
 deficiency")
Last Updated: June 3, 2026
Introduction to MCAD Deficiency
The acyl-CoA dehydrogenases are a family of enzymes involved in the first step of the mitochondrial β-oxidation of fatty acids. Medium-chain acyl-CoA dehydrogenase (MCAD), which is also called acyl-CoA dehydrogenase, medium-chain (ACADM), is so called because of the size range of its fatty acyl-CoA substrates. MCAD acts on saturated fatty acyl-CoA molecules that range in size from 12 down to 4 carbons in length.
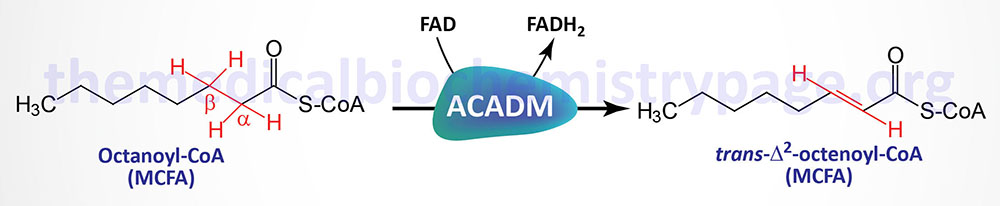
Deficiency in MCAD is the most common defect observed in the process of mitochondrial β-oxidation of fatty acids. In fact, MCAD deficiency is one of the most common inherited disorders of metabolism occurring with a frequency of approximately 1 in 10,000 live births. This disorder has been described in populations world-wide with the most frequently affected individuals being of northwestern European origin.
MCAD deficiency (MCADD) is an autosomal recessive disorder that satisfies the criteria for newborn screening and is in fact one of the diseases that is screened for in the US in all newborns. MCAD deficiency is a common inherited disorder with a frequency that approaches that of PKU. The disease can result in life threatening complications and relatively simple dietary intervention can avert the clinical phenotype of MCAD deficiency.
Molecular Biology of MCAD Deficiency
The MCAD enzyme is encoded by the acyl-CoA dehydrogenase, medium chain (ACADM) gene. The ACADM gene resides on chromosome 1p31.1 spanning 13 exons that generate five alternatively spliced mRNAs each of which encode a distinct precursor isoform of the MCAD enzyme. These different MCAD precursor isoforms are 421 amino acids (isoform a), 425 amino acids (isoform b), 385 amino acids (isoform c; translation starts at alternative start codon), 454 amino acids (isoform d), and 232 amino acids (isoform e; translation initiates at a downstream AUG).
At least 340 mutations in the ACADM gene have been identified. By far, the most prevalent mutation (89%) found in MCAD deficiency patients is a single nucleotide substitution at position 985. This substitution is an A for G change that converts amino acid 329 from a lysine to a glutamic acid (K329E). This mutation alters the α-helical domain of the C-terminal portion of the enzyme. In addition to the A985G substitution mutation several additional nucleotide substitution mutations and insertion and deletion mutations have been identified in MCAD deficiency patients.
Clinical Features of MCAD Deficiency
The most common symptom of MCAD deficiency is episodic hypoketotic hypoglycemia brought on by fasting. Symptoms appear within the first 2 years of life. Clinical crisis is characterized by an infant presenting with episodes of vomiting and lethargy that may progress to seizures and ultimately coma. A prior upper respiratory or gastrointestinal viral infection will lead to reduced oral intake in these infants which can precipitate the acute crisis. The first episode in an infant may be fatal and the death ascribed to sudden infant death syndrome (SIDS). Autopsy results will often find marked fatty liver (hepatic steatosis) and cerebral edema. These findings are sometimes misdiagnosed as Reye syndrome especially in the circumstance where there is reported a prior infection. Because the capacity of the gluconeogenesis pathway is limited in newborn infants, they are highly susceptible to the lack of brain energy from the ketones that would normally be derived from fatty acid oxidation.
The defect in fatty acid oxidation leads to the presence, in the plasma and urine, of toxic metabolic intermediates such as dicarboxylic acids as well as medium-chain acylcarnitines. The presence of the medium chain dicarboxylic acids results from excess medium chain fatty acids being oxidized via the microsomal ω-oxidation pathway. Normally dicarboxylic acids are oxidized in the peroxisomes but medium chain dicarboxylic acids are not good substrates for the pathway and so these lipids end up accumulating in the blood and urine.
Characteristic and diagnostic of MCAD deficiency is the presence of octanoylcarnitine (a medium-chain length acylcarnitine). Diagnosis can be made within 24hr to 48hr in specialized laboratories using tandem mass spectrometry (MS) of the blood for specific acylcarnitines. In MCAD deficiency the acylcarnitines that are found are highly diagnostic and specific for this disorder and include C6:0-, 4-cis-, and 5-cis-C8:1, C8:0, and 4-cis-C10:1 acylcarnitine species. The nomenclature C6:0, for example, refers to a fatty acid 6 carbons in length with no sites of unsaturation, whereas, C8:1 refers to a 8 carbon fatty acid with 1 site of unsaturation.
When medium-chain length fatty acids are oxidized in the mitochondria there is no requirement for them first being attached to carnitine as is the case for long-chain length fatty acids. The lack of requirement for carnitine attachment is also true for short-chain length fatty acid oxidation. The mechanism that leads to the accumulation of medium-chain acylcarnitines in MCAD deficiency stems from the need for the mitochondria to replenish the CoASH pool for continued oxidation of long-chain fatty acids. When a long-chain fatty acyl-CoA is oxidized down to medium-chain length it can no longer be further oxidized due to the deficiency in MCAD. This results in the “trapping” of CoA as medium-chain acyl-CoAs. The inner mitochondrial membrane localized carnitine palmitoyltransferase 2 (CPT2) will exchange the CoA, from these accumulating medium-chain acyl-CoAs, for carnitine, thereby replenishing the CoASH pool for use by long-chain fatty acids. This mechanism results in the accumulation of the medium-chain acylcarnitines typically seen in MCAD deficient patient.
Treatment of MCAD Deficiency Patients
The primary goal of treatment for MCAD deficiency patients is to provide adequate caloric intake, the avoidance of fasting, IV glucose to treat acute episodes, and aggressive therapy during periods of infection. Anorexia can develop during infections and fever leading to mobilization of stored fatty acids. The effect of increased lipid mobilization is the production of toxic intermediates from the accumulating medium-chain fatty acids, medium-chain acylcarnitines, and dicarboxylic acids which can lead to vomiting, lethargy, coma, and even death. Treatment with oral carnitine increases the removal of these toxic intermediates.