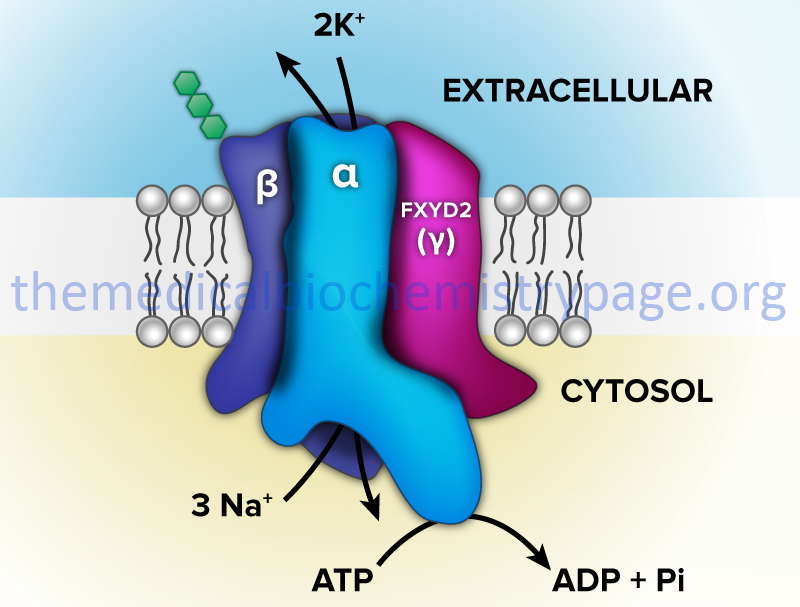
The Na+/K+-ATPases belong to the P2 class and specifically to the P2C subclass of ATPases. These ATPases are composed of two subunits (α and β). The α-subunit (≈113 kD) binds ATP and both Na+ and K+ ions and contains the phosphorylation sites typical of the P-type ATPases. The autophosphorylation site is the P domain. As discussed below, P-type ATPases are also subject to additional phosphorylation events via other kinases. The smaller β-subunit (≈35 kDa glycoprotein) is absolutely necessary for activity of the complex. It appears to be critical in facilitating the plasma membrane localization and activation of the α-subunit.
Several isoforms of both α- and β-subunits have been identified that exhibit different kinetic parameters and tissue distribution. There are four α-subunit genes (ATP1A1, ATP1A2, ATP1A3, and ATP1A4) and three β-subunit genes in humans (ATP1B1, ATP1B2, and ATP1B3). Another gene identified as ATP1B4 does not encode a β-subunit of Na+/K+-ATPases in placental mammals.
The α1 isoform (encoded by ATP1A1) is the predominant form in the kidney but is also ubiquitously expressed. The α2 isoform (encoded by ATP1A2) is primarily expressed in muscle tissues (skeletal, smooth, and cardiac) as well as in adipose tissue, brain, and lung. The α3 isoform (encoded by ATP1A3) is expressed primarily in the heart and neurons. The α4 isoform (encoded by ATP1A4) is only expressed in the testes.
The β1 isoform (encoded by ATP1B1) is ubiquitously expressed and is associated with the α1 subunit in the ubiquitously expressed α1β1 Na+/K+-ATPase complex. The β2 isoform (encoded by ATP1B2) is predominantly expressed in neurons and heart cells. The β3 isoform (encoded by ATP1B3) is expressed in testes but has also been detected in early developing neurons.
In addition to the ability to form numerous complexes through the interactions of different α- and β-subunits, the Na+/K+-ATPases also associate with a family of small single transmembrane-spanning proteins termed the FXYD (fix-id) proteins. These proteins get their name from the fact that they all share a signature 35-amino acid homology domain that contains the invariant five amino acid motif: PFXYD derived from the single letter amino acid code. Although the designation X indicates any amino acid can occupy that position, it is in fact usually tyrosine (Y), but can also be glutamate (E), threonine (T), or histidine (H).
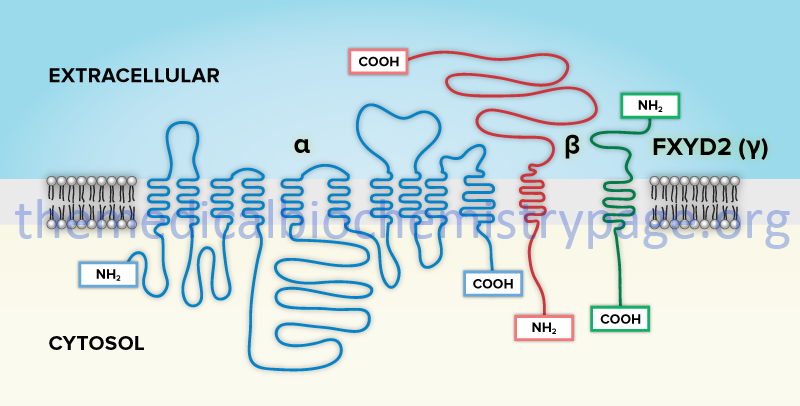
The human FXYD family of proteins is composed of at least seven members identified as FXYD1–FXYD7. Five members of this family, including FXYD1 (also known as phospholemman), FXYD2 (also known as the γ-subunit of Na+/K+-ATPase), FXYD3 (also known as MAT8), FXYD4 (also known as corticosteroid hormone-induced factor, CHIF), and FXYD7, are auxiliary subunits of Na+/K+-ATPases and they regulate Na+/K+-ATPase activity in a tissue- and isoform-specific way. FXYD5 is also known as dysadherin and FXYD6 is also known as phosphohippolin.
When Na+/K+-ATPases have ATP bound they can bind intracellular Na+ ions. The hydrolysis of the ATP results in the phosphorylation of an Asp residue in the conserved DKTGT motif present in the P domain of the α-subunit of all P-type ATPases. Autophosphorylation of P-type ATPases occurs as a function of the nucleotide binding (N domain) domain. The phosphorylation of this Asp residue results in release of ADP.
The phosphorylation of the transporter results in a conformational change which exposes the Na+ ions to the outside of the cell and they are released. The transporter then binds two extracellular K+ ions which stimulates dephosphorylation of the α-subunit that in turn allows the transporter to bind ATP again. Dephosphorylation results as a consequence of the P domain interacting with the actuator domain (A domain) which contains a catalytic Glu residue in a conserved TGE motif. The dephosphorylation and ATP binding causes the transporter to reorient to its original conformation and releases the K+ ions inside the cell. At this point the transporter is capable of the release of Na+ ions again.
In addition to the autophosphorylation site in the P domain of Na+/K+-ATPases, these transporters are subject to additional regulatory phosphorylation events catalyzed by PKA and PKC. The stimulus for PKA and PKC-mediated phosphorylation of Na+/K+-ATPases is activation of associated receptors. Adrenergic, cholinergic, and dopaminergic receptor agonists result in PKA-mediated phosphorylation of the transporter. Activation of the prostaglandin E receptors has also been shown to lead to PKA-mediated phosphorylation of neuronal Na+/K+-ATPases. The most studied site of PKA phosphorylation is a serine residue (Ser943) that is found in a domain between two transmembrane domains in the α1 isoform of the transporter.
PKA-mediated phosphorylation results in reduced activity of the transporter but also results in increased levels of the transporter in the plasma membrane. The N-terminus of the α1 isoform has been shown to be phosphorylated by PKC although no obvious conserved consensus site is present. Serine 16 is the only conserved serine residue in the N-terminus of the α1 subunit and mutation of this residue abolishes PKC-mediated phosphorylation.
As indicated above, several members of the P-type ATPases form complexes with proteins of the FXYD family. In the heart and in skeletal muscle FXYD1 has been shown to be a target for PKA and PKC phosphorylation. Whereas, PKA-mediated phosphorylation of the α1 subunit of the transporter reduces its transport activity, PKA-mediated phosphorylation of FXYD1 results in increased activity of associated Na+/K+-ATPases. This indicates that unmodified FXYD1 serves as an inhibitor of the transporter.
The Na+/K+-ATPases are also receptors for the endogenous cardiotonic steroids as well as certain toxins from plants and amphibians. Binding of these various compounds to the transporter results in activation of various kinases such as SRC and PI3K resulting in modulation of cell adhesion and growth. Endogenous cardiotonic steroids (also referred to as cardiac glycosides) are specific inhibitors of Na+/K+-ATPases and have been isolated from adrenal glands, heart tissue, the hypothalamus, and cataractous lenses.
Pregnenolone and progesterone are the precursors in the biosynthesis of endogenous ouabain (also identified as g-strophanthin) and endogenous digoxin. Ouabain and digoxin are referred to as cardenolides. Exogenous ouabain is a poisonous compound found in the ripe seeds of the African plant Strophanthus gratus and the bark of Acokanthera ouabaio. Another class of endogenous cardiotonic steroids is the bufadienolides which includes marinobufagenin, marinobufotoxin (the C3-site arginine-suberoyl ester of marinobufagenin), telocinobufagin (the reduced form of marinobufagenin), and 19-norbufalin. There are indications that many more endogenous cardiotonic steroids may exist in mammals. Unlike its role in endogenous oubain synthesis progesterone does not appear to be a precursor of marinobufagenin. However, mevastatin (a statin drug that inhibits HMG-CoA reductase), reduces the biosynthesis of marinobufagenin, indicating that cholesterol is a precursor of bufadienolides in mammals.
Ion Channels
Ion channels, including those that are ligand- or voltage-gated, are found in all cellular membranes including the plasma membrane and intracellular organelle membranes. The ion channels represent a large class of proteins, and multiprotein complexes, that form pores within membranes that allow the flow of ions across the membrane. The ion channels are broadly classified into six large families of channel proteins or channel complexes. These six families consist of the calcium channels, the chloride channels, the potassium channels, the sodium channels, the gap junction proteins, and the porins (includes the aquaporins discussed above). Within the broad context of the six large families of ion channels, there are numerous distinct subfamilies.
The calcium channel family includes the cation channels sperm associated (CatSper) subfamily, the voltage-gated calcium channels, the IP3 receptors, the ryanodine receptors, and the two pore segment channels. The chloride channel family includes the calcium-activated chloride channels, the voltage-gated chloride channels, the intracellular chloride channels, and the ATP-gated chloride channel (CFTR). The gap junction protein channel family includes the connexins and the pannexins. The potassium channel family includes the calcium-activated potassium channels, the voltage-gated potassium channels, the inwardly rectifying potassium subfamily J channels, the sodium-activated potassium subfamily T channels, and the two pore domain potassium subfamily K channels. The porin channel family includes the aquaporins and the voltage-dependent anion channels. The sodium channel family includes the acid sensing ion channel subunits, the epithelial sodium channels, the sodium leak channels, and the voltage-gated sodium channels.
Not all ion channels are gated such as is the case for certain K+ and Cl– channels, the TRP (transient receptor potential) superfamily of cation channels, the ryanodine receptors and the IP3 receptors. However, many ion channels are gated such as is the case for most Na+, K+ Ca2+ and some Cl– channels which are all gated by voltage. The non-voltage-gated ion channels can, however, be gated but the gating is controlled by second messengers and other intracellular and/or extracellular mediators. The clinical significance of ion channels is without question and currently, this large class of membrane transporter represents one of the largest targets for existing drugs, second only to G-protein coupled receptor (GPCR) targeting drugs.
In addition to the voltage-gated ion channels there is another large family of gated ion channels defined as the ligand-gated channels. These channels are regulated in response to ligand binding such as is the case for numerous neurotransmitters. These ligand gated neurotransmitter receptors are referred to as ionotropic receptors. Numerous neurotransmitters bind to ionotropic receptors such as glutamate, acetylcholine, glycine, GABA, and serotonin. More details on the actions of these neurotransmitters and their respective receptors can be found in the Biochemistry of Nerve Transmission page.
Table of Several Ion Channel Family Members
| Ion Channel Family | Family Member Genes | Comments |
| Acid-sensing (proton-gated): ASIC | ASIC1, ASIC2, ASIC3, ASIC4, ASIC5 | these genes encode the acid sensing subunits of a subfamily of sodium channels; primarily expressed in central and peripheral neurons; involved in neuronal sensitivity to acidosis; found associated with receptors for nociception (pain) and taste, with photoreceptors, and in cochlear hair cells, testis, pituitary gland, lung epithelial cells, urothelial cells, adipose cells, vascular smooth muscle cells, immune cells, and bone; activation of ASIC channels in the brain is associated with the response to neuronal injury caused by focal ischemia and to axonal degeneration in autoimmune inflammation; ASIC4 and ASIC5 are family members but may not be proton-gated as for ASIC1–ASIC3 |
| Aquaporins | AQP0, AQP1, APQ2, APQ3, APQ4, APQ5, APQ6, APQ7, APQ8, APQ9, APQ10 | represent a subfamily of the porin channel family; discussed above |
| Calcium channels | numerous genes in several subfamilies | the calcium channels represent a large diverse family of proteins involved in calcium ion transport across a membrane; there are five subfamilies with more details presented in a separate section below 1: calcium channels sperm associated: CatSper (see next row this Table) 2: voltage-gated calcium channel (Cav) subunits (details below): 26 genes comprising primary alpha1(α1)-subunits (ten genes) and auxiliary beta(β) subunits (four genes); alpha2/delta(α2δ) subunits (four genes); gamma(γ) subunits (eight genes) 3: ryanodine receptors: RYR1, RYR2, RYR3 (see below in this Table and in Calcium Channels section) 4: inositol-1,4,5-trisphosphate (IP3) receptors; three genes: ITPR1, ITPR2, ITPR3; these receptors are ligand-gated Ca2+ channels (details in Signal Transduction Pathways: Phospholipids page) 5: two pore segment channels: TPCN1, TPCN2 |
| CatSper | CATSPER1, CATSPER2, CATSPER3, CATSPER4 | represent a subfamily of voltage-gated calcium channels; required for proper movement of spermatozoa; in addition to the four primary genes there are three auxiliary subunit genes: CATSPERB (β-subunit), CATSPERD (δ-subunit), CATSPERG (γ-subunit) |
| Chloride channels | numerous genes in several subfamilies | the chloride channels represent a large diverse family of proteins involved in chloride ion transport across a membrane; there are five subfamilies 1: calcium-activated channels: the anoctamins (10 genes: ANO1–ANO10) and the bestrophins (four genes: BEST1–BEST4) 2: voltage-gated chloride channels: 9 channel coding genes (CLCN1–CLCN7, CLCNKA, CLCNKB); CLCNKA and CLCNKB proteins function in association with a β-subunit called barttin encoded by the BSND gene 3: intracellular chloride channels: six channel coding genes CLIC1–CLIC6 4: chloride channel ATP-gated: more commonly known as the cystic fibrosis transmembrane conductance regulator: CFTR 5: volume-regulated anion channels (VRAC): these channels are members of the leucine-rich repeat containing 8 (LRRC8) family; five channel coding genes (LRRC8A, LRRC8B, LRRC8C, LRRC8D, and LRRC8E) |
| Connexins and Pannexins | 21 connexin genes (designated with GJ) and 3 pannexin genes | represent the gap junction ion channel family connexin family composed of alpha(α) proteins (seven genes, GJA), beta(β) proteins (seven genes, GJB), gamma(γ) proteins (three genes, GJC), delta(δ) proteins (three genes, GJD), epsilon(ε) proteins (one gene GJE1) the pannexins (PANX1, PANX2, PANX3) are related to the invertebrate gap junction proteins identified as the innexins |
| Cyclic nucleotide-gated channels | CNGA1, CNGA2, CNGA3, CNGA4, CNGB1, CNGB3, HCN1, HCN2, HCN3, HCN4 | non-selective cation channels that are responsible for signaling in the primary sensory neurons of the visual (photoreceptors) and olfactory systems (olfactory sensory neurons, OSN) as well as activation of action potentials in cardiac muscle alpha(α)-subunit genes are CNGA1–CNGA4 beta(β)-subunit genes are CNGB1 and CNGB2 HCN genes encode the hyperpolarization-activated cyclic nucleotide-gated ion channels; control the rhythmic pacemaker depolarizations in cardiac and neuronal cells HCN2 functions in Na+ influx at the initiation of action potentials in the sinoatrial (SA) node of the heart HCN4 functions in Na+ influx at the initiation of action potentials in the atrioventricular (AV) node of the heart |
| IP3 receptors | ITPR1, ITPR2, ITPR3 | specialized calcium channels that are present in the membranes of the endoplasmic reticulum, ER; activated in response to the binding of inositol-1,4,5-trisphosphate (IP3) which is released from membrane phosphatidylinositol-4,5-bisphosphate (PIP2) following GPCR-activation of phospholipase Cβ, PLCβ |
| Potassium channels | numerous genes in several subfamilies | the potassium channels represent a large family of diverse channels that are divided into five distinct functional families, the details of which are covered in a separate section below: 1: calcium-activated potassium channels (Kca): KCNMA1, KCNN1, KCNN2, KCNN3, KCNN4, KCNU1; although the KCNU1 gene is included in the Ca2+-activated K+ channel subfamily based on sequence identity it is not activated by Ca2+ but is activated by high pH 2: voltage-gated potassium channels (Kv): 40 genes comprising subfamilies A (eight genes), B (two genes), C (four genes), D (three genes), F (one gene), G (four genes), H (eight genes), Q (five genes), S (three genes), V (two genes) 3: inwardly rectifying potassium subfamily J channels (Kir): 16 genes KCNJ1–KCNJ6, KCNJ8–KCNJ16, KCNJ18 4: sodium-activated potassium subfamily T channels (KNa): KCNT1, KCNT2 5: two pore domain potassium subfamily K+ channels (also known as potassium leak channels): 15 genes KCNK1–KCNK7, KCNK9, KCNK10, KCNK12, KCNK13, KCNK15–KCNK18 Regulatory Subunits: 15 human genes: DPP6, DPP10, KCNAB1, KCNAB2, KCNAB3, KCNE1, KCNE1B, KCNE2, KCNE3, KCNE4, KCNE5, KCNIP1, KCNIP2, KCNIP3, KCNIP4 |
| Ryanodine receptors | RYR1, RYR2, RYR3 | specialized intracellular ligand-gated calcium release channels; the ligand for these receptors is Ca2+ itself; involved in the release of sarcoplasmic reticulum (SR) stored Ca2+ in response to muscle stimulation; also present in numerous other cell types and participate in neurotransmission and secretory processes RYR1: predominates in skeletal muscle RYR2: predominates in cardiac muscle RYR3: predominates in skeletal muscle |
| Sodium channels | numerous genes in several subfamilies | the sodium channels represent a large family of diverse channels that are divided into four subfamilies: 1. voltage-gated sodium channels: composed of large pore-forming α-subunit and small accessory β-subunit that modulates activity of the α-subunit; nine α-subunit genes SCN1A–SCN5A, SCN8A–SCN11A; four β-subunit genes SCNB1–SCNB4 2. acid sensing ion channel subunits; five genes ASIC1–ASCI5 (more information included above in this Table) 3. epithelial sodium channels (ENaC); four genes SCNN1A (α-subunit: ENaCα), SCNN1B (β-subunit: ENaCβ), SCNN1D (δ-subunit: ENaCδ), SCNN1G (γ-subunit: ENaCγ); these channels mediate Na+ reabsorption in the aldosterone-sensitive collecting duct of the kidney; the ENaC channels function as heterotrimers composed of either α-, β-, and γ-subunits or δ-, β-, and γ-subunits 4. sodium leak channel; one gene NALCN |
| Transient receptor potential (TRP) channels | represents a superfamily of voltage- and/or ligand-gated cation channels involved in physical and chemical sensory processing; divided into six subfamilies: TRPC, TRPV, TRPA, TRPM, TRPML, TRPP | TRPC: canonical; seven genes: TRPC1–TRPC7 (TRPC2 is a pseudogene in humans) TRPV: vanilloid; six genes: TRPV1–TRPV6 TRPA: ankyrin; one gene: TRPA1 TRPM: melastatin; eight genes: TRPM1–TRPM8 TRPML: mucolipin (genes designated MCOLN); three genes: MCOLN1–MCOLN3 TRPP: polycystin (genes designated PKD); three genes: PKD2, PKD2L1, PKD2L2 |
| Voltage-dependent anion channels | VDAC1, VDAC2, VDAC3 | represent a subfamily of porin channels; these channels are located on the outer mitochondrial membrane (OMM) and the plasma membrane; in the OMM they facilitate small molecule and ion exchange between the mitochondria and the cytosol; in the plasma membrane they are involved in cell volume regulation and apoptosis |
| Volume-regulated anion channel subunits | LRRC8A–LRRC8E | represent a subfamily of chloride ion channels; these channels are members of the leucine-rich repeat containing 8 (LRRC8) family; included in the overview of chloride channels above in this Table |
Ligand-Gated Ion Channels
The ligand-gated ion channel (LGIC) superfamily of ion channels represents a large family of channel proteins/protein complexes that mediate the regulated flow of selected ions across the plasma membrane in response to ligand-specific binding. Ion movement through the channel is a passive process driven by the energy of the electrochemical gradient for the ion. As the name implies, the LGIC are opened, or gated, by the binding of an appropriate ligand, which is, most often, a neurotransmitter. In addition to gating of the channels in response to ligand binding, there is modulation of the gating process by the binding of endogenous, or exogenous, modulators to allosteric sites.
The large LGIC superfamily of channels that are all activated (gated) by extracellular ligands can be subdivided into ten families of structurally related channels, several of which are also outlined in the section above covering ion channels. These ten families are the ionotropic 5-hydroxytryptamine (5-HT3: serotonin) receptors, nicotinic acetylcholine receptors (nAChR), the ionotropic glutamate receptors, the GABA-A receptors, the glycine receptors (GlyR), the IP3 receptors, the ATP-gated channel (CFTR), the ryanodine receptors (RYR), the purinergic (P2X) receptors, and the zinc-activated channel (ZAC).
Several ligand-gated channels have a common motif in their extracellular domains that is the result of an intrachain disulfide bond. These channels are referred to as the Cys-loop family of channels. Members of the Cys-loop family include the nAChR, the GlyR, the 5-HT3 receptors, the GABA-A receptor subunits, and the zinc-activated channel. The ionotropic glutamate receptors are the AMPA (GluA), NMDA (GluN), kainate (GluK), and delta (GluD) receptors.
Not included in the following Table (but is included in the Table of ion channels above) are the proton-gated acid sensing ion channel (ASIC) subunits.
Table of Ligand-Gated Ion Channel Family Members
| Ligand-Gated Channel/Receptor Family | Family Member Genes | Comments |
| Serotonin: 5-HT3 receptors | HTR3A, HTR3B, HTR3C, HTR3D, HTR3E | see the Biochemistry of Nerve Transmission page |
| GABA-A receptor subunit | 18 different subunit genes: six alpha(α)-subunit genes [GABRA1–GABRA6]; three beta(β)-subunit genes [GABRB1–GABRB3]; one delta(δ)-subunit gene [GABRD]; one epsilon(ε)-subunit gene [GABRE]; three gamma(γ)-subunit genes [GABRG1–GABRG3]; one pi(π)-subunit gene [GABRP]; one theta(θ)-subunit gene [GABRQ]; two rho(ρ)-subunit genes [GABRR1, GABRR2] | the minimal composition for a functional GABA-responsive receptor is the inclusion of an α- and a β-subunit; the most common GABA-A receptor in the brain is a heteropentameric structure: α2β2γ details covered in the page |
| Glycine receptors | four α-subunit genes: GLRA1, GLRA2, GLRA3, GLRA4; 1 β-subunit gene: GLRB | see the Biochemistry of Nerve Transmission page |
| Glutamate receptors: ionotropic | four receptor families: AMPA (GluA) four genes, NMDA (GluN) seven genes, Kainate (GluK) five genes, and Delta (GluD) two genes | AMPA: GRIA1, GRIA2, GRIA3, GRIA4 NMDA: GRIN1, GRIN2A, GRIN2B, GRIN2C, GRIN2D, GRIN3A, GRIN3B Kainate: GRIK1, GRIK2, GRIK3, GRIK4, GRIK5 Delta: GRID1, GRID2 details covered in the Biochemistry of Nerve Transmission page |
| Nicotinic acetylcholine receptors | 16 different subunit genes: nine alpha(α)-subunit genes [CHRNA1–CHRNA7, CHRNA9, CHRNA10]; four beta(β)-subunit genes [CHRNB1, CHRNB2, CHRNB3, CHRNB4]; one delta(δ)-subunit gene [CHRND]; one epsilon(ε)-subunit gene [CHRNE]; one gamma(γ)-subunit gene [CHRNG] | see the Biochemistry of Nerve Transmission page |
| Purinergic (P2X) receptors | P2RX1, P2RX2, P2RX3, P2RX4, P2RX5, P2RX6, P2RX7 | represent a family of ATP-gated non-selective channels; transport Na+, K+, and Ca2+ ions; form homo- and heterotrimeric channels; channels are expressed in numerous excitatory and non-excitatory cells that includes neurons, glia cells, platelets, epithelial cells, and macrophages; involved in many physiological and pathological processes, including synaptic transmission, nociception (pain perception), inflammation, cardiovascular modulation, immunomodulation, and tumorigenesis another family of purinergic receptors is the P2Y family which is composed of nucleotide-activated G-protein coupled receptors, GPCR |
| IP3 receptors | ITPR1, ITPR2, ITPR3 | see the Signal Transduction Pathways: Phospholipids page |
| Ryanodine receptors | RYR1, RYR2, RYR3 | see the Skeletal and Cardiac Muscle Excitability page |
| ATP-gated receptor (CFTR) | CFTR | expressed in cells that are responsible for the production of mucus, sweat, saliva, tears, and digestive enzymes; mutations in the gene responsible for cystic fibrosis |
| Zinc-activated channel (ZAC) | ZACN | channel is a member of the Cys-loop superfamily of ligand-gated ion channels; primarily activated by Zn2+ but may also be spontaneously activated |
Calcium Channels and Transporters
Calcium is a critical metal (as the Ca2+ ion) in numerous biochemical and physiological processes. In order for Ca2+ to exert its multiple effects, its entry into cells and storage within cells must be tightly regulated.
Voltage-Gated Calcium Channels
Much of the membrane transport of Ca2+ into and out of cells, and into and out of intracellular organelles is controlled by a large family of voltage-gated calcium channels. In addition, neuronal and striated skeletal muscle excitation involves local depolarization of the plasma membrane (termed the sarcolemma in muscle cells) and the subsequent activation of voltage-gated calcium channels allows Ca2+ ion movement across the membrane. The voltage-gated calcium channels (also called voltage-dependent calcium channels, VDCC) are divided into three distinct families, each with multiple members. These channel families are termed the Cav1.x, Cav2.x, and Cav3.x families. The Cav1.x and Cav2.x families represent the high voltage activated (HVA) channels while the Cav3.x family represents the low voltage activated (LVA) family.
Nearly all of the voltage-gated calcium channels are pentameric structures composed of five different protein subunits identified as the α1-, α2-, β-, γ, and δ-subunits. Some voltage-gated calcium channels are composed of only four subunits and lack the γ subunit. These latter four subunit channels are typical of cardiac voltage-gated calcium channels. The α1-subunit is the actual transmembrane channel through which the Ca2+ ions flow.
Humans express 10 genes encoding α1-subunit proteins. It is the distinct α1-subunits that define the various Cav1.x, Cav2.x, and Cav3.x channel types. The α2- and the δ-subunits are derived from a single preproprotein that, following cleavage, forms an extracellular disulfide bonded glycoprotein dimer designated α2/δ. There are four α2/δ preproprotein genes in humans identified as CACNA2D1, CACNA2D2, CACNA2D3, and CACNA2D4. An alternative splice variant mRNA from the CACNA2D1 gene lacks the δ-subunit portion of the preproprotein. The γ-subunit is a transmembrane glycoprotein and the β-subunit is an intracellular protein. There are eight genes encoding γ-subunit proteins which are identified as the CACNG1–CACNG8 genes. There are four different β-subunit encoding genes identified as CACNB1, CACNB2, CACNB3, and CACNB4.
Cav1 Family
There are four members of the Cav1 channel family (also referred to as L-type calcium channels) identified as Cav1.1, Cav1.2, Cav1.3, and Cav1.4. The designation of these calcium channels as L-type refers to the initial characterization of these channels being Long-lasting channels. A high degree of variability exists in the Cav1 channel family due to multiple genes encoding the various protein subunits as well as due to alternative splicing of the mRNAs derived from these genes.
Humans express four distinct α1-subunit genes that form the various Cav1 calcium channels. These four genes are identified as CACNA1S (forms the core of the Cav1.1 channel), CACNA1C (forms the core of the Cav1.2 channel), CACNA1D (forms the core of the Cav1.3 channel), and CACNA1F (forms the core of the Cav1.4 channel). One function of the CACNA1C encoded protein is in the L-type Ca2+ channels that are found in the ventricular cardiomyocytes of the heart. This ventricular cell channel contributes to the typical plateau phase (phase 2) of ventricular action potentials. One function of the CACNA1D encoded protein is in the L-type Ca2+ channels that are found in the sinoatrial (SA) node cells of the heart.
Alterations in smooth muscle cytosolic calcium levels also occur via voltage-dependent activation processes and also by receptor-mediated processes. The voltage-mediated processes involve the activation of plasma membrane Cav1.x channels as in the case of striated muscle cells. In vascular and visceral smooth muscle cells the predominant α1 subunit of the channel is α1C (encoded by the CACNA1C gene) forming the Cav1.2 type calcium channel. However, in some smooth muscle types the α1D isoform (encoded by the CACNA1D gene) is also expressed forming the Cav1.3 type calcium channel.
Cav2 Family
All of the Cav2.x family member proteins are found within the central nervous system (CNS). There are three Cav2.x family member channels identified as Cav2.1 (also called the P/Q-type channel), Cav2.2 (N-type channel), and Cav2.3 (R-type channel). The designation of P/Q-type calcium channels refers to the initial characterization of these channels in Purkinje cells. The Q of the P/Q-type channels refers to an ion current similar to that identified in the P channels but that is pharmacologically distinct channel originally found in cerebellar granule neurons. The N-type calcium channels are so-called since they are enriched in Neuronal cells. The designation of R-type channels refers to the fact that the current through these channels was Resistant to blockade by pharmacological agents known to inhibit the L-type, N-type, and P/Q-type Ca2+ channels.
The three α1-subunit genes that encode the three unique proteins of the Cav2.x channels are identified as CACNA1A (forms the core of the Cav2.1 channel), CACNA1B (forms the core of the Cav2.2 channel), and CACNA1E (forms the core of the Cav2.3 channel). All of the Cav2 type Ca2+ channels are responsible for the initiation of synaptic transmission at fast synapses in the nervous system.
Cav3 Family
There are three members of the Cav3.x family (Cav3.1, Cav3.2, and Cav3.3) which are also referred to as the T-type calcium channels. The designation of these calcium channels as T-type refers to the initial characterization of these channels being Transient opening channels. The three α1-subunit genes that encode the three unique proteins of the Cav3.x channels are identified as CACNA1G (forms the core of the Cav3.1 channel), CACNA1H (forms the core of the Cav3.2 channel), and CACNA1I (forms the core of the Cav3.3 channel). The Cav3.x channels are expressed in the brain, kidney, and heart. These calcium channels are involved in many important physiological functions including neuronal firing, smooth muscle contraction, hormone secretion, and cardiac muscle cell activity. The Cav3 channels (specifically Cav3.1) in the heart are abundant in sinoatrial (SA) node cells and Purkinje fibers. The neuronal Cav3 channels generate low-threshold action potentials that lead to nerve transmission oscillations that are prominent in the thalamus.
Calcium Release Channels
Within striated muscle cells, Ca2+ entry across the sarcolemma causes the initiating neural stimulus to spread to the associated T-tubule (transverse tubule) system and deep into the interior of the myofiber. T-tubule depolarization spreads to the sarcoplasmic reticulum (SR: muscle form of the endoplasmic reticulum, ER), with the effect of the opening of calcium release channels in the SR membranes. These calcium release channels belong to the family of ligand-gated ion channels where the ligand is Ca2+.
The SR calcium release channels are also known as the ryanodine receptor (RYR) due to the fact that they were originally identified by their high affinity for the plant alkaloid ryanodine. There are three RYR genes in humans with the RYR1 gene being the predominantly expressed member in skeletal muscle. The RYR2 gene is highly expressed in cardiac myocytes. The activation of the SR calcium release channel leads to a massive, rapid movement of calcium from the lumen (cisternal compartment) of the SR into the sarcoplasm (muscle cell cytoplasm) close to nearby myofilaments. The appearance of calcium very close to the TnC subunit of the troponin complex results in the production of multiple myosin power strokes, as long as the available calcium concentration remains greater than about 1μM–5μM.
Calcium Reuptake Transporters
Calcium is also critical in the cessation of contractile activity and the accompanying state of relaxation in skeletal and cardiac muscle. When the initiating signal for muscle contraction is removed, the myocytes need to reverse the localization of the activating calcium from the sarcoplasm back into the SR. Sarcoplasmic calcium is pumped back into the lumen of the SR by an extremely active ATP–driven calcium pump, which comprises one of the main proteins of the SR membrane. This SR calcium pump is a member of a family of Ca2+-ATPases identified as sarco/endoplasmic reticulum Ca2+–ATPases (SERCA) as outlined in the section covering ATPases above. The human SERCA proteins are encoded by a family of three genes identified as the ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting (ATP2A) genes. These three genes are identified as ATP2A1 (SERCA1), ATP2A2 (SERCA2), and ATP2A3 (SERCA3). For each ATP hydrolyzed by the SR Ca2+-ATPase, two calcium ions are moved out of the sarcoplasm (cytoplasm) into the SR lumen.
Potassium Channels
Potassium (K+) is a key circulating electrolyte as well as being involved in the regulation of ATP-dependent channels along with sodium (Na+) ion. These channels are referred to as Na+/K+-ATPases and their primary function is in the regulation of electrochemical gradients between the inside of cells and the interstitial spaces particularly in the brain and the kidney tubule. Numerous other forms of potassium channels utilize this ion to regulate action potential propagation in the context of the transmission of nerve impulses in the brain and in the control of cardiac muscle and skeletal muscle activity. Potassium ions represent approximately 5% of the total electrolyte pool in the human body. The majority of potassium ion in the body is found intracellularly. The average intracellular potassium concentration in around 150mM, whereas the concentration of potassium in the blood is only around 3.5mM–5mM.
The potassium channel proteins have multiple domains that are critical for their highly selective transport of K+ ions (this is referred to as the P region), passage of the channel proteins across the membrane (transmembrane domains, TM), and interaction sites for regulatory molecules. Based upon structural features the potassium channels can be divided into four distinct classes. These four classes include those channel proteins that possess two TM domains and one P region, those that possess four TM domains and two P regions, those that contain six TM domains and one P region, and those that contain seven TM domains and one P region.
The potassium channels that possess two TM domains and a single P region include the inwardly rectifying channels that are designated Kir and are encoded by the KCNJ family genes. The ATP-sensitive potassium channels that are also encoded by KCNJ family genes.
Within the inwardly rectifying potassium channel family there are four channels whose activities are regulated by the βγ-subunits of heterotrimeric G-proteins and as such are designated G protein-coupled inwardly-rectifying potassium channels (GIRK).
The potassium channels that possess four TM domains and two P regions are the two pore potassium channels that are encoded by genes of the KCNK family.
The potassium channels that possess six TM domains and a single P region include the voltage-gated potassium channels that are designated Kv and encoded by nine different gene families.
The small/intermediate conductance calcium-activated potassium channels are designated KCa and encoded by members of the KCNN gene family.
The potassium channel that possesses seven TM domains and a single P region is the large conductance calcium- and voltage-activated potassium channel (designated KCa1.1 and encoded by the KCNMA1 gene). The KCNMA1 encoded potassium channel is also known as MaxiK or BK (big potassium).
All potassium channels, except the two pore subfamily, form homotetrameric or heterotetrameric complexes in the membrane. The two pore subfamily form homodimers.
In addition to the channel proteins themselves there are 15 genes that encode regulatory subunits. The regulatory subunits are divided into those that are cytosolic and those that are transmembrane. The cytosolic regulatory proteins include the Kvβ proteins (encoded by the KCNAB1–KCNAB3 genes) and the K+ channel-interacting proteins (designated KChIP and encoded by the KCNIP genes: four genes in family).
The transmembrane potassium channel regulators are encoded by the KCNE gene family (six genes). Two additional regulatory subunits have been identified that are members of the dipeptidylpeptidase (DPP) family, the DPP6 and DPP10 encoded proteins. Although these proteins have sequence identity to other DPP family members they themselves do not possess any enzymatic activity.
Voltage-Gated Potassium Channels
The first voltage-gated potassium channel gene to be identified was isolated from the fruit fly (Drosophila melanogaster) mutant called shaker, Sh. Subsequently three related genes were isolated and identified as shab, shaw, and shal. Numerous human homologs of each of the fly genes have been identified. The mammalian voltage-gated potassium channel proteins are designated Kv or Kv. The actual conductance pore of the Kv channels are formed from α-subunits (often designated as Kvα).
There have been 40 voltage-gated potassium channel Kvα genes identified in humans and all are designated with KCN as the first three letters of the gene designation. These 40 genes are divided into 10 subfamilies designated KCNA (8 genes), KCNB (2 genes), KCNC (4 genes), KCND (3 genes), KCNF (1 gene), KCNG (4 genes), KCNH (8 genes), KCNQ (5 genes), KCNS (3 genes), and KCNV (2 genes). Members of the KCNA subfamily are related to the fly shaker gene with KCNA3 being the closest relative. Members of the KCNB subfamily are related to the fly shab gene. Members of the KCNC subfamily are related to the fly shaw gene. Members of the KCND subfamily are related to the fly shal gene. The KCNG, KCNS, and KCNV genes do not encode proteins that form functional potassium channels but instead act as modifiers when in heteromeric complexes with other Kvα subunit proteins.
The voltage-gated potassium channels are classified by various functional characteristics that include rapidly inactivating, slowly inactivating, slowly activating, outward rectifying, inward rectifying (distinct from the large family of inwardly rectifying potassium channels that are not voltage-gated), and modifying/silencing. The rapidly inactivating voltage-gated K+ channels are referred to as A-type channels since their K+ currents resemble the fast acting voltage-gated Na+ channel currents. The slow inactivating voltage-gated K+ channels are delayed rectifying channels that allow sustained K+ efflux from a cell following a delay after initial membrane depolarization. Delayed rectifier potassium channels are involved in the process of membrane repolarization in excitable cells. The voltage-gated inwardly rectifying K+ channels are related to the Drosophila melanogaster ether-a-go-go gene and as such are also associated with the designations hERG (KCNH2), hERG2 (KCNH6), and hERG3 (KCNH7). The ether-a-go-go gene was identified in flies whose legs would start to shake when they were anesthetized with ether.
Table of the Voltage-Gated Potassium Channels
| Gene Symbol | Channel Designation | Functional Class | Functions/Comments |
| KCNA1 | Kv1.1 | delayed rectifier | forms functional homotetrameric and heterotetrameric channels, heteromeric channels formed with KCNA2, KCNA4, KCNA5, KCNA6, KCNA7; functions of KCNA1 containing channels are modulated by cytoplasmic β-subunits; primary site of localization is the endoplasmic reticulum (ER); expression is near exclusive to the brain, very low levels seen in digestive system, kidney, and thyroid; clustered on chromosome 12 with KCNA5 and KCNA6 genes |
| KCNA2 | Kv1.2 | delayed rectifier | forms homotetrameric and heterotetrameric channels; heteromeric channels formed with KCNA1, KCNA4, KCNA5, KCNA6, and KCNA7; functions of KCNA2 channels are modulated by cytoplasmic β-subunits; highest levels of expression are in the brain; clustered on chromosome 1 with KCNA3 and KCNA10 genes; entire protein coding region contained in a single exon |
| KCNA3 | Kv1.3 | delayed rectifier | broadly expressed in numerous tissues, highest levels in lymph followed by gastrointestinal system; clustered on chromosome 1 with KCNA2 and KCNA10 genes; entire protein coding region contained in a single exon |
| KCNA4 | Kv1.4 | rapidly inactivating | constitutes the Ito,s potassium current that contributes to the latter part of the plateau phase (phase 2; fast repolarization phase) of ventricular cardiomyocyte action potential propagation; highest levels of expression in adrenals, brain and heart; lower levels in several other tissues |
| KCNA5 | Kv1.5 | delayed rectifier | constitutes the atrial specific delayed rectifier IKur potassium current; can form homotetrameric and heterotetrameric channels; heteromeric channels contain variable proportions of KCNA1, KCNA2, and KCNA4; function of these channels modified by association with cytoplasmic regulatory β-subunits; clustered on chromosome 12 with KCNA1 and KCNA6 genes; mutations in KCNA5 result in familial atrial fibrillation type 7 (ATFB7) |
| KCNA6 | Kv1.6 | delayed rectifier | can form homotetrameric and heterotetrameric channels; heteromeric channels contain variable proportions of KCNA1, KCNA2, and KCNA4; function of these channels modified by association with cytoplasmic regulatory β-subunits; clustered on chromosome 12 with KCNA1 and KCNA5 genes |
| KCNA7 | Kv1.7 | delayed rectifier | preferentially expressed in heart, skeletal muscle, kidney, and pancreatic islet cells |
| KCNA10 | Kv1.8 | delayed rectifier | cGMP regulated channel; clustered on chromosome 1 with KCNA2 and KCNA3 genes; |
| KCNB1 | Kv2.1 | delayed rectifier | in addition to functional homotetramers, the protein forms heterotetramers with KCNB2; channel function modulated through association with regulator subunits encoded by the KCNE1, KCNE2, and KCNE3 gene; also forms functional heterotetrameric channels with KCNF1, KCNG1, KCNG3, KCNG4, KCNH1, KCNH2, KCNS1, KCNS2, KCNS3 and KCNV1 where these proteins alone do not form functional potassium channels |
| KCNB2 | Kv2.2 | delayed rectifier | in addition to functional homotetramers the protein forms heterotetramers with KCNB1; can also form functional heterotetramers with the modifier proteins encoded by the KCNS1 and KCNS2 genes; preferentially expressed in gastrointestinal smooth muscle |
| KCNC1 | Kv3.1 | delayed rectifier | in addition to functional homotetramers the protein forms heterotetramers with KCNC2 |
| KCNC2 | Kv3.2 | delayed rectifier | in addition to functional homotetramers the protein forms heterotetramers with KCNC1; preferentially expressed in the brain |
| KCNC3 | Kv3.3 | rapidly inactivating | involved in repolarization of fast-firing brain neurons |
| KCNC4 | Kv3.4 | rapidly inactivating | predominantly expressed in neurons and skeletal muscle; involved in repolarization |
| KCND1 | Kv4.1 | rapidly inactivating | may play a role in the Ito type K+ currents in ventricular cardiomyocytes |
| KCND2 | Kv4.2 | rapidly inactivating | forms homotetrameric and heterotetrameric channels; heteromeric channels formed with KCND3; preferentially expressed in the brain, next highest levels in endometrium, ovary, testis, and gall bladder; involved in neural signals downstream of the metabotropic glutamate receptor, mGluR5 (encoded by the GRM5 gene); channel proteins interact with members of the KCNIP regulatory subunits |
| KCND3 | Kv4.3 | rapidly inactivating | forms homotetrameric and heterotetrameric channels; heteromeric channels formed with KCND2; constitutes the Ito,f potassium current that contributes to the early part of the plateau phase (phase 2; fast repolarization phase) of ventricular cardiomyocyte action potential propagation; expressed in numerous tissues |
| KCNF1 | Kv5.1 | modifier/silencer | primarily expressed in the brain; does not function as a channel without forming heteromeric complexes with other members of the voltage-gated potassium channel family |
| KCNG1 | Kv6.1 | modifier/silencer | forms functional heterotetrameric channels with KCNB1 |
| KCNG2 | Kv6.2 | modifier/silencer | primarily expressed in the heart and smooth muscle; does not function as a channel without forming heteromeric complexes with other members of the voltage-gated potassium channel family |
| KCNG3 | Kv6.3 | modifier/silencer | forms functional heterotetrameric channels with KCNB1 |
| KCNG4 | Kv6.4 | modifier/silencer | forms functional heterotetrameric channels with KCNB1; highest levels of expression in adrenals then brain |
| KCNH1 | Kv10.1 | delayed rectifier | human homolog of the Drospohila melanogaster ether-a-go-go gene (hEAG); involved in osteogenic and adipogenic differentiation; channel is inhibited by Ca2+-bound calmodulin as well as by phosphatidylinositol-4,5-bisphosphate (PIP) and phosphatidylinositol-3,5-bisphosphate; highly expressed in the brain and myoblasts |
| KCNH2 | Kv11.1 | inward rectifying | forms a heteromeric complex with the regulatory subunit encoded by the KCNE2 gene; constitutes the delayed rectifier IKr potassium current that contributes to the early part of the repolarization (phase 3) of ventricular cardiomyocyte action potential propagation; also known as the human ether-a-go-go-related gene (hERG); inactivating mutations in gene associated with long QT syndrome (LQTS) type 2 that is associated with Torsade de Pointes ventricular arrhythmia and sudden cardiac death; gain-of-function mutations associated with short QT syndrome (SQTS) variant 1 |
| KCNH3 | Kv12.2 | slow inactivating | expression near exclusive to the brain |
| KCNH4 | Kv12.3 | slow inactivating | expression near exclusive to the brain |
| KCNH5 | Kv10.2 | outward rectifying | primarily expressed in brain |
| KCNH6 | Kv11.2 | inward rectifying | also known as the human ether-a-go-go-related gene 2(hERG2); |
| KCNH7 | Kv11.3 | inward rectifying | also known as the human ether-a-go-go-related gene 3(hERG3); |
| KCNH8 | Kv12.1 | slow inactivating | predominantly expressed in the forebrain; can form heteromeric complexes with KCNH3 and KCNH4 |
| KCNQ1 | Kv7.1 | delayed rectifier | forms a heteromeric complex with the regulatory subunit encoded by the KCNE1 gene; can also form heteromeric complexes with the KCNE3 regulatory subunit; constitutes the delayed rectifier IKs potassium current that contributes to the latter part of the repolarization (phase 3) of ventricular cardiomyocyte action potential propagation; inactivating mutations in gene associated with long QT syndrome (LQTS) type 1 hence the gene is also known as KvLQT1; gain-of-function mutations associated with short QT syndrome (SQTS) variant 2; expression regulated by tissue specific imprinting |
| KCNQ2 | Kv7.2 | delayed rectifier | forms a heterotetrameric channel with KCNQ3; the KCNQ2/KCNQ3 channel is the target for retigabine (anticonvulsant); mutations in gene result in benign familial neonatal convulsions type 1 (BFNC) |
| KCNQ3 | Kv7.3 | delayed rectifier | forms a heterotetrameric channel with KCNQ3 or KCNQ5; the KCNQ2/KCNQ3 channel is the target for retigabine (anticonvulsant); mutations in gene result in benign familial neonatal convulsions type 2 (BFNC2) |
| KCNQ4 | Kv7.4 | delayed rectifier | primarily expressed in the cochlea (inner ear); mutations in gene are associated with autosomal dominant nonsyndromic hearing loss (DFNA2) |
| KCNQ5 | Kv7.5 | delayed rectifier | in addition to forming homotetrameric channels the protein forms heteromeric complexes with the KCNQ3 protein |
| KCNS1 | Kv9.1 | modifier/silencer | encoded protein can form heterotetrameric complexes with the KCNB1 or KCNB2 encoded proteins and thereby modulates the delayed rectifier activation and deactivation rates of the channel |
| KCNS2 | Kv9.2 | modifier/silencer | encoded protein can form heterotetrameric complexes with the KCNB1 or KCNB2 encoded proteins and thereby modulates the delayed rectifier activation and deactivation rates of the channel |
| KCNS3 | Kv9.3 | modifier/silencer | encoded protein can form heterotetrameric complexes with the KCNB1 encoded protein |
| KCNV1 | Kv8.1 | modifier/silencer | encoded protein can form heterotetrameric complexes with the KCNB1 or KCNB2 encoded proteins and in so doing shift the threshold for inactivation to a more negative values which in turn slows the rate of channel deactivation; KCNV1 protein can also down-regulate the channel activity of KCNB1, KCNB2, KCNC4 and KCND1; expression restricted to the brain |
| KCNV2 | Kv8.2 | modifier/silencer | encoded protein can form heterotetrameric complexes with the KCNB1 or KCNB2 encoded proteins and in so doing shift the threshold for inactivation to a more negative values which in turn slows the rate of channel deactivation; expression predominates in the testes |
Inwardly Rectifying Potassium Channels
Inwardly rectifying potassium (Kir or Kir) currents were initially identified in skeletal muscle through observations that demonstrated that K+ movement, instead of the Nernst equation prediction of outward rectification, flowed inward. These initial observations were described as “anomalous” rectifier K+ currents. The functional characteristics of these K+ channels did not follow Hodgkin-Huxley kinetics as was typified by voltage-gated K+ (Kv or Kv) channel currents. The behavior of these channels was found to depend on the electrochemical gradient for K+ across the membrane. Inwardly rectifying K+ channels have subsequently been found in a wide variety of different cell types including cardiac myocytes, endothelial cells, epithelial cells, neurons, glial cells, blood cells, and osteoclasts.
To date 16 Kir channel protein encoding genes have been characterized in humans: KCNJ1–KCNJ6, KCNJ8–KCNJ16, and KCNJ18. The Kir proteins are divided into seven subfamilies identified as Kir1.x–Kir7.x. In addition, these seven Kir subfamilies are divided into four functional groups that includes the constitutively active channels, also referred to as the classic Kir channels (Kir2.1–Kir2.4, and Kir2.6), the G-protein-gated K+ channels, (Kir3.1–Kir3.4), the ATP-sensitive channels (Kir6.1 and Kir6.2), and the K+ transport channels (Kir1.1, Kir4.1, Kir4.2, Kir5.1, and Kir7.1). The G protein-gated K+ channels, which are activated by the β/γ-subunits of Gi-type G-proteins, are also referred to as G protein-coupled inwardly-rectifying potassium channels (GIRK). The ATP-sensitive K+ (KATP) channels were originally defined as being opened by a decrease in intracellular ATP.
The protein subunits of the Kir channels allow for both homomeric and heteromeric combinations. Heteromerization generally occurs between members of the same subfamily, for example, Kir2.1 can associate with any one of other Kir2.x subfamily members and Kir3.1 can form heteromeric complexes with other members of the Kir3.x family. One exception to family specific heteromeric complexes involves Kir4.1 associating with Kir5.1. The formation of heteromeric channel protein assemblies allows for distinct properties to the particular channels, can determine their location in a cell, and can extend the functional range of Kir channels in different cell types.
Kir family channels function through mechanisms that include channel location in the cell, ion flux, and the regulatory controls exerted on pore opening. The major mechanisms that regulate Kir pore opening and K+ ion flux include phospholipids, such as phosphatidylinositol-4,5-bisphosphate (PIP2), ions such as Mg2+, polyamines, nucleotides, and a variety of intracellular proteins. The inward movement of K+ ions through Kir channels is controlled by Mg2+ ions and polyamines interacting with the lining of the channel pore.
Table of the Inwardly Rectifying Potassium Channels
| Gene Symbol | Channel Designation | Functional Class | Functions/Comments |
| KCNJ1 | Kir1.1 | K+ transport | three isoforms due to alternative splicing: Kir1.1a, Kir1.1b, and Kir1.1c; more commonly called renal outer medullary potassium (ROMK) channel; present in the thick ascending limb (TAL) of the loop of Henle and in the connecting tubules (CNT); within the CNT ROMK is responsible for efflux of K+ from renal epithelial cells to the lumen of the nephron; within the CNT aldosterone activates ROMK by inducing its phosphorylation by SGK1 (serum/glucocorticoid regulated kinase 1) which results in potassium secretion; activity of channel inhibited by PKC-mediated phosphorylation; mutations in gene are the cause of Bartter syndrome type 2 |
| KCNJ2 | Kir2.1 | constitutively active | was originally identified as the human homolog of the mouse IRK (inwardly rectifying K+) channel; constitutes the IK1 potassium current that represents phase 4 of ventricular cardiomyocyte action potential propagation; this channel responsible for the resting membrane potential of non-nodal cardiomyocytes |
| KCNJ3 | Kir3.1 | GIRK | also referred to as GIRK1; forms a heteromeric complex with the KCNJ5 encoded protein; constitutes the IKACh potassium current that represents phase 4 of nodal cardiomyocyte action potential propagation; this channel responsible for the resting membrane potential of nodal cardiomyocytes; is a target of the βγ-subunits of parasympathetic acetylcholine activated muscarinic M2 receptor |
| KCNJ4 | Kir2.3 | constitutively active | expressed in heart, skeletal muscle, and brain (hippocampus and forebrain); forms tetrameric channels with KCNJ2 |
| KCNJ5 | Kir3.4 | GIRK | also referred to as GIRK4; forms a heteromeric complex with the KCNJ3 encoded protein; constitutes the IKACh potassium current that represents phase 4 of nodal cardiomyocyte action potential propagation; this channel responsible for the resting membrane potential of nodal cardiomyocytes; is a target of the βγ-subunits of parasympathetic acetylcholine activated muscarinic M2 receptor |
| KCNJ6 | Kir3.2 | GIRK | also referred to as GIRK2; increased activity of channel by direct association with βγ-subunits of Gi-type G-proteins |
| KCNJ8 | Kir6.1 | ATP-sensitive | smooth muscle ATP-sensitive K+ channel; forms heteromeric complexes with the regulatory protein SUR2 (encoded by the ABCC9 gene) |
| KCNJ9 | Kir3.3 | GIRK | also referred to as GIRK3; increased activity of channel by direct association with βγ-subunits of Gi-type G-proteins |
| KCNJ10 | Kir4.1 | K+ transport | functions with KCNJ16 in the basal lateral membranes of renal distal tubule cells to regulate K+ recycling |
| KCNJ11 | Kir6.2 | ATP-sensitive | forms the core of the ATP-sensitive K+ channel responsible for regulated secretion of insulin from pancreatic β-cells; the channel in β-cells is a heterooctameric complex formed from four subunits of the KCNJ11 encoded protein and four subunits from the ABCC8 (SUR1) encoded protein |
| KCNJ12 | Kir2.2 | constitutively active | constitutes the IK1 potassium current that represents phase 4 of ventricular cardiomyocyte action potential propagation; this channel responsible for the resting membrane potential of non-nodal cardiomyocytes |
| KCNJ13 | Kir7.1 | K+ transport | may be functionally coupled to the ubiquitous Na+/K+-ATPases; predominantly expressed in thyroid follicular cells |
| KCNJ14 | Kir2.4 | constitutively active | expressed in retina, brain, spinal cord; elevated expression observed in numerous cancers; high expression correlates with poor prognosis in colorectal carcinoma and lung cancer |
| KCNJ15 | Kir4.2 | K+ transport | highly expressed in intestinal mucosa, brain, pancreas, and renal tubule cells |
| KCNJ16 | Kir5.1 | K+ transport | functions with KCNJ10 in the basal lateral membranes of renal distal tubule cells to regulate K+ recycling |
| KCNJ18 | Kir2.6 | constitutively active | expression regulated by thyroid hormone |
Calcium-Activated Potassium Channels
The Ca2+-activated K+ channels (designated KCa or KCa) are a distinct family of K+ channels that are capable of directly relaying changes in cellular Ca2+ to changes in membrane potential via K+ conductance. There are three distinct subfamilies of KCa channels defined, not only by their potassium conductance, but also based upon pharmacological properties and nucleotide sequence. Based upon their potassium conductance these three subfamilies are identified as big conductance channels (BK or Maxi-K), intermediate conductance channels (IK), and small conductance (SK) channels.
Humans express a total of six KCa α-subunit encoding genes, five of which are functionally defined by their potassium conductance as BK (Maxi-K), IK, or SK. The BK and IK channels are voltage sensitive (i.e. voltage-gated) whereas the SK channels are voltage insensitive. The BK channels are formed from the KCNMA1 encoded proteins which are identified as KCa1.1. The human KCNMA1 gene is related to the fruitfly slowpoke (Slo) gene and as such the human BK channels are also identified Slo1. The IK channels are formed from the KCNN4 encoded protein which is identified as KCa3.1. The SK channels are formed from one of three genes, KCNN1, KCNN2, or KCNN3 where the proteins are identified as KCa2.1, KCa2.2, and KCa2.3, respectively. The other KCa channel, encoded by the KCNU1 (KCa5.1) gene, is included only due to amino acid sequence similarity but is not activated by Ca2+ but instead by pH. In addition to the α-subunit encoding genes humans express four β-subunit encoding genes identified as KCNMB1, KCNMB2, KCNMB3, and KCNMB4. The β-subunits only associate with the homotetrameric KCNMA1 encoded α-subunit channel.
The BK channels encoded by the KCNMA1 gene are the most well studied channels of the KCa family. The KCNMA1 gene is expressed in smooth muscle cells throughout the peripheral and cerebral vasculatures. The KCNMA1 gene undergoes extensive alternative splicing such that 13 different functional BK type channels can be generated, each of which can associate with one of the four different β-subunit proteins. The primary function of the BK channels is the regulation of vascular tone. IK channels are also involved in vascular functions as well as being involved in the regulation of the cell cycle in stem cells, lymphocytes, and cancer cells. The SK channels are predominantly expressed in neurons where they are involved in the regulation of nerve cell firing frequency.
Vasodilators, which include those that function via activation of PKA and PKG, activate BK channels which results in smooth muscle membrane hyperpolarization and vasodilation. Both PKA and PKG activate BK channels through two primary pathways, one is increased release of Ca2+ from the sarcoplasmic reticulum (SR; muscle cell endoplasmic reticulum) and the other is through increasing the sensitivity of BK channels to Ca2+. On the other hand vasoconstrictors, such as serotonin and thromboxane A2 (TXA2), whose smooth muscle cell receptors are coupled to Gq-type G-proteins, activate phospholipase Cβ (PLCβ). Activated PLCβ hydrolyzes membrane phosphatidylinositol 4,5-bisphosphate (PIP2) to diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). The released DAG activates PKC which directly reduces BK channel activity and also reduces the release of Ca2+ from the SR. These effects, of PKC on BK channels, result in smooth muscle cell depolarization and vasoconstriction.
Two-Pore Domain Potassium Channels
The two pore (also called tandem pore) domain potassium channels (designated K2P or K2P) are so-called because the channel α-subunit proteins possess two pore loops (domains) unlike the other members of the K+ channel family whose α-subunit proteins possess a single pore loop. Because these channel proteins possess two pore domains they can form functional K+ channels through dimerization instead of the tetrameric structure of the other K+ channels. Although these channels are called “two pore” because their α-subunit proteins possess two pore loops, the functional channels do not have two pores through which K+ ions move. The K2P channels are also referred to as “leak” channels given that they contribute to leak currents in both excitable and non-excitable cells.
The two-pore domain potassium channel encoding genes are all designated by KCNK and humans express a total of 15 genes in this subfamily of potassium channels. Historical nomenclature of these potassium channels is based upon both pharmacological and biophysical properties and these names are still commonly utilized. Based upon the historical nomenclature the two-pore domain potassium channels can be classified into seven subfamilies, TWIK, TREK, TASK, TRAAK, THIK, TALK, and TRESK. TWIK is derived from Tandem of P-domains in a Weak Inward rectifying K+ channel. TREK is derived from TWIK-RElated K+ channel. TASK is derived from Two-pore domain, Acid-Sensitive K+ channel. TRAAK is derived from Two-pore domain-Related Arachidonic Acid-activated K+ channel. THIK is derived from Two-pore domain Halothane-Inhibited K+ channel. TALK is derived from Two-pore domain ALkaline-activated K+ channel. TRESK is derived from TWIK-RElated Spinal cord K+ channel.
Table of the Two-Pore Domain Potassium Channels
| Gene Symbol | Channel Designation | Alias | Functions/Comments |
| KCNK1 | K2P1.1 | TWIK1 | expressed in several tissues including kidney, heart, brain, and lung; within kidney KCNK1 is expressed in cells of the thick ascending limb (TAL) of the loop of Henle where it likely functions K+ recycling; expressed along the auditory and vestibular sensory system, including neurons and nonsensory epithelia |
| KCNK2 | K2P2.1 | TREK1 | activated by arachidonic acid similar to TRAAK subfamily K2P channels; lysophospholipids (LysoPL) activate the channel more rapidly than arachidonic acid; PKA phosphorylation inhibits the channel; interacts with proteins of the Popeye domain containing (Popdc) family whose activities are regulated by cAMP |
| KCNK3 | K2P3.1 | TASK1 | as the historical name of this channel implies it is sensitive to acidic extracellular pH; |
| KCNK4 | K2P4.1 | TRAAK | as the historical name of this channel implies it is activated by arachidonic acid; additional polysunsaturated fatty acids (PUFA) such as docosahexaenoic acid (DHA), linolenic acid, and linoleic acid also directly activate the channel; the effect of PUFA is to sensitize the channel to mechanical stimulation; lysophospholipids (LysoPL) activate the channel more rapidly than arachidonic acid |
| KCNK5 | K2P5.1 | TASK2 | although originally referred to as TASK2 this channel is not sensitive to acid but to alkaline pH and belongs to the TALK subfamily; expressed in the kidney, pancreas, and liver; in the pancreas the channel is found in both endocrine and exocrine cells |
| KCNK6 | K2P6.1 | TWIK2 | |
| KCNK7 | K2P7.1 | although a member of the TWIK subfamily, based upon amino acid sequence, the protein contains an unconventional sequence in the second K+ channel pore suggesting that the human KCNK7 protein may not function as a K+ channel | |
| KCNK9 | K2P9.1 | TASK3 | as the historical name of this channel implies it is sensitive to acidic extracellular pH; |
| KCNK10 | K2P10.1 | TREK2 | activated by arachidonic acid similar to TRAAK subfamily K2P channels; lysophospholipids (LysoPL) activate the channel more rapidly than arachidonic acid |
| KCNK12 | K2P12.1 | THIK2 | abundant expression in the heart, skeletal muscle, and pancreas, also expressed in kidney; within the kidney KCNK12 channels are found in the same cells as ROMK (KCNJ1) |
| KCNK13 | K2P13.1 | THIK1 | expressed in numerous tissues; within the kidney KCNK13 channels are found in the same cells as ROMK (KCNJ1) |
| KCNK15 | K2P15.1 | TASK5 | |
| KCNK16 | K2P16.1 | TALK1 | as the historical name of this channel implies it is sensitive to alkaline extracellular pH; highest level of expression in pancreas with restriction to exocrine cells |
| KCNK17 | K2P17.1 | TALK2 | although originally referred to as TASK4 this channel is not sensitive to acid but to alkaline pH and belongs to the TALK subfamily; highest level expression in pancreas with restriction to exocrine cells |
| KCNK18 | K2P18.1 | TRESK | regulated by Ca2+ in a manner distinct from the role of this ion in regulating the KCa family of K+ channels; Ca2+ exerts its effects through the regulation of the Ca2+/calmodulin-dependent phosphatase, calcineurin; Ca2+-mediated dephosphorylation activates the channel |
Sodium-Activated Potassium Channels
Humans express two sodium-activated potassium channels (KNa). These two channels were originally classified as members of the calcium-activated potassium channel family, and identified as KCa4.1 and KCa4.2, based upon amino acid sequence homology. However, these two channels are not activated by Ca2+ but by Na+. These two channels are encoded by the KCNT1 and KCNT2 genes, respectively. The KCNT1 encoded protein is also referred to as Slack (Sequence Like A Calcium-activated K+ channel) and the KCNT2 encoded protein is also referred to as Slick (Sequence Like an Intermediate Calcium-activated K+ channel). Both KCNT1 and KCNT2 genes are expressed throughout the brain where the encoded proteins are involved in the adaptation of neuronal firing rates and to slow after hyperpolarization (AHP) that follows repetitive firing.
The ABC Family of Transporters
The ABC transporters comprise the ATP-binding cassette transporter superfamily. All members of this superfamily of membrane proteins contain a conserved ATP-binding domain and use the energy of ATP hydrolysis to drive the transport of various molecules across all cell membranes. There are 48 known members of the ABC transporter superfamily and they are divided into seven subfamilies based upon phylogenetic analyses. These seven subfamilies are designated ABCA (ABC1), ABCB (also called TAP/MDR for transporter, ATP cassette), ABCC (also called MRP for multidrug resistance protein), ABCD (also called ALD for adrenoleukodystrophy), ABCE (also called OABP for oligoadenylate-binding protein), ABCF (also called GCN20), and ABCG (also called White). Each member of a given subfamily is distinguished with numbers (e.g. ABCA1).
The ABCA subfamily comprises 12 genes identified as ABCA1–ABCA10, ABCA12, and ABCA13 (ABCA11 and ABCA17 are pseudogenes). The ABCB subfamily comprises 11 genes identified as ABCB1, ABCB4-ABCB11, TAP1 and TAP2 (also known as ABCB2 and ABCB3, respectively). The ABCC subfamily comprises 13 genes identified as ABCC1–ABCC6, ABCC8–ABCC13, and CFTR (less commonly identified as ABCC7). The ABCD subfamily comprises 4 genes identified as ABCD1–ABCD4. The ABCE subfamily contains a single gene, ABCE1. The ABCF subfamily comprises three genes identified as ABCF1–ABCF3. The ABCG subfamily comprises five genes identified as ABCG1, ABCG2, ABCG4, ABCG5, and ABCG8.
Table of ABC Family Transporters
| Gene Symbol | Other Names | Gene Location/Structure | Functions/Comments |
| ABCA1 | ABC1 | 9q31.1 / 58 exons | involved in cellular cholesterol efflux; transfers cellular cholesterol and phospholipids to HDLs (reverse cholesterol transport); also involved in transport of retinyl esters from intestinal enterocytes into the portal circulation; defects in gene associated with development of Tangier disease |
| ABCA2 | ABC2 | 9q34.3 / 50 exons | role in delivery of LDL-derived free cholesterol to the endoplasmic reticulum for esterification; involved in protection against reactive oxygen species, drug resistance |
| ABCA3 | ABC3 | 16p13.3 / 33 exons | role in delivery of LDL-derived free cholesterol to the endoplasmic reticulum for esterification; involved in protection against reactive oxygen species, drug resistance |
| ABCA4 | ABCR, ABC10 | 1p22.1 / 50 exons | expressed exclusively in retinal photoreceptors; toxic metabolites can accumulate during the process of phototransduction and this transporter is involved in the efflux of the toxic compound N-retinylidene-phosphatidylethanolamine from photoreceptor cells; mutations in this gene are associated with Stargardt disease which is an autosomal recessive form of macular degeneration |
| ABCA5 | ABC13 | 17q24.3 / 39 exons | clustered on chromosome 17 with ABCA6, ABCA8, ABCA9, and ABCA10; cholesterol efflux transporter in macrophages; present in lysosomal membranes as well |
| ABCA6 | 17q24.2-q24.3 / 44 exons | clustered on chromosome 17 with ABCA5, ABCA8, ABCA9, and ABCA10; may be involved in lipid homeostasis in macrophages | |
| ABCA7 | ABCX | 19p13.3 / 47 exons | may be involved in lipid homeostasis of immune cells |
| ABCA8 | 17q24.2 / 42 exons | clustered on chromosome 17 with ABCA5, ABCA6, ABCA9, and ABCA10; may be involved in myelin maintenance | |
| ABCA9 | 17q24.2 / 42 exons | clustered on chromosome 17 with ABCA5, ABCA6, ABCA8, and ABCA10; cholesterol transporter in the endoplasmic reticulum (ER); may function as a tumor suppressor in breast cancer | |
| ABCA10 | 17q24.2 / 40 exons | clustered on chromosome 17 with ABCA5, ABCA6, ABCA8, and ABCA9; may function as a cholesterol efflux transporter | |
| ABCA12 | 2q35 / 55 exons | involved in ceramide transport in keratinocytes | |
| ABCA13 | 7p12.3 / 75 exons | involved in ganglioside and cholesterol transport in the brain | |
| ABCB1 | P-GP, PGY1, MDR1 | 7p21.12 / 29 exons | PGY1: P-glycoprotein 1 MDR1: multidrug resistance protein 1 encoded protein is a multidrug resistance P-glycoprotein; is an integral component of the blood-brain barrier, transports a number of drugs from the brain back into the blood |
| TAP1 | ABCB2, PSF1, APT1 | 6p21.32 / 12 exons | TAP1: transporter, ATP-binding cassette, major histocompatibility complex (MHC) 1 PSF1: peptide supply factor 1 APT1: antigen peptide transporter1 functions as a heterodimer with TAP2/ABCB3 to transport cytosolic peptide fragments across the ER into the membrane compartment where MHC class I molecules assemble |
| TAP2 | ABCB3, PSF2, APT2 | 6p21.32 / 13 exons | TAP2: transporter, ATP-binding cassette, major histocompatibility complex (MHC) 2 PSF2: peptide supply factor 2 APT2: antigen peptide transporter1 functions as a heterodimer with TAP1/ABCB2 to transport cytosolic peptide fragments across the ER into the membrane compartment where MHC class I molecules assemble |
| ABCB4 | PGY3, MDR3 | 17q21.12 / 31 exons | PGY3: P-glycoprotein 3 MDR3: multidrug resistance protein 3 is a class III multidrug resistance P-glycoprotein; canalicular phospholipid translocator; biliary phosphatidylcholine transport; defects in gene associated with 6 liver diseases: progressive familial intrahepatic cholestasis type 3 (PFIC3), adult biliary cirrhosis, transient neonatal cholestasis, drug-induced cholestasis, intrahepatic cholestasis of pregnancy, and low phospholipid-associated cholelithiasis syndrome |
| ABCB5 | 7p21.1 / 31 exons | expressed in limbal stem cells; required for corneal development; originally identified as a marker for melanoma stem cells and skin progenitor cells | |
| ABCB6 | MTABC3 | 2q35 / 19 exons | mitochondrial transporter involved in heme biosynthesis; transports porphyrins into mitochondria; represents the Langereis (Lan) blood group antigen |
| ABCB7 | ASAT | Xq13.3 / 16 exons | transport of heme from mitochondria to the cytosol; iron-sulfur (Fe/S) cluster transport; ASAT is X-linked sideroblastic anemia with ataxia |
| ABCB8 | MABC1 | 7q36.1 / 17 exons | MABC1: mitochondrial ATP-binding cassette 1; present in the inner mitochondrial membrane; involved in organic and inorganic molecule transport out of the mitochondria |
| ABCB9 | TAPL | 12q24.31 / 20 exons | TAPL: TAP-like; transports a broad spectrum of peptides from the cytosol into the lumen of the lysosomes; transported peptides range from 6 to 59 amino acids with optimum size shown to be 23 amino acids |
| ABCB10 | 1q42.13 / 15 exons | present in the inner mitochondrial membrane; facilitates mitochondrial iron import; may be involved in mitochondrial heme transport | |
| ABCB11 | BSEP, SPGP | 2q31.1 / 30 exons | BSEP: bile salt export protein, bile salt transport out of hepatocytes SPGP: sister of P-glycoprotein mutations in ABCB11 gene associated with progressive familial intrahepatic cholestasis type 2 (PFIC2) |
| ABCC1 | MRP1 | 16p13.11 / 34 exons | MRP1: multidrug resistance associated protein 1; transport of cobalamin from intestinal enterocytes to the blood; sphingosine-1-phosphate (S1P) release from mast cells which enhances their migration; uses glutathione as a co-factor in mediating resistance to heavy metal oxyanions |
| ABCC2 | MRP2, CMOAT | 10q24.2 / 34 exons | MRP2: multidrug resistance associated protein 2 CMOAT: canalicular multi-specific organic anion transporter; biliary excretion of many non-bile organic anions; gene defects result in Dubin-Johnson syndrome |
| ABCC3 | MRP3, CMOAT2, MOATD | 17q21.33 / 32 exons | MRP3: multidrug resistance associated protein 3 CMOAT2: canalicular multi-specific organic anion transporter; involved in drug resistance MOATD: multi-specific organic anion transporter D |
| ABCC4 | MRP4, MOATB | 13q32.1 / 36 exons | MRP4: multidrug resistance associated protein 4 MOATB: multi-specific organic anion transporter B; enriched in prostate; regulator of intracellular cyclic nucleotide levels; mediator of cAMP-dependent signal transduction to the nucleus |
| ABCC5 | MRP5, MOATC | 3q27.1 / 36 exons | MRP5: multidrug resistance associated protein 5 MOATC: multi-specific organic anion transporter C resistance to thiopurines and antiretroviral nucleoside analogs; overexpression is correlated to age-related development of type 2 diabetes; knock-out in mice results in increased GLP-1 release, increase insulin sensitivity, decreased fat mass, and increased activity |
| ABCC6 | MRP6, MOATE, PXE | 16p13.11 / 34 exons | MRP6: multidrug resistance associated protein 6 MOATE: multi-specific organic anion transporter E PXE: pseudoxanthoma elasticum, a rare disorder in which the skin, eyes, heart, and other soft tissues become calcified |
| CFTR | ABCC7 | 7q31.2 / 32 exons | CFTR: cystic fibrosis transmembrane conductance regulator; chloride ion channel; gene defects result in cystic fibrosis (CF) with the most common mutation being a three base deletion that removes the Phe residue at position 507, identified as the ΔF507 mutation |
| ABCC8 | SUR | 11p15.1 / 39 exons | SUR: sulfonylurea receptor; four copies of the ABCC8 encoded proteins associate with four copies of the ATP-sensitive potassium channel encoded by the KCNJ11 gene; the ABCC8 encoded protein is the target of the type 2 diabetes drugs such as glipizide |
| ABCC9 | SUR2 | 12p12.1 / 42 exons | forms part of an ATP-sensitive potassium channel in cardiac, skeletal, and smooth muscle cells; alternative splicing yields two major isoforms, SUR2A includes exon 38A while SUR2B includes exon 38B; mutations in ABCC9 gene associated with cardiomyopathy dilated type 1O |
| ABCC10 | MRP7 | 6p21.1 / 26 exons | MRP7: multidrug resistance associated protein 7; likely to be involved in lipophilic anion efflux |
| ABCC11 | MRP8 | 16q12.1 / 39 exons | MRP8: multidrug resistance associated protein 8; involved in bile acid (glycocholate and taurocholate) uptake, glutathione S-conjugated steroid uptake, and cyclic nucleotide efflux; ABCC11 and ABCC12 derived via gene duplication, both reside at 16q12.1 |
| ABCC12 | MRP9 | 16q12.1 / 29 exons | MRP9: multidrug resistance associated protein 9; ABCC11 and ABCC12 derived via gene duplication, both reside at 16q12.1; function unknown |
| ABCC13 | 21q11.2 / 7 exons | likely a pseudogene in humans | |
| ABCD1 | ALD | Xq28 / 11 exons | ALD: adrenoleukodystrophy; involved in the import and/or anchoring of very long-chain fatty acid-CoA synthetase (VLCFA-CoA synthetase) to the peroxisomes; gene defects result in X-linked adrenoleukodystrophy (XALD) |
| ABCD2 | ALDR | 12q12 / 13 exons | ALDR: adrenoleukodystrophy-related protein; also found in peroxisomal membranes; modifier that contributes to phenotypic variability seen in XALD; can restore peroxisomal fatty acid oxidation defect of XALD liver cells |
| ABCD3 | PMP70, PXMP1 | 1p21.3 / 28 exons | PMP70: peroxisomal membrane protein 70 kDa; also called peroxisomal membrane protein 1 (PXPM1); mutation associated with Zellweger syndrome 2 (ZWS2); transports branched-chain fatty acids and C-27 bile acids into the peroxisomes |
| ABCD4 | PMP69, PXMP1L | 14q24.3 / 20 exons | PMP69: peroxisomal membrane protein 69 kDa; related to the other ABCD family members but localized to ER membranes; also called peroxisomal membrane protein 1-like (PXMP1L); mutations increase severity of XALD; may be involved in release of vitamin B12 from the lysosomes to the cytosol |
| ABCE1 | OABP, RNS4I | 4q31.21 / 18 exons | OABP: oligoadenylate binding protein RNS4I: ribonuclease 4 inhibitor may be involved in the regulation of mRNA turnover; antagonizes the anti-viral effects of the interferon-regulated 5′-phosphorylated 2′,5′-linked oligoadenylates that bind and activate RNaseL |
| ABCF1 | 6q21.33 / 25 exons | may be a ribosomal protein involved in mRNA translation | |
| ABCF2 | 7q36.1 / 17 exons | may be involved in the regulation of mRNA translation; overexpression is associated with resistance of certain cancers to drug-induced apoptosis | |
| ABCF3 | 3q27.1 / 21 exons | may exert antiviral effects against flaviviruses such as West Nile virus | |
| ABCG1 | White1 | 21q22.3 / 20 exons | involved in mobilization and efflux of intracellular cholesterol; responsible for approximately 20% of cholesterol efflux from macrophages to HDL (reverse cholesterol transport, RCT) |
| ABCG2 | ABCP, MXR, BCRP | 4q22.1 / 20 exons | ABCP: ATP-binding cassette transporter, placenta-specific MXR: mitoxantrone-resistance protein BCRP: breast cancer resistance protein xenobiotic transporter; plays a major role in multidrug resistance; heme and protoporphyrin IX export from mitochondria; is a major uric acid secretion transporter in the small intestine; involved in kidney uric acid excretion as well |
| ABCG4 | White2 | 11q23.3 / 17 exons | expression restricted to astrocytes and neurons; transfers cholesterol to HDL-like particles in the CNS; may function in sterol transport with ABCG1 in cells where the two genes are co-expressed; may increase lipidation of apoE in Alzheimer disease |
| ABCG5 | Sterolin-1 | 2p21 / 15 exons | forms an obligate heterodimer with ABCG8; expressed in intestinal enterocytes and hepatocytes; functions to limit plant sterol and cholesterol absorption from the diet by facilitating efflux out of enterocytes into the intestinal lumen and out of hepatocytes into the bile canaliculi; tandemly arrayed in head-to-tail orientation with ABCG8 gene |
| ABCG8 | Sterolin-2 | 2p21 / 14 exons | see above for ABCG5 |
The Solute Carrier Family of Transporters
The solute carrier (SLC) family of transporters includes over 450 proteins functionally grouped into 76 families. The SLC family of transporters includes facilitative transporters, primary and secondary active transporters, and ion channels. Given the scope of this discussion it is not possible to cover all of the transporters in each of the 76 families.
Listed in the Table below are all of the families of SLC transporters and within each family is a description of several member proteins. All of the members of a particular family are not included due to space limitations. Focus is primarily on solute carriers discussed on other web pages in this site or due to known clinical significance.
With respect to the numerous amino acid transporters represented in the SLC superfamily of transporters, there is a set of historical nomenclature designations. Neutral amino acid transporters that prefer leucine and other large hydrophobic neutral amino acids are called system L transporters, those that prefer alanine and other small and polar neutral amino acids are called the system A transporters, and those that prefer alanine, serine, and cysteine are called the system ASC transporters. A distinct nomenclature has been used for systems mediating transport of cationic amino acids [system y+ or written y(+)] and those transporting anionic amino acids (system X–). Amino acid transporters that are Na+-independent are named with lowercase acronyms, whereas Na+-dependent transporters are named with uppercase acronyms.
The mitochondrial carrier family of SLC transporters (SLC25 family) is the largest family consisting of 53 members. These transporters are responsible for the transport of substances across the impermeable inner mitochondrial membrane, IMM. These IMM transporters serve critical functions related to whole body energy homeostasis by supporting the mitochondrial oxidation of fatty acid, carbohydrates, and amino acids. In addition, transport of metabolites into the mitochondria supports thermogenesis, heme biosynthesis, iron-sulfur center synthesis, and amino acid interconversions.
Table of SLC Family Transporters
| SLC Family | Functional Class | Gene Designations / Comments |
| 1 | high affinity glutamate and neutral amino acid transporters | SLC1A – SLC1A7 SLC1A1: also called Excitatory Amino Acid Transporter 3 (EAAT3); prevalent in apical membranes of intestinal enterocytes; is a high affinity glutamate transporter; also transports aspartate and cystine SLC1A2: also called EAAT2; clears glutamate from the synaptic cleft of glutamatergic neurons; decreased expression is associated with amyotrophic lateral sclerosis (ALS: Lou Gehrig disease); also transports aspartate SLC1A3: high affinity glutamate transporter in glutamatergic neurons; also known as EAAT1; also transports aspartate SLC1A4: neutral amino acid transporter; also known as ASCT1 (derived from AlaSerCys Transporter 1); transports alanine, glycine, serine, and cysteine SLC1A5: neutral amino acid transporter primarily responsible for plasma membrane glutamine transport; also known as ASCT2; also transports alanine, glycine, serine, threonine, and cysteine; mitochondria-localized variant, SLC1A5_var (MTS-SLC1A5) transports glutamine into and out of the mitochondria; prevalent in apical membranes of intestinal enterocytes SLC1A6: also called EAAT4; is a high affinity glutamate transporter; also transports aspartate; expressed in Purkinje cells of cerebellum SLC1A7: also called EAAT5; is a high affinity glutamate transporter; also transports aspartate; expressed in bipolar cells and retinal rod cells |
| 2 | facilitative GLUT transporters | SLC2A1 – SLC2A14 SLC2A1: common name is GLUT1; glucose and dehydroascorbate (vitamin C) transporter; ubiquitously expressed in various tissues but only at low levels in liver and skeletal muscle; is the primary glucose transporter in brain, placenta, and erythrocytes; is primarily responsible for glucose transport across the blood-brain-barrier (BBB); mutations in the SLC2A1 gene result in GLUT1 deficiency syndrome SCL2A2: common name is GLUT2; this glucose transporter is expressed predominantly in the liver, kidney, and intestines SCL2A3: common name as GLUT3; glucose and dehydroascorbate (vitamin C) transporter; primarily expressed in neurons and possess the lowest Km for glucose of any of the glucose transporters SLC2A4: common name is GLUT4; glucose and dehydroascorbate (vitamin C); predominantly expressed in insulin-responsive tissues such as skeletal muscle and adipose tissue SLC2A5: common name is GLUT5; the major fructose transporter SLC2A6: common name is GLUT6; glucose transporter SLC2A7: common name is GLUT7; glucose and fructose transporter SLC2A8: common name is GLUT8; transports glucose and fructose; highly expressed in oxidative tissues such as the liver; is essential in the development of fructose-induced macrosteatosis in the liver; sensitive to insulin like GLUT4; also transports dehydroascorbate (vitamin C) SLC2A9: common name is GLUT9; also known as URATv1; is a major uric acid transporter in the liver and kidneys; two alternatively spliced mRNAs generate two protein isoforms, GLUT9a and GLUT9b SLC2A10: common name is GLUT10; glucose and galactose transporter SLC2A11: common name is GLUT11; glucose and fructose transporter SLC2A12: common name is GLUT12; glucose transporter; sensitive to insulin like GLUT4 SCL2A13: also called the proton (H+) myo–inositol cotransporter, HMIT SLC2A14: common name is GLUT14; glucose transporter |
| 3 | heavy subunits of heteromeric amino acid transporters activator of dibasic and neutral amino acid transporters | SLC3A1, SLC3A2 SLC3A1: encodes one of the heavy chain subunits of heteromeric amino acid transporters; also known as rBAT; forms the renal cystine transporter necessary for cystine reabsorption by the proximal tubules of the kidney along with the light chain subunit encoded by the SLC7A9 gene; defects in this transporter can found in type I cystinurias SLC3A2: encodes a heavy chain subunit of heteromeric amino acid transporters; also known as 4F2hc; forms heterodimers with the light chain subunits SLC7A5(LAT1); SLC7A6(y+LAT2); SLC7A7(y+LAT1); SLC7A10(ASC1); SLC7A11(xCT) |
| 4 | sodium bicarbonate transporters (NBC); anion exchangers (AE) | SLC4A1 – SLC4A5, SLC4A7 – SLC4A11 SLC4A1: also known as anion exchanger 1 (AE1) and BND3 (band 3 of red cell membranes); responsible for Cl– and HCO3– exchange in the connecting tubules of kidney and in erythrocytes (the “chloride shift”); mutations associated with autosomal recessive and dominant forms of renal tubular acidosis (type 1 RTA); functions in erythrocyte oxalate transport SLC4A2: also known as AE2; involved in biliary bicarbonate excretion SLC4A3: also known as AE3 SLC4A4 (NBC1): multiple splice variants identified as NBCe1-A (proximal tubule of kidney: also identified as kNBC1), NBCe1-B (pancreas: also identified as pNBC1), and NBCe1-C (brain); mutations associated with renal tubular acidosis (type 2 RTA) SLC4A5: also known as NBC4 and NBCe2 SLC4A7 (mNBC3 and NBC2 variant): was formerly identified as SLC4A6 and so the SLC4A6 identity is no longer used SLC4A8 (NBC3): regulation of pH in neurons SLC4A9 (AE4): expression predominates in kidney; also found in apical membrane of intestinal enterocytes SLC4A10: regulates intracellular pH of neurons SLC4A11: Na+-borate co-transporter; mutations associated with corneal endothelial dystrophy |
| 5 | sodium glucose co–transporters (SGLT) | SLC5A1 – SLC5A12 SLC5A1: commonly called SGLT1; is a Na+-dependent intestinal glucose and galactose transporter; H+-coupled glucose transport can also be carried out SLC5A2: commonly called SGLT2 which is responsible for the majority of Na+-dependent glucose re-absorption by the kidneys and as such is a current target of therapeutic intervention in the hyperglycemia associated with type 2 diabetes; is a low affinity but high capacity glucose transporter SLC5A3: is a Na+-myoinositol glucose co-transporter (SMIT); ubiquitously expressed with highest levels in endothelial cells, kidney, and thyroid gland SLC5A4: also known as the Na+-dependent amino acid transporter 1 (SAAT1); also identified as SGLT3; expressed in cholinergic neurons, the small intestine, and skeletal muscle; acts as a glucose sensor which depolarizes the membrane when at high concentration SLC5A5: Na+-iodide co-transporter commonly identified as NIS; mutations associated with thyroid dyshormonogenesis SLC5A6: Na+-dependent pantothenate, biotin, and lipoic acid transporter; also identified as Na+-multivitamin transporter (SMVT) SLC5A7: Na+– and Cl–-dependent choline transporter for choline uptake into cholinergic neurons; also known as CHT1 (choline transporter 1) SLC5A8: lactate, monocarboxylate, and short-chain fatty acid transporter; also known as Na+-monocarboxylate transporter 1 (SMCT1); present in apical membranes of proximal tubule epithelial cells; also transports nicotinic acid; is a tumor suppressor that is silenced by hypermethylation in certain colorectal carcinomas SLC5A9: commonly called SGLT4; is a Na+-dependent mannose and fructose transporter; highly expressed in small intestine and kidney SLC5A10: commonly called SGLT5; Na+-dependent sugar transporter; high affinity for mannose and fructose; low affinity for glucose and galactose; expressed in the kidney SLC5A11: commonly called SGLT6; myo-inositol and glucose transporter; highly expressed in the brain SLC5A12: lactate transporter also known as the Na+-monocarboxylate transporter 2 (SMCT2); present in apical membrane of proximal tubule epithelial cells |
| 6 | sodium– and chloride–dependent neurotransmitter transporters | SLC6A1 – SLC6A20 SLC6A1: GABA transporter in GABAergic neurons and elsewhere in the brain; commonly called GAT1 (GABA Transporter 1) SLC6A2: major norepinephrine transporter in the brain and in adrenal medullary chromaffin cells; also transports dopamine SLC6A3: major dopamine transporter in dopaminergic neurons SLC6A4: is the serotonin transporter; defects in this gene are associated with obsessive-compulsive disorder and anxiety-related traits; the tricyclic antidepressants function principally by inhibiting this transporter in the brain SLC6A5: glycine transporter in glycinergic neurons and other tissues in the periphery; commonly called GLYT2 SLC6A6: taurine and β-alanine transporter; prevalent in apical membranes of intestinal enterocytes SLC6A7: proline transporter SLC6A8: is the creatine transporter by which neurons and skeletal muscle cells take up the compound that was produced in the liver SLC6A9: glycine transporter; commonly called GLYT1 SLC6A11: GABA transporter; commonly called GAT3 SLC6A13: GABA transporter; commonly called GAT2; also involved in taurine uptake into the brain SLC6A14: neutral and cationic amino acid uptake transporter coupled to Na+ (two ions) and Cl– (one ion) uptake; plasma membrane glutamate and glutamine uptake transporter; major transporter for serine uptake in cancer cells such as colorectal carcinomas; also identified as ATB0,+ SLC6A15: proline, leucine, valine, isoleucine, and methionine transporter; protein also called system B0 neutral amino acid transporter 2 (B0AT2, or B0AT2) SLC6A18: glycine, alanine, methionine, serine, cysteine transporter; protein also called system B0 neutral amino acid transporter 3 (B0AT3, or B0AT3) SLC6A19: involved in neutral amino acid transport; prevalent in apical membranes of intestinal enterocytes; deficiency results in Hartnup disorder; protein also called system B0 neutral amino acid transporter 1 (B0AT1, or B0AT1) SLC6A20: proline, hydroxyproline, and betaine transporter |
| 7 | cationic amino acid transporters [y+ system] and the glycoprotein-associated amino acid transporters | SLC7A1 – SLC7A11, SLC7A13, SLC7A14 SLC7A1: prevalent in apical membranes of intestinal enterocytes; primary arginine import transporter; essential for lysine and arginine uptake; may also transport ornithine; also called cationic amino acid transporter 1, CAT1 SLC7A2: lysine and ornithine transporter; also called CAT2; may transport arginine SLC7A3: lysine and ornithine transporter; also called CAT3; may transport arginine SLC7A4: also called CAT4; may transport arginine SLC7A5: also called L-type (or light subunit) amino acid transporter 1, LAT1; functions in the transport of Phe, Tyr, and Trp into the brain; transport of methionine into T cells; major methionine transporter in cancer cells SLC7A6: primary arginine efflux transporter; also called y+L-type amino acid transporter 2, y+LAT2 SLC7A7: also called y+L-type amino acid transporter 1, y+LAT1; major glutamine influx transporter in macrophages; mutations in the SLC7A7 gene are the cause of lysinuric protein intolerance, SLC7A8: also called L-type amino acid transporter 2, LAT2 LAT1, y+LAT1, and y+LAT2 are the light chains of the heterodimeric amino acid transporters that are disulfide bonded to the heavy chain subunit identified as 4F2hc (encoded by the SLC3A2 gene) SLC7A9: encodes the light chain subunit [b(0,+)AT] of the renal cystine transporter that, in association with the heavy chain subunit called rBAT (encoded by the SLC3A1 gene), forms the transporter responsible for cystine reabsorption in the proximal tubules of the kidney and can be defective in cystinurias (referred to as non-type I) SLC7A10: encodes the light chain subunit (ASC-1) that associates with 4F2hc (SLC3A1) forming a heteromeric neutral amino acid transporter; transports alanine, cysteine, glycine, serine, and threonine SLC7A11: encodes the light chain subunit (xCT) of the heteromeric acidic amino acid transporter designated Xc–; is a Na+-independent, Cl–-dependent cystine-glutamate antiporter; primary transporter for cystine import; also transports aspartate; endothelial cells increase expression in preparation for proliferation; is also upregulated in many cancer cells |
| 8 | K+-dependent Na+/Ca2+ exchangers (NCX) | SLC8A1(NCX1), SLC8A2(NCX2), SLC8A3(NCX3), SLC8B1(NCLX) typical function is to transport one Ca2+ ion out of a cell while transporting three Na+ ions into the cell; at less negative membrane potentials, such as in depolarizing non-nodal cardiomyocytes, the transporter is capable of switching the direction of ion transport so as to transport Ca+ into and Na+ out of the cell SLC8A1: primary transporter for Ca2+ extrusion from depolarized cardiomyocytes allowing return to the resting state SLC8A3: regulates electrogenic Na+ and Ca2+ exchange across plasma membrane and intracellular membranes SLC8B1: formerly identified as SLC24A6 |
| 9 | Na+/H+ exchangers (NHE) | SLC9A1(NHE1), SLC9A2(NHE2), SLC9A3(NHE3), SCL9A4(NHE4), SLC9A5(NHE5), SLC9A6(NHE6), SLC9A7(NHE7), SLC9A8(NHE8), SLC9A9(NHE9) SLC9A1: expressed in proximal tubule of kidney and intestines SLC9A2: involved in Na+ uptake from the colon SLC9A3: major proximal tubule transporter for Na+ and HCO3– reabsorption with H+ efflux; excretion of H+ from intestinal cells; mutations associated with congenital secretory sodium diarrhea SLC9A4: expressed in proximal tubule of kidney and intestines SLC9A6: localized to endosomal membranes where it regulates pH and volume; mutations associated with X-linked Christianson-type syndromic intellectual impairment SLC9A8: expressed in proximal tubule of kidney and intestines; highest level of expression in neonate SLC9A9: mutations associated with ADHD and autism susceptibility 16 |
| 10 | sodium bile salt co-transporters | SLC10A1, SLC10A2, SLC10A3, SLC10A4, SLC10A5, SLC10A6(SOAT), SLC10A7 SLC10A1: also called NTCP for Na+-taurocholate co-transporting polypeptide; involved in hepatic uptake of bile acids through the sinusoidal/basolateral membrane SLC10A2: also called ASBT for apical sodium bile salt co-transporter; primary bile acid transporter for uptake into enterocytes of the distal ileum SLC10A3, SLC10A4, and SLC10A5 are considered orphan transporters SLC10A6: commonly identified as Na+-dependent organic anion transporter (SOAT); transporter of sulfated steroids and sulfated bile acids |
| 11 | proton-coupled metal ion transporters | SLC11A1, SLC11A2, SLC11A3 SLC11A1: divalent metal transporter; primarily iron and manganese; involved in iron homeostasis; involved in host resistance to pathogens SLC11A2: also known as the divalent metal-ion transporter-1 (DMT1); encoded splice variant mRNA contains iron-responsive elements (IREs) that allow translation to be regulated by the levels of iron SLC11A3: now referred to as SLC40A1, this protein is more commonly called ferroportin, but is also known as iron-regulated gene 1 (IREG1), and reticuloendothelial iron transporter (MTP1) |
| 12 | electroneutral cation/Cl– co-transporters | SLC12A1 – SLC12A9 SLC12A1: expressed almost exclusively in the thick ascending limb of the loop of Henle in the kidney; commonly identified as Na+-K+-Cl– co-transporter type 2 (NKCC2) SLC12A2: encodes the Na+-K+-Cl– co-transporter type 1; commonly identified as NKCC1; ubiquitously expressed; functions in the distal convoluted tubule and the collecting ducts in the kidney SLC12A3: renal thiazide (diuretic used to treat hypertension)-sensitive Na+-Cl– co-transporter (commonly identified as NCC) expressed in the distal convoluted tubule of kidney; mutations in gene cause Gitelman syndrome (hypokalemic alkalosis with hypomagnesemia) SLC12A4: facilitates serine uptake particularly in colorectal carcinomas |
| 13 | Na+–sulfate/carboxylate co-transporters | SLC13A1, SLC13A2, SLC13A3, SLC13A4, SLC13A5 SLC13A1: apical membrane transporter; involved in renal sulfate homeostasis in proximal tubule of kidney; also identified as NaS1 (Na+-sulfate transporter 1) SLC13A2: citrate transporter in proximal tubule of kidney and intestine; also identified as NaDC1 (Na+-dicarboxylate transporter 1) SLC13A3: also called Na+-dicarboxylate cotransporter 3 (NaDC3); transports itaconate, citrate, succinate, 2-oxoglutarate (α-ketoglutarate) in proximal tubule of kidney, liver, brain, and pancreas |
| 14 | urea transporters | SLC14A1 (UT-B): erythrocyte urea transporter; UT-B1 and UT-B2 isoforms SLC14A2 (UT-A): renal Na+-independent urea transporter in the collecting duct; six protein isoforms in mammals with UT-A1, UT-A2, UT-A3, and UT-A6 isoforms expressed in humans |
| 15 | proton oligopeptide co-transporters | SLC15A1, SLC15A2, SLC15A3, SLC15A4 SLC15A1: prevalent in apical membranes of intestinal enterocytes; transport di- and tripeptides (but not free amino acids) from small intestine into enterocytes and peptide reabsorption in proximal tubule of kidney; also identified as PepT1 SLC15A2: peptide and peptide-like drug transporter expressed in apical membranes of proximal tubule cells; also identified as PepT2 PepT1 and PepT2 transport 400 di-peptides and 8000 tri-peptides as well as synthetic drugs; PepT1 shows higher affinities than does PepT1; peptide transport is coupled to Na+/H+ exchange SLC15A3 (PHT2) and SLC15A4 (PHT1): mediate egress of bacterial peptides from dendritic cells of immune system |
| 16 | monocarboxylate transporters (MCT) | SLC16A1 (MCT1), SLC16A2 (MCT8), SLC16A3 (MCT4), SLC16A4 (MCT5), SLC16A5 (MCT6), SLC16A6 (MCT7), SLC16A7 (MCT2), SLC16A8 (MCT3), SLC16A9 (MCT9), SLC16A10 (MCT10), SLC16A11 (MCT11), SLC16A12 (MCT12), SLC16A13 (MCT13), SLC16A14 (MCT14) SLC16A1 (MCT1), SLC16A3 (MCT4), SLC16A4 (MCT5), SLC16A5 (MCT6), SLC16A6 (MCT7), SLC16A7 (MCT2), SLC16A8 (MCT3): all are plasma membrane lactate, pyruvate, ketone body, and branched-chain oxo acid (derived from branched-chain amino acids) transporters SLC16A1 (MCT1): present in plasma membrane and inner mitochondrial membrane; primary lactate uptake transporter; also transports branched-chain α-keto acids across plasma and inner mitochondrial membranes; responsible for efflux of succinate from cells in response to intracellular acidification; also responsible for transport of succinate into brown adipocytes; increased expression of SLC16A1 gene associated with metastasis of melanoma and poor prognosis SLC16A2 (MCT8): specific thyroid hormone uptake transporter SLC16A3 (MCT4): primary lactate efflux transporter; also transports branched-chain α-keto acids SLC16A7 (MCT2): primary lactate uptake transporter SLC16A8 (MCT3): primarily expressed in retina and epithelial cells of the eye; is responsible for efflux of lactate SLC16A9 (MCT9): lactate transporter; also functions as a carnitine efflux transporter SLC16A10 (TAT1): prevalent in basolateral membranes of intestinal enterocytes; T-type amino acid transporter): Na+-independent aromatic amino acid transporter SLC16A11 (MCT11): plasma membrane pyruvate transporter; polymorphisms in gene associated with increased risk of type 2 diabetes SLC16A12: betaine/γ-aminobutyric acid (GABA) transporter 1 (BGT-1), also known as Na+/Cl–-dependent betaine transporter |
| 17 | organic anion transporters; originally identified as type I Na+–phosphate co-transporters (NPT) | SLC17A1 – SLC17A9 SLC17A1 (NPT1): expressed in apical membrane of proximal tubule cells in kidney; involved in uric acid efflux into urine; also involved in the efflux of the exercise-induced suppressor food intake and body weight, Lac-Phe, to the urine SLC17A2 (NPT3): was misidentified as NPT2 which is encoded by the SLC34A1 gene SLC17A3 (NPT4): expressed in apical membrane of proximal tubule cells in kidney; involved in uric acid efflux into urine; also involved in the efflux of the exercise-induced suppressor food intake and body weight, Lac-Phe, to the urine SLC17A4: expressed in apical membrane of intestinal enterocytes; involved in luminal phosphate uptake SLC17A5: lysosomal sialic acid efflux transporter; mutations in gene associated with two lysosomal storage disorders, one that is a severe infantile disorder called infantile sialic acid storage disease (ISSD) and a less severe disorder called Salla disease SLC17A6, SLC17A7, and SCL17A8 are glutamate transporters in the brain SLC17A9: involved in vesicular uptake, storage, and secretion of ATP |
| 18 | vesicular amine transporters | SLC18A1, SLC18A2, SLC18A3 SLC18A1and SLC18A2: both function in vesicular accumulation of monoamines such as dopamine and epinephrine; both are targets of reserpine (anti-hypertensive and antipsychotic) and tetrabenazine (treatment of tremors in Huntington disease) SLC18A3: functions to accumulate acetylcholine into secretory vesicles |
| 19 | folate/thiamine transporters | SLC19A1, SLC19A2, SLC19A3 SLC19A1: cellular uptake of reduced folate and thiamine mono- and di-phosphate but not free thiamine; protein is commonly known reduced folate transporter 1, RFT1 SLC19A2: intestinal uptake of thiamine SLC19A3: cellular uptake of thiamine; mutations in gene associated with a Wernicke-like encephalopathy |
| 20 | type III Na+–phosphate co-transporters | SLC20A1, SLC20A2 SLC20A1: commonly called Pit-1 [inorganic (Pi) phosphate transporter 1] SLC20A2: commonly called Pit-2 |
| 21 SLCO | organic anion transporting polypeptides (OATP) | the SLC21 genes are all identified as SLCO genes; there are 12 human SLCO family member genes divided into 6 subfamilies identified as 1 through 6; these transporter genes have the nomenclature SLCO followed by the family number, subfamily letter, and member number; the proteins encoded by the SLCO genes are identified as organic anion transporting polypeptides (OATP) SLCO1A2 (SLC21A3): protein is OATP1A2; Na+-independent bile acid transporter in kidney, intestines, liver, and brain; also transports folate; formerly OATPA SLCO1B1 (SLC21A6): protein is OATP1B1; basolateral membrane Na+-independent liver-specific bile acid transporter; also transports folate; formerly OATPC; also transports the dicarboxylic acids tetradecanedioic acid and hexadecanedioic acid SLCO1B3 (SLC21A8): protein is OATP1B3; liver-specific Na+-independent liver-specific bile acid and bilirubin transporter as well as xenobiotics SLCO1B7 (SLC21A21): SLCO1C1 (SLC21A14): protein is OATP1C1; Na+-independent thyroid hormone uptake into brain; formerly OATPF SLCO2A1 (SLC21A2): protein is OATP2A1; prostaglandin transporter SLCO2B1 (SLC21A9): protein is OATP2B1; formerly OATPB; enriched in microglia; heme import transporter SLCO3A1 (SLC21A11): protein is OATP3A1; formerly OATPD SLCO4A1 (SLC21A12); protein is OATP4A1; formerly OATPE SLCO4C1 (SLC21A20): protein is OATP4C1; transports bile acids, peptides, certain drugs, conjugated steroids, thyroid hormones; formerly OATPX SLCO5A1 (SLC21A15): protein is OATP5A1; formerly OATPJ SLCO6A1: protein is OATP6A1; formerly OATPY |
| 22 | organic cation transporters (OCT), zwitterion/cation transporters (OCTN) and organic anion transporters (OAT) | SLC22A1 (OCT1), SLC22A2 (OCT2), SLC22A3 (OCT3), SLC22A4 (OCTN1), SLC22A5 (OCTN2), SLC22A6 (OAT1), SLC22A7 (OAT2), SLC22A8 (OAT3), SLC22A9 (OAT7), SLC22A10 (OAT5), SLC22A11 (OAT4), SLC22A12 (URAT1), SLC22A13 (OAT10), SLC22A14 (OCTL2), SLC22A15, SLC22A16, SLC22A17, SLC22A18, SLC22A20 (OAT6), SLC22A23, SLC22A24, SLC22A25, SLC22A31 SLC22A1 (OCT1): Na+-independent bidirectional transporter of endogenous bioactive amines (such as the trace amines), choline, cationic drugs, and xenobiotics; highest levels of expression are in the basolateral membranes of hepatocytes; also transports acylcarnitines SLC22A2 (OCT2): Na+-independent bidirectional transporter of bioactive amines (such as the trace amines), choline, cationic drugs, and xenobiotics; highest levels of expression are in the liver and the proximal tubules of the kidney SLC22A4 (OCTN1): organic cation transport in exchange for H+ uptake from lumen of gut and kidney tubules; reabsorption of carnitine, ergothioneine (thiourea derivative of histidine made by bacteria), and acetylcholine SLC22A5 (OCTN2): required for active cellular uptake of carnitine SLC22A6 (OAT1): uric acid transporter involved in uric acid uptake from blood into proximal tubule cells of kidney SLC22A7 (OAT2): uric acid transporter involved in uric acid uptake from blood into proximal tubule cells of kidney SLC22A8 (OAT3): uric acid transporter involved in uric acid uptake from blood into proximal tubule cells of kidney SLC22A11 (OAT4): present in apical membrane of cells in the proximal tubule of kidney; involved in uric acid reabsorption SLC22A12 (URAT1): the major uric acid transporter for urate reabsorption in the apical membrane of cells in the proximal tubule of kidney; target for uricosuric drugs (e.g. probenecid) SLC22A13 (OAT10): uric acid transporter present in apical membrane of cells in the proximal tubule of kidney; also involved in nicotinic acid exchange in the nephron; nicotinic acid and nicotinamide uptake transporter in the small intestines SLC22A15: ergothioneine (thiourea derivative of histidine made by bacteria) transporter SLC22A16: also called carnitine transporter 2, CT2 SLC22A18: plays a role in pancreatic β-cell insulin secretion in response to glucose; found in the imprinted region of chromosome 11 associated with Beckwith-Wiedemann syndrome (BWS) SLC22A24: transporter of steroid conjugates, bile acids, and other dicarboxylic acids |
| 23 | Na+–dependent vitamin C cotransporters (SVCT) | SLC23A1(SVCT1), SLC23A2(SVCT2), SLC23A3(SVCT3) SLC23A1: found in intestinal and renal epithelial cells where it is responsible for ascorbic acid uptake from the diet and reabsorption within the proximal tubules of the kidneys, respectively SLC23A2: present in the plasma membrane of all nucleated cells, responsible for ascorbate uptake from the blood |
| 24 | Na+/(Ca2+–K+) exchangers (NCKX) | SLC24A1(NCKX1), SLC24A2(NCKX2), SLC24A3(NCKX3), SLC24A4(NCKX4), SLC24A5(NCKX5) SLC24A1: expressed in platelets and rod photoreceptors; mutations cause congenital stationary night blindness SLC24A2: expressed in retinal cone cells and brain; mediates light-induced decreases in free Ca+ SLC24A4: involved in skin, hair, and eye pigmentation SLC24A5: involved in skin, hair, and eye pigmentation; mutations are associated with oculocutaneous albinism type VI |
| 25 | mitochondrial carriers | SLC25A1 – SLC25A53 SLC25A1: is an inner mitochondrial membrane citrate transporter; transports citrate out of mitochondria in exchange for transport of malate into mitochondria; also identified as the mitochondrial citrate (tricarboxylate) carrier (CiC) SLC25A2: is commonly referred to as ORNT2; ornithine transporter SLC25A3: is an inner mitochondrial membrane copper transporter; also functions as a phosphate-proton (H+) symporter; also identified as PIC (Pi Carrier) SLC25A4, SLC25A5, and SLC25A6: all three are mitochondrial ADP/ATP (adenine nucleotide) transporters; transport ADP into matrix and ATP out to cytosol; also identified as AAC1 (ADP/ATP Carrier 1), AAC2, and AAC3, respectively SLC25A7: is uncoupling protein 1 (UCP1) SLC25A8: is uncoupling protein 2 (UCP2) SLC25A9: is uncoupling protein 3 (UCP3) SLC25A10: is a mitochondrial dicarboxylic acid transporter; transports succinate to the inner membrane space and then transporters of the VDAC family transport the succinate across the outer mitochondrial membrane to the cytosol; also identified as dicarboxylate carrier (DIC) SLC25A11: is an inner mitochondrial membrane malate and 2-oxoglutarate (α-ketoglutarate) transporter; transports 2-oxoglutarate out of mitochondria in exchange for transport of malate into mitochondria; also identified as oxoglutarate carrier (OGC) SLC25A12: is commonly called aralar; transports aspartate out of the mitochondria in exchange for glutamate; also called aspartate-glutamate carrier 1 (AGC1) SLC25A13: is commonly called citrin, transports aspartate out of (or into) mitochondria in exchange for glutamate; also called aspartate-glutamate carrier 2 (AGC2); mutations in gene associated with type II citrullinemia (CTLN2) SLC25A14: is a UCP-related protein identified as brain mitochondrial carrier protein or UCP5 SLC25A15: is commonly identified as ORNT1; is an ornithine transporter; also transports citrulline, arginine, and lysine; mutations in gene are associated with hyperornithinemia-hyperammonemia-homocitrullinemia (HHH) syndrome; major mitochondrial serine transporter particularly in colorectal carcinomas SLC25A16: inner mitochondrial membrane coenzyme A (CoASH) transporter; is also known as a Graves disease autoantigen SLC25A18: inner mitochondrial membrane glutamate/H+ symporters SLC25A19: transports thiamine pyrophosphate (TPP) into mitochondria; also identified as mitochondrial thiamine pyrophosphate transporter (MTPPT) SLC25A20: is also called carnitine-acylcarnitine translocase (CACT) as well as carnitine-acylcarnitine carrier (CAC) SLC25A21: oxoadipic acid carrier (ODC) SLC25A22: inner mitochondrial membrane glutamate/H+ symporters SLC25A23, SLC25A24, and SLC25A25: all three are ATP-Mg/Pi transporters that carry out the electroneutral antiport of ATP-Mg (but also ATP, ADP, and AMP) and Pi; all three possess an N-terminal Ca2+-regulatory domain; are also identified as APC2 (ATP Phosphate Carrier 2), APC1, and APC3, respectively SLC25A26: transporter of S-adenosylmethionine (SAM or AdoMet); also identified as SAM carrier (SAMC) SLC25A27: is uncoupling protein 4 (UCP4) SLC25A28: is required for transport of ferrous (Fe2+) iron across the inner mitochondrial membrane where is can be used for heme biosynthesis; transporter is also known as mitoferrin 2 (MFRN2) SLC25A29: is mitochondrial ornithine transporter 3 (ORNT3); also identified as BAC (Basic Amino acid Carrier) SLC25A31: is a mitochondrial ADP/ATP (adenine nucleotide) transporter; transports ADP into matrix and ATP out to cytosol; also identified as AAC4 SLC25A32: is likely to be a mitochondrial formate transporter SLC25A35: recently (2026) identified as a mitochondrial phosphoenolpyruvate (PEP) exporter and as such promotes triglyceride synthesis SLC25A37: is required for transport of ferrous (Fe2+) iron across the inner mitochondrial membrane where it can be used for heme biosynthesis; transporter is also known as mitoferrin 1 (MFRN1); may also be involved in import of glutathione into the mitochondria SLC25A38: is required for transport of glycine across the inner mitochondrial membrane where it can be used for heme biosynthesis; also identified as glycine carrier (GLYC) SLC25A39: is required for import of glutathione into the mitochondria SLC25A40: likely involved in import of glutathione into the mitochondria SLC25A41: is an ATP-Mg/Pi transporter that carry out the electroneutral antiport of ATP-Mg (but also ATP, ADP, and AMP) and Pi; lacks the N-terminal Ca2+-regulatory domain present in SLC25A23, SLC25A24, and SLC25A25; also identified as APC4 SLC25A42: inner mitochondrial membrane coenzyme A (CoASH) transporter; mutation of Asn 291 to Asp (N291D) is found in several cases of mitochondrial encephalomyopathy SLC25A44: brown adipose tissue mitochondrial import transporter for branched-chain amino acids (BCAA); this facilitates the utilization of BCAA for the process of thermogenesis SLC25A45: mitochondrial import transporter for methylated amino acid derivatives such as trimethyllysine (TML) and asymmetric dimethylarginine (ADMA) SLC25A48: highly expressed in brown adipose tissue, liver, and kidney; required for mitochondrial import of choline which is required for the synthesis of betaine (trimethylglycine) that is involved in one-carbon metabolism; only SLC25 family transporter upregulated in response to high-fat diet SLC25A49: is mitochondrial carrier 1 which is encoded by the MTCH1 gene; also known as presenilin-associated protein SLC25A51: also known as mitochondrial carrier triple repeat protein 1 (MCART1); is the mitochondrial NAD+ import transporter; critical for mitochondrial electron transport functions and TCA cycle activity |
| 26 | multifunctional anion exchangers | SLC26A1 – SLC26A11; SLC26A10 is pseudogene SLC26A1: also known as SAT1 (sulfate anion transporter 1); hepatic, renal, and intestinal sulfate, oxalate, and glyoxalate transporter; mutations in gene cause hyperoxaluria and formation of calcium oxalate kidney stones SLC26A2: chloride, sulfate, and oxalate transporter which is mutated in diastrophic dysplasia (DTD) which is associated with reduced sulfation of proteoglycans in cartilage matrices SLC26A3: essential for intestinal chloride uptake; also known as DRA (down regulated in adenoma); mutations in gene are associated congenital chloride diarrhea, as disorder characterized by extensive loss of chloride in acidic stools accompanied by metabolic alkalosis SLC26A4: also called pendrin, expressed at significant levels in the thyroid gland where it is involved in iodine homeostasis and thyroid hormone biosynthesis; also functions in chloride and bicarbonate transport in kidney and ear epithelial cells; mutations in gene result in Pendred syndrome (PDS) which is associated with the most common form of syndromal deafness, also results in enlarged vestibular aqueduct and goiter SLC26A5: also called prestin, is a motor protein of cochlear outer hair cells; chloride, sulfate, oxalate, and formate transporter SLC26A6: apical membrane localized chloride, bicarbonate, oxalate, and formate transporter in kidney, intestines, and pancreas; major oxalate efflux transporter of intestines and kidneys SLC26A7: chloride, sulfate, and bicarbonate transporter in gastric parietal cells SLC26A8: expression restricted to spermatocytes SLC26A9: chloride and bicarbonate transporter in gastric parietal cells, airway epithelial cells, and kidney |
| 27 | fatty acid transporters (FATP) | SLC27A1 (FATP1), SLC27A2 (FATP2), SLC27A3 (FATP3), SLC27A4 (FATP4), SLC27A5 (FATP5), SLC27A6 (FATP6) SLC27A1/FATP1: is also known as acyl-CoA synthetase very long-chain family, member 5 (ACSVL5); broad specificity for long and very long chain fatty acids; transports C16:0, C18:1, C20:4, and C24:0 SLC27A2/FATP2: is also known as acyl-CoA synthetase very long-chain family, member 1 (ACSVL1) and very long-chain acyl-CoA synthetase (VLACS or VLCS); transports C16:0, C24:0, bile acids, and cholestanoic acids SLC27A3/FATP3: is also known as acyl-CoA synthetase very long-chain family, member 3 (ACSVL3); transports multiple long chain fatty acids SLC27A4/FATP4: is also known as acyl-CoA synthetase very long-chain family, member 4 (ACSVL4); transports C18:1 and C20:4 SLC27A5/FATP5: is also known as acyl-CoA synthetase very long-chain family, member 6 (ACSVL6) and very long-chain acyl-CoA synthetase-related protein (VLACSR) or very long-chain acyl-CoA synthetase homolog 2 (VLCSH2); also known as bile acid-CoA synthetase (BACS); transports multiple long chain fatty acids; catalyzes the activation of bile acids via formation of bile acid-CoA thioesters; exclusive to the liver; localized to the endoplasmic reticulum (ER) SLC27A6/FATP6: is also known as as acyl-CoA synthetase very long-chain family, member 2 (ACSVL2) and very long-chain acyl-CoA synthetase homolog 1 (VLCSH1); transport preference for C16:0, C18:1, and C18:2; will not transport fats less than 10 carbons FATP function is discussed in the Lipolysis and the Oxidation of Fatty Acids page |
| 28 | Na+–dependent concentrative nucleoside transport (CNT) | SLC28A1, SLC28A2, SLC28A3 SLC28A1: also identified as CNT1; import transporter for uridine, cytidine, and adenosine; is essential for nucleotide synthesis via salvage pathways SLC28A2: also identified as CNT2; purine (primarily adenosine) selective import transporter SLC28A3: also identified as CNT3; nucleoside import transporter |
| 29 | equilibrative nucleoside transporters (ENT) | SLC29A1(ENT1), SLC29A2(ENT2), SLC29A3(ENT3), SLC29A4(ENT4) SLC29A1(ENT1): highest preference for adenosine; present in the plasma membrane and mitochondrial membrane SLC29A2(ENT2): transports both purine and pyrimidine nucleosides and also hypoxanthine SLC29A3(ENT3): transports adenosine, adenine, and uridine as well as several nucleoside analog drugs SLC29A4(ENT4): organic cation transporter; transports dopamine, serotonin, epinephrine, and norepinephrine; does not transport nucleosides or nucleoside analogs |
| 30 | efflux and compartmentalization of zinc (ZNT) | SLC30A1 – SLC30A10 SLC30A1: encodes the zinc efflux transporter ZnT1; also involved in the uptake of copper (Cu2+) into cells SLC30A5: zinc transporter SLC30A6: zinc transporter SLC30A7: zinc transporter SLC30A8: encodes the zinc efflux transporter identified as ZnT8; ZnT8 autoantibodies are common in type 1 diabetes; polymorphisms in the gene encoding SLC30A8 are associated with increased diabetes risk |
| 31 | copper transporters (CTR) | SLC31A1(CTR1), SLC31A2(CTR2): these mediate uptake of dietary copper ATP7A and ATP7B are related copper transporting P-type ATPases that mediate copper uptake and export ATP7A is defective in Menkes disease ATP7B is defective in Wilson disease |
| 32 | vesicular inhibitory amino acid transporter (VIAAT) | SLC32A1: also called vesicular GABA transporter (VGAT) |
| 33 | acetyl-CoA transporter (ACATN) | SLC33A1: transports acetyl-CoA into the endoplasmic reticulum (ER); transports oxidized glutathione (GSSG) out of the ER which helps to prevent ER-mediated stress responses; mutation in gene result in lethal disorder identified as Huppke-Brendel syndrome (HPBDS); HPBDS is associated with severe psychomotor impairment, congenital hearing loss, bilateral cataracts, and low serum ceruloplasmin and copper |
| 34 | type II Na+–phosphate co-transporters (NPT) | SLC34A1: also identified as NPT2a; major transporter involved in renal phosphate reabsorption; primarily expressed in the proximal tubule SLC34A2: also identified as NPT2b; involved in intestinal phosphate uptake from the lumen; primarily expressed in intestinal enterocytes, liver, and testis SLC34A3: also identified as NPT2c; major transporter involved in renal phosphate reabsorption; primarily expressed in the proximal tubule |
| 35 | nucleoside sugar transporters | 30 family members in humans divided into seven subfamilies identified as A through G SLC35C1: also identified as the GDP-fucose transporter (gene symbol: FUCT1); defects in gene result in the congenital disorder of glycosylation type IIc more commonly called leukocyte adhesion deficiency syndrome II (LAD II) |
| 36 | proton–coupled amino acid transporters | SLC36A1, SLC36A2, SLC36A3, SLC36A4 SLC36A1: prevalent in apical membranes of intestinal enterocytes; alanine, glycine, and proline transporter; also known as proton-coupled amino acid transporter 1 (PAT1); can also transport GABA SLC36A2: glycine and proline transporter; also known as PAT2 SLC36A3: precise function unknown; also known as PAT3 SLC36A4: alanine, proline, and tryptophan transporter; also known as PAT4 |
| 37 | sugar–phosphate/phosphate exchangers (SPX) | SLC37A1, SLC37A2, SLC37A3, SLC37A4 SLC37A4: also known as glucose-6-phosphate transporter-1 (G6PT1) which is defective in glycogen storage disease type1b |
| 38 | sodium–coupled neutral amino acid (system N and system A) transporters (SNAT) | System A family includes SLC38A1, SLC38A2, SLC38A4 SLC38A1 (SNAT1; ATA1): plasma membrane-localized for amino acid uptake; mediates Na+–coupled transport of neutral amino acids with preference for alanine, asparagine, cysteine, glutamine, histidine, and methionine; also transports glycine and serine into cells SLC38A2 (SNAT2; ATA2): prevalent glutamine transporter in basolateral membranes of intestinal enterocytes for delivery to the portal circulation; plasma membrane glutamate transporter; also transports serine, glycine, and alanine into cells SLC38A4 (SNAT4; ATA3): Na+-dependent neutral amino acid transporter with broad specificity; may also transport arginine and lysine in a Na+– and pH–independent process; prevalent in apical membranes of intestinal enterocytes System N family includes SLC38A3, SLC38A5, SLC38A7 SLC38A3 (SNAT3; SN1): expressed at highest levels in the liver, pancreas, and skeletal muscle; also expressed in brain, heart, and kidney; involved in both efflux and uptake of glutamine; also transports asparagine and histidine SLC38A5 (SNAT5: SN2): prevalent in apical membranes of intestinal enterocytes; mediates Na+–coupled transport of neutral amino acids with preference for glutamine, asparagine, and histidine SLC38A7 (SNAT7): expressed at high levels in both excitatory and inhibitory neurons and exhibits a preference for glutamine SLC38A9 (SNAT9): is amino acid sensitive regulator of the late endosome and lysosome (LEL)-localized mTORC1 through its interaction with a protein of the complex identified as ragulator SLC38A6 (SNAT6): an orphan member of the SLC38 family not yet fully characterized as to whether it is in the system A or N sub-family; there are 4 additional recently described orphan members SLC38A8, SLC38A10, SLC38A11 |
| 39 | metal ion transporters (ZIP) | SLC39A1 – SLC39A14 SLC39A8: commonly identified as ZIP8; the ZIP family of transporters are so-called from Zinc-regulated transporter, Iron-regulated transporter-like Proteins; transports zinc, iron, cadmium, and manganese; mutations in the SLC39A8: gene are associated with manganese deficiency SLC39A11: also known as ZIP11; facilitates Zn2+ uptake; associated with pancreatic β-cell insulin secretion in response to glucose SLC39A14: also identified as ZIP14; functions in the transport of zinc, iron, cadmium, and manganese; highest level of expression are in the liver; mutations in the SLC39A14 gene are associated with manganese overload |
| 40 | basolateral iron transporter | SLC40A1: more commonly known as as ferroportin, but is also known as iron-regulated gene 1 (IREG1) or reticuloendothelial iron transporter (MTP1); was also identified as SLC11A3 which is no longer used |
| 41 | MgtE–like magnesium transporters | SLC41A1, SLC41A2, SLC41A3 SLC41A1: also known as MgtE where MgtE is a divalent cation transporter first identified in the bacteria Chlamydomonas reinhardtii |
| 42 | Rh ammonia and ammonium ion transporters | SLC42A1(RhAG), SLC42A2(RhBG), SLC42A3(RhCG) these transporters are named for the Rh blood–group antigens; e.g. RhAG is encoded by the RHAG gene which is also designated as the CD241 gene (cluster of differentiation 241) RHBG (SLC42A2): ammonia uptake from blood in kidney RHCG (SLC42A3): ammonia uptake from blood in kidney |
| 43 | Na+–independent, system L amino acid transporters L-type amino acid transporters, LAT | SLC43A1 (LAT3), SLC43A2 (LAT4), SLC43A3 SLC43A1: Na+-independent transport of large neutral amino acids with preferences for isoleucine, leucine, phenylalanine, and valine SLC43A2: Na+-, Cl–-, and pH-independent neutral amino acid transporter; prevalent in apical membranes of intestinal enterocytes; preference for isoleucine, leucine, methionine, phenylalanine, and valine; major methionine transporter in cancer cells SLC43A3: import transporter for purine nucleobases adenine, guanine, and hypoxanthine |
| 44 | choline–like transporters | SLC44A1, SLC44A2, SLC44A3, SLC44A4, SLC44A5 SLC44A1: Na+-dependent choline transporter; also known as CTL1 SLC44A2: Na+-dependent choline transporter; may also transport ethanolamine; also known as CTL2; localized to plasma and mitochondrial membranes SLC44A2: Na+-dependent choline transporter; may also transport ethanolamine; also known as CTL3; localized to plasma and mitochondrial membranes; also known as ENBT1 (equilibrative nucleobase transporter 1) |
| 45 | glycoside-pentoside-hexuronide cation symporters | SLC45A1, SLC45A2, SLC54A3, SLC45A4 SCL45A4: transporter for N-acetylglucosamine (GlcNAc) and uptake of extracellular polyamines |
| 46 | folate transporter family | SLC46A1, SLC46A2 SLC46A1: is a H+-coupled folate transporter; also functions as a heme transporter for heme uptake in the duodenum and as such is also called heme carrier protein 1 (HCP1) |
| 47 | multidrug and toxin extrusion (MATE) | SLC47A1(MATE1), SLC47A2(MATE2-K) SLC47A1: creatine and organic anion efflux from proximal tubule cells of kidney SLC47A2: creatine and organic anion efflux from proximal tubule cells of kidney |
| 48 | heme transporters | SLC48A1 (HRG-1): essential for heme-iron recycling in reticuloendothelial macrophages during erythrophagocytosis |
| 49 | major facilitator transporters, heme transporters | first family member originally identified as feline leukemia virus subgroup C cellular receptor 1 (FLVCR1) SLC49A1 – SLC49A4 SLC49A1: official gene symbol is FLVCR1; regulates erythropoiesis by controlling the rate of heme efflux from the mitochondria to the cytosol; protects erythroid cells from heme toxicity; also transports extracellular choline into cells SLC49A2: official gene symbol is FLVCR2; is a Ca2+ transporter; involved in brain vascular endothelial cell development; unlike FLVCR1-encoded protein, this protein does not bind feline leukemia virus envelope protein; also transports extracellular choline into cells |
| 50 | sugar efflux transporters | SLC50A1: glucose transporter; also identified as SWEET1 |
| 51 | steroid-derived molecule transporters | SLC51A, SLC51B SLC51A: encodes organic solute transporter α-chain (OSTA, OSTα); functions as heterodimer with SLC51B; is a 7-transmembrane domain protein SLC51B: encodes organic solute transporter β-chain (OSTB, OSTβ); functions as heterodimer with SLC51A; is a single pass transmembrane protein the SLC51A:SLC51B heterodimer is expressed in basolateral membranes of cells in nearly all tissues with highest levels in small intestine, liver, kidney, and steroidogenic tissues; transports bile acids, steroids, and prostaglandin E2 (PGE2); can act as efflux or uptake transporter dependent upon substrate concentration |
| 52 | riboflavin transporter family (RFVT) | SLC52A1, SLC52A2, SLC52A3 SLC52A1: also known as GPR172B; mutations in SLC52A1 cause maternal riboflavin deficiency resulting in neonatal glutaric acidemia type 2 SLC52A2: mutations in SLC52A2 are associated with Brown-Vialetto-van Laere syndrome type 2 (BVVLS2); is a disorder that primarily affects motor neurons and causes sensorineural hearing loss, bulbar palsy (weakness in face/throat), and respiratory issues SLC52A3: mutations in SLC52A3 are associated with Brown-Vialetto-van Laere syndrome type 1 (BVVLS1); is a disorder that primarily affects motor neurons and causes sensorineural hearing loss, bulbar palsy (weakness in face/throat), and respiratory issues |
| 53 | phosphate carriers | SLC53A1: protein was originally identified as the receptor for the xenotropic and polytropic classes of murine leukemia viruses and thus termed XPR1 |
| 54 | mitochondrial pyruvate carriers (MPC) | SLC54A1, SLC54A2, SLC54A3 SLC54A1: protein is mitochondrial pyruvate carrier 1 (MPC1) SLC54A2: protein is MPC2 SLC54A3: protein is MPC1-like (MPC1L) |
| 55 | mitochondrial cation/proton exchangers | SLC55A1, SLC55A2, SLC55A3 SLC55A1: protein is LETM1 (leucine zipper and EF-hand containing transmembrane protein 1) SLC55A2: protein is LETM2 SLC55A3: protein is LETMD1 (LETM1-domain containing 1) |
| 56 | sideroflexins | SLC56A1, SLC56A2, SLC56A3, SLC56A4, SLC56A5 SLC56A1: protein is sideroflexin 1; gene identified as SFXN1; functions as a mitochondrial alanine and serine transporter SLC56A2: gene identified as SFXN2 SLC56A3: gene identified as SFXN3; functions as a mitochondrial serine transporter SLC56A4: gene identified as SFXN4 SLC56A5: gene identified as SFXN5 |
| 57 | NiPA-like magnesium transporter family | SLC57A1, SLC57A2, SLC57A3, SLC57A4, SLC57A5, SLC57A6 SLC57A1: protein is identified as NIPA1 (non-imprinted in Prader-Willi/Angelman syndrome region protein 1) SLC57A2: protein is NIPA2 SLC57A3: protein is NIPA-like 1 (NIPAL1) SLC57A4: protein is NIPAL2 SLC57A5: protein is NIPAL3 SLC57A6: protein is NIPAL4 |
| 58 | MagT-like magnesium transporter family | SLC58A1, SLC58A2 SLC58A1: encoded protein is MAGT1 (MAGnesium Transporter 1) SLC58A2: encoded protein is tumor suppressor candidate 1 (TUSC1) |
| 59 | Sodium-dependent lysophosphatidylcholine symporter family | SLC59A1, SLC59A2, SCL59B1 SCL59A1: gene name is MFSD2A (Major Facilitator Superfamily Domain containing 2A); involved in the transport of docosahexaenoic acid (DHA) across blood brain barrier (BBB) SLC59A2: gene name is MFSD2B; transport sphingosine-1-phosphate (S1P) SLC59B1: gene name is MFSD12; transports cysteine |
| 60 | glucose transporters | SLC60A1, SLC60A2 SLC60A1: also known as MFSD4A (Major Facilitator Superfamily Domain containing 4A) SLC60A2: also known as MFSD4B |
| 61 | Molybdate transporter family | SLC61A1: protein identified as MFSD5 (Major Facilitator Superfamily Domain containing 5) |
| 62 | Pyrophosphate transporters | SLC62A1: originally identified as progressive ANKylosis protein Homolog (ANKH) |
| 63 | Sphingosine-phosphate transporters | SLC63A1, SLC63A2, SLC63A3 SLC63A1: gene identified by original identification nomenclature which is Spinster homolog lysolipid transporter 1 (SPNS1) where Spinster is a lifespan regulating gene in Drosophila melanogaster; lysosomal transporter of lysophosphatidylcholine, lysoPC SLC63A2: gene identified as SPN2 SLC63A3: gene identified as SPN3 |
| 64 | Golgi Ca2+/H+ exchangers | SLC64A1: originally identified as transmembrane protein 165 (TMEM165) |
| 65 | NPC-type cholesterol transporters | SLC65A1, SLC65A2 SLC65A1: more commonly identified as NPC1 (Niemann-Pick type C1) SLC65A2: more commonly identified as NPC1L1 (Niemann-Pick type C1 like; the NPC1L1 transporter is the target of the drug, ezetimibe, used to treat hypercholesterolemia |
| 66 | PQ-loop amino acid transporters | SLC66A1, SLC66A2, SLC66A3, CTNS, MPDU1 CTNS: cystinosin, lysosomal cystine transporter; mutations in CTNS gene is the cause of the lysosomal storage disorder termed cystinosis; three forms of disorder identified with infantile being the most common MPDU1: mannose-P-dolichol utilization defect 1; mutations in MPDU1 gene are the causes of the congenital disorder of glycosylation (CDG) identified as CDG-1f; disorder is associated with dwarfism, hypotonia, frequent seizures, severe psychomotor impairment and cerebral atrophy; affected individuals have severe failure to thrive after birth |
| 67 | organic cation transporter | SLC67A1 |
| 68 | SLC68A1 | |
| 69 | vitamin A receptor/transporter family | SLC69A1 (STRA6) gene is identified as STRA6 (signaling receptor and transporter of retinol STRA6); serves as a receptor for complexes of retinol and retinol binding protein to facilitate transport of retinol into cells |
| 70 | cyclin and CBS domain divalent metal cation transport mediators (CNNM) | SLC70A1, SLC70A2, SLC70A3, SLC70A4 SLC70A1: gene is identified as CNNM1 (cyclin and CBS domain divalent metal cation transport mediator 1) SLC70A2: gene is identified as CNNM2 SLC70A3: gene is identified as CNNM3 SLC70A4: gene is identified as CNNM4 |
| 71 | putative ammonia transporter family | SLC71A1, SLC71A2 |
| 72 | lysosomal solute carrier family, MFSD1 family | SLC72A1 (MFSD1) gene is identified as MFSD1 (Major Facilitator Superfamily Domain containing 1) |
| 73 | MFSD6 transporter family | SLC73A1, SLC73A2 SLC73A1: gene is MFSD6 (Major Facilitator Superfamily Domain containing 6) SLC73A2: gene is MFSD6L (Major Facilitator Superfamily Domain containing 6 like) |
| 74 | lysosomal chloride channel family | SLC74A1 (MFSD8) gene is identified as MFSD8 (Major Facilitator Superfamily Domain containing 8); mutations in the MFSD8 gene are the cause of the lysosomal disorder identified as ceroid-lipofuscinoses neuronal 7 (CNL7) |
| 75 | tetracycline transporter-like family | SLC75A1 (MFSD10) also known as MFSD10 (Major Facilitator Superfamily Domain containing 10) |
| 76 | glycolipid translocator family | SLC76A1 (RFT1) gene is identified as RFT1 (RFT1 glycolipid translocator homolog 1); RFT1 is the human homolog of the yeast gene called “requiring fifty-three” because mutations in the gene could be suppressed by human tumor suppressor protein p53; mutations in the RFT1 gene are the cause of the congenital disorder of glycosylation (CDG) identified as CDG-1n |
Pharmacogenomics of SLC Family Transporters
The genes encoding SLC family transporters have been shown to harbor numerous single nucleotide polymorphisms (SNPs), many of which result in changes to the amino acid sequences of the encoded proteins. Many of these SNPs in SCL genes result in clinically relevant pharmacogenetic alterations in the functions of the encoded transporters. These clinically relevant alterations include effects that modify individual patient responses to analgesics, antidiabetic medications, anticoagulants, and chemotherapeutic drugs.
The SLC19A1 encoded transporter is responsible for the uptake of reduced folate and thiamine and as such is also known as the reduced folate transporter 1, RFT1. Polymorphisms in the SLC19A1 gene are associated with increased risk for toxicity associated with the use of methotrexate, particularly when used to treat juvenile forms of acute lymphoblastic leukemia (ALL).
The SLC22A1 encoded transporter (organic cation transporter-1: OCT1) is a Na+-independent transporter that transports a number of organic compounds, both endogenous and exogenous. Polymorphisms in the SCL22A1 gene are the most common of all the organic cation transporter (OCT) encoding genes. One important drug, used to treat the hyperglycemia of type 2 diabetes, that is transported by OCT1 is metformin. Common SNPs in SLC22A1 have been repeatedly linked to altered drug disposition and efficacy of metformin. Four variants of OCT1 (R61C, G401S, M420del, and G465R), are associated with reduced metformin transport. All four of these SLC22A1 polymorphisms have been found at frequencies as high as 22% in most populations excluding East Asians where they have not been identified. These SLC22A1 polymorphisms are all associated with reduced glucose-lowering effects of metformin. In addition to metformin, these SLC22A1 polymorphisms are associated with altered responses to the chemotherapeutic, imatinib (Gleevec), used in the treatment of leukemias, in particular chronic myelogenous leukemia (CML).
Another SLC gene whose encoded protein plays a role in the efficacy of drugs used to treat type 2 diabetes is SLC30A8. A polymorphism that is common in East Asians and Africans is rs16889462 (defines a Reference SNP cluster identity) which is a missense mutation of the Arg residue at position 325 for a Glu residue (R325Q). This polymorphism is associated with increased efficacy of the drug repaglinide (Prandin). Repaglinide is a member of the meglitinide family of non-sulfonylurea drugs that are classified as insulin secretagogues.
Clinical Significances of Transporter Defects
As might be expected, defects in the expression and/or function of membrane transporters leads to the manifestation of numerous clinical disorders. It is not the intention of this section to cover all disorders related to defects in membrane transporters but to highlight several with emphasis on diseases that have been mentioned throughout the pages of this web site or in specific disease discussion pages such as the Cystinuria page and the Hartnup disorder page.
ABCA1 is involved in the transport of cholesterol out of cells when HDLs are bound to their cell surface receptor, SR-B1 (see the Lipoproteins, Blood Lipids, and Lipoprotein Metabolism page for more details). One important consequence of the activity of ABCA1 in macrophages is that the efflux of cholesterol results in a suppression of inflammatory responses triggered by macrophages that have become foam cells due to cholesterol uptake. Defects in ABCA1 result in Tangier disease which is characterized by two clinical hallmarks; enlarged lipid-laden tonsils and low serum HDL.
ABCB4 is a member of the P-glycoprotein family of multidrug resistance transporters. Defects in the ABCB4 gene are associated with 6 liver diseases: progressive familial intrahepatic cholestasis type 3 (PFIC3), adult biliary cirrhosis, transient neonatal cholestasis, drug-induced cholestasis, intrahepatic cholestasis of pregnancy, and low phospholipid-associated cholelithiasis syndrome.
ABCG5 and ABCG8 form an obligate heterodimer pair that function to limit plant sterol and cholesterol uptake by the gut and mediate cholesterol efflux from the liver into the bile. Mutations in either ABCG5 or ABCG8 result in a rare genetic disorder identified as sitosterolemia (also called phytosterolemia). This disorder is characterized by unrestricted absorption of plant sterols (such as sitosterol) and cholesterol. Individuals afflicted with this disorder manifest with very high levels of plant sterols in the plasma and develop tendon and tuberous xanthomas, accelerated atherosclerosis, and premature coronary artery disease.
ABCB7 is a protein localized to the inner mitochondrial membrane and is involved in iron homeostasis. Defects in the gene are associated with X-linked sideroblastic anemia with ataxia (XSAT) which is characterized by an infantile to early childhood onset of non-progressive cerebellar ataxia and mild anemia with hypochromia and microcytosis.
ABCB11 is also known as bile salt export protein (BSEP) which is involved in bile salt transport out of hepatocytes. Defects in the gene are associated with progressive familial intrahepatic cholestasis type 2 (PFIC2).
ABCC2 was first identified as the canalicular multispecific organic anion transporter (CMOAT) and is also called multidrug resistance associated protein 2 (MRP2). Defects in the gene encoding ABCC2 result in Dubin-Johnson syndrome which is a form of conjugated hyperbilirubinemia.
ABCD1 is involved in the import and/or anchoring of very long-chain fatty acid-CoA synthetase (VLCFA-CoA synthetase) to the peroxisomes. Defects in the gene result in X-linked adrenoleukodystrophy (XALD).
ATP7A and ATP7B are copper transporting ATPases that are related to SLC31A1. Defects in ATP7A result in Menkes disease and defects in ATP7B are associated with Wilson disease.
Defects in many members of the SLC6 family are associated with intellectual impairment, affective disorders, and other neurological dysfunctions. SLC6A1 defects are associated with epilepsy and schizophrenia. SLC6A2 defects are associated with depression and anorexia nervosa. SLC6A3 defects are associated with Parkinsonism, Tourette syndrome, ADHD, and substance abuse. SLC6A4 defects are associated with anxiety disorder, depression , autism, and substance abuse.
SLC5A8 is a Na+-dependent transporter of short-chain fatty acids, lactate, and monocarboxylic acids. The SLC5A8 gene was originally identified as a tumor suppressor gene whose expression was silenced by hypermethylation in various colorectal carcinomas. Subsequent studies demonstrated that SLC5A8 is a major transporter of the short-chain fatty acids, in particular butyrate and propionate, produced through colonic bacterial fermentation of soluble fiber. Butyrate and propionate have been shown to be inhibitors of histone deacetylases, HDAC. Given that HDAC inhibitors are known anticancer compounds, the tumor suppressor of the SLC5A8 protein is most likely related to its role in colonic epithelial cell uptake of butyrate and propionate.
SLC6A19 is also called system B(0) neutral amino acid transporter 1 [B(0)AT1, or B0AT1]. This transporter is involved in neutral amino acid transport with highest levels of expression in the kidney and small intestine. Deficiency in SLC6A19 leads to Hartnup disorder which results from impaired transport of neutral amino acids across epithelial cells in renal proximal tubules and intestinal mucosa. Symptoms include transient manifestations of pellagra-like light sensitive rash, cerebellar ataxia, and psychosis.
SLC11A2, which is also known as DMT1 (divalent metal-ion transporter-1), is involved in the uptake of iron by the apical surface of the duodenum. In addition to iron, DMT1 is involved in manganese, cobalt, cadmium, nickel, copper, and zinc transport. Defects in DMT1 activity are associated with hypochromic microcytic anemia with iron overload.
SLC12A6 defects are associated with Andermann syndrome which is also referred to as agenesis of corpus callosum and peripheral neuropathy (ACCPN). The disorder gets its name from the fact that afflicted individuals have sensory and motor neuropathy associated
SLC16A2 (MCT8) is defective in Allan-Herndon-Dudley syndrome (AHDS) which is characterized by marked hypotonia, weakness, excessive drooling, reduced muscle mass, and delay of developmental milestones in infancy and early childhood. Afflicted individuals have severe impairment of cognitive development but do not exhibit any major malformations.
SLC22A18 is found in the imprinted region of chromosome 11 associated with Beckwith-Wiedemann syndrome (BWS).
SLC30A8 is involved in efflux of zinc and polymorphisms in the gene encoding this transporter are associated with increased risk of the development of type 2 diabetes. The functional defect in this protein results in impaired pancreatic β-cell function leading to defects in insulin secretion.
SLC35C1 is also known as the GDP-fucose transporter (gene symbol: FUCT1). Defects in the FUCT1 gene result in a congenital disorder of glycosylation (CDG). Specifically the disorder is a type II CDG identified as CDG-IIc. Type II CDGs result from defects in the processing of the carbohydrate structures on N-linked glycoproteins. CDG-IIc is also called leukocyte adhesion deficiency syndrome II (LAD II). LAD II is a primary immunodeficiency syndrome which manifests due to leukocyte dysfunction. Symptoms of LAD II include unique facial features (flat face with a depressed nasal bridge, antiverted nostrils, and long eyelashes), recurrent infections, persistent leukocytosis, defective neutrophil chemotaxis, and severe growth and intellectual impairment.
SLC40A1 is a multiple transmembrane spanning protein involved in iron transport. The protein is highly expressed in the intestine, liver, and reticuloendothelial cells. SLC40A1 is more commonly known as ferroportin or iron-regulated transporter 1 (IREG1). The protein is required for the transport of dietary iron across the basolateral membranes of intestinal enterocytes. Defects in the SLC40A1 gene are associated with type 4 hemochromatosis.