
Last Updated: March 6, 2026
Intestinal Uptake of Lipids
In order for the body to make use of dietary lipids, they must first be absorbed from the small intestine. A complete discussion of lipid digestion and absorption is presented in the Digestion and Digestive Processes page. The predominant form of dietary lipid in the human diet is triglyceride. Since these molecules are oils, they are essentially insoluble in the aqueous environment of the intestine. The solubilization (or emulsification) of dietary lipids is accomplished principally in the small intestine by means of the bile acids. Bile acids are synthesized from cholesterol in the liver and then stored in the gallbladder. Following the ingestion of food, bile acids are released and secreted into the gut. Some lipid emulsification occurs in the stomach due to the churning action in this organ which renders some of the lipid accessible to gastric lipase.
The emulsification of dietary fats renders them accessible to various pancreatic lipases in the small intestine. These lipases, pancreatic lipase and pancreatic phospholipase A2 (PLA2), generate free fatty acids and a mixture of mono- and diglycerides from dietary triglycerides. Pancreatic lipase degrades triglyceride at the sn-1 and sn-3 positions sequentially to generate 1,2-diacylglycerides (DAG) and 2-acylglycerides. Phospholipids are degraded at the sn-2 position by pancreatic PLA2 releasing a free fatty acid and a lysophospholipid. The products of pancreatic lipases then enter the intestinal epithelial cells via the action of various transporters as well as by simple diffusion. Within the enterocyte the lipids are used for re-synthesis of triglycerides.
Dietary triglyceride and cholesterol, as well as triglyceride and cholesterol synthesized by the liver, are solubilized in lipid-protein complexes. These complexes contain triglyceride lipid droplets and cholesteryl esters surrounded by the polar phospholipids and proteins identified as apolipoproteins. These lipid-protein complexes, termed lipoproteins, vary in their content of lipid and protein.
There are three major classes of lipoproteins, one of which is dietary in origin and the other two are considered endogenous lipoproteins. Dietary lipoproteins are the chylomicrons. The endogenous lipoproteins are those produced by the liver (very low density lipoprotein: VLDL) and those produced in the circulation (high density lipoprotein: HDL). The assembly of chylomicrons and VLDL occurs within the lumen of the endoplasmic reticulum (ER) in enterocytes and hepatocytes, respectively.
During the course of transport in the vasculature, coupled with the action of various vascular lipases (predominantly lipoprotein lipase, LPL), VLDL are converted to intermediate density lipoproteins (IDL) and then to low density lipoproteins (LDL). As a result of the changes in composition and size, vascular lipoproteins are often indicated to exist in five major groups; chylomicron, VLDL, IDL, LDL, and HDL. Because chylomicrons and VLDL contain a large amount of triglyceride these lipoproteins are commonly classified as the triglyceride-rich lipoproteins, TRL.
Another lipid-protein complex in the vasculature is composed of the free fatty acids released from adipose tissue bound to albumin, although these complexes are not strictly defined as lipoproteins.
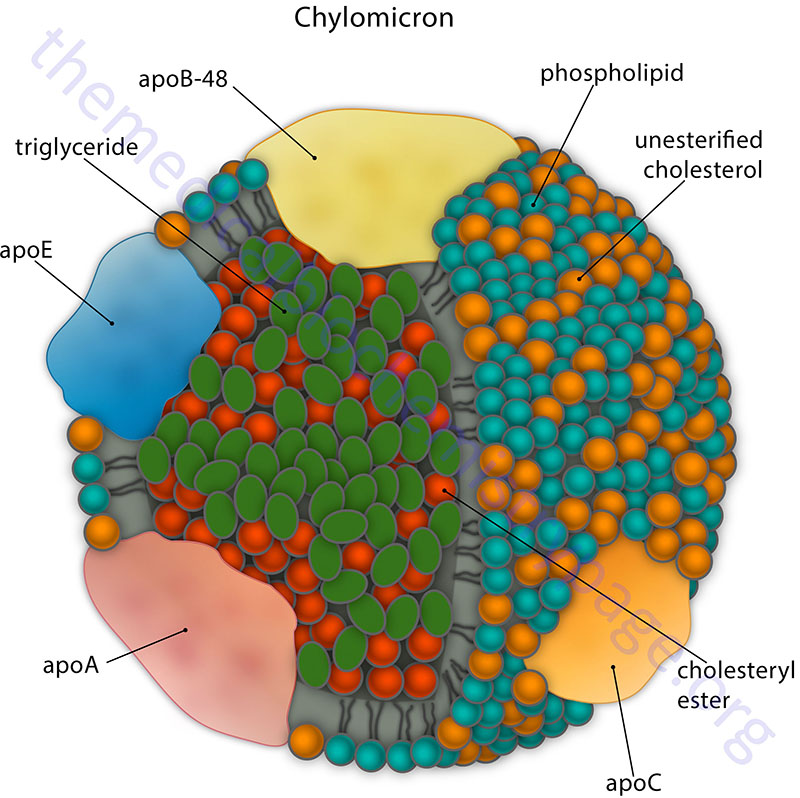
Table of the Composition of Lipoprotein Complexes
| Complex | Source | Density (g/ml) | %Protein | %TGa | %PLb | %CEc | %Cd | %FFAe |
| Chylomicron | Intestine | <0.95 | 1-2 | 85-88 | 8 | 3 | 1 | 0 |
| VLDL | Liver | 0.95-1.006 | 7-10 | 50-55 | 18-20 | 12-15 | 8-10 | 1 |
| IDL | VLDL | 1.006-1.019 | 10-12 | 25-30 | 25-27 | 32-35 | 8-10 | 1 |
| LDL | VLDL | 1.019-1.063 | 20-22 | 10-15 | 20-28 | 37-48 | 8-10 | 1 |
| *HDL2 | Intestine, liver (chylomicrons and VLDL) | 1.063-1.125 | 33-35 | 5-15 | 32-43 | 20-30 | 5-10 | 0 |
| *HDL3 | Intestine, liver (chylomicrons and VLDL) | 1.125-1.21 | 55-57 | 3-13 | 26-46 | 15-30 | 2-6 | 6 |
| ++Albumin-FFA | Adipose tissue | >1.281 | 99 | 0 | 0 | 0 | 0 | 100 |
aTriglycerides, bPhospholipids, cCholesteryl esters, dFree cholesterol, eFree fatty acids
*HDL2 (HDL2) and HDL3 (HDL3) are derived from nascent HDL as a result of the acquisition of numerous proteins (such as apolipoproteins), cholesteryl esters, and triglycerides
++ Free fatty acids transported bound to albumin are strictly not classified as lipoprotein particles
Lipid Profile Values
Standard fasting blood tests for cholesterol and lipid profiles will include values for total cholesterol, HDL cholesterol (so-called “good” cholesterol), LDL cholesterol (so-called “bad” cholesterol) and triglycerides. Family history and life style, including factors such as blood pressure and whether or not one smokes, affect what would be considered ideal versus non-ideal values for fasting blood lipid profiles. Included here are the values for various lipids that indicate low to high risk for coronary artery disease.
Total Serum Cholesterol
- <200mg/dL = desired values
- 200–239mg/dL = borderline to high risk
- 240mg/dL and above = high risk
HDL Cholesterol
- With HDL cholesterol the higher the better
- <40mg/dL for men and <50mg/dL for women = higher risk
- 40–50mg/dL for men and 50–60mg/dL for woman = normal values
- >60mg/dL is associated with some level of protection against heart disease
LDL Cholesterol
- With LDL cholesterol the lower the better
- <100mg/dL = optimal values
- 100mg/dL–129mg/dL = optimal to near optimal
- 130mg/dL–159mg/dL = borderline high risk
- 160mg/dL–189mg/dL = high risk
- 190mg/dL and higher = very high risk
Triglycerides
- With triglycerides the lower the better
- <150mg/dL = normal
- 150mg/dL–199mg/dL = borderline to high risk
- 200mg/dL–499mg/dL = high risk
- >500mg/dL = very high risk
Apolipoproteins
The proteins that are components of the various lipoprotein complexes are referred to as apolipoproteins. Apolipoproteins bind to and aid in the transport of lipids such as triglycerides, cholesterol, phospholipids, and fat soluble vitamins. Apolipoproteins also interact with lipoprotein receptors on cells as well as other lipid transport proteins. Most apolipoproteins are associated with HDL which serves as the reservoir and which function, in part, to facilitate transfer of some apolipoproteins to VLDL and chylomicrons. Humans express 21 genes that are functionally classified as apolipoproteins, several of which are covered in the following Table.
Table of the Major Apolipoprotein Classifications*
| Apoprotein | Gene Name and Structure | Lipoprotein Association | Function and Comments |
| apoA-I | APOA1: 11q23.3; four exons; four alternatively spliced mRNAs; two protein isoforms: 267 and 158 amino acid preproproteins | Chylomicrons, HDL | APOA1, APOA4, APOA5, and APOC3 genes are clustered at 11q23.3; major protein of HDL, binds ABCA1 on macrophages to facilitate cholesterol uptake by HDL, critical anti-oxidant protein of HDL, activates lecithin:cholesterol acyltransferase, LCAT |
| apoA-II | APOA2: 1q23.3; four exons; 100 amino acid preproprotein | Chylomicrons, HDL | near exclusive to HDL, represents the second-most abundant protein in HDL, enhances hepatic lipase activity, extrahepatic expression as well as misfolding of proprotein contribute to senile amyloidosis |
| apoA-IV | APOA4: 11q23.3; four exons; 396 amino acid precursor protein | Chylomicrons, HDL | APOA4, APOA1, APOA5, and APOC3 genes are clustered at 11q23.3; present in triglyceride rich lipoproteins; synthesized in small intestine, synthesis activated by PYY, acts in central nervous system to inhibit food intake, the details of which are discussed in the Regulation of Feeding Behaviors page |
| apoA-V | APOA5: 11q23.3; four exons; two alternatively spliced mRNAs; both encode same 366 amino acid precursor protein | Chylomicrons, HDL, VLDL, but not LDL | APOA5, APOA1, APOA4, and APOC3 genes are clustered at 11q23.3; expression is restricted to the liver; likely protects the liver from lipid excess during the early period of liver regeneration; associates with heparin, heparan sulfate proteoglycans (HSPG), and glycosylphosphatidylinositol-anchored HDL binding protein 1 (GPIHBP1); activates lipoprotein lipase but only in association with GPIHBP1 and HSPG; protects LPL from inhibition by ANGPTL3/ANGPTL8 complexes; increases intracellular concentration of triglycerides in hepatocytes through interaction with lipid droplets; is taken up by adipocytes through interaction with the LDL receptor family member protein, LRP1; decreases triglyceride concentration in adipocytes by increasing lipolysis, in part via increased expression of uncoupling protein 1, UCP1 (also known as thermogenin); mutations in the APOA5 gene are associated with familial chylomicronemia syndrome, FCS |
| apoB-48 | APOB: 2p24.1; 29 exons; 2179 amino acid precursor due to RNA editing | Chylomicrons | exclusively found in chylomicrons, derived from APOB gene by RNA editing in intestinal epithelium; lacks the LDL receptor-binding domain of apoB-100; the designation of apoB-48 refers to the fact that this protein contains 48% of the full-length apoB-100 protein |
| apoB-100 | APOB: 2p24.1; 29 exons; 4563 amino acid precursor protein | VLDL, IDL, LDL | major protein of VLDL, IDL, and LDL, binds to LDL receptor; one of the longest known proteins in humans; the designation of apoB-100 refers to the fact that this protein contains 100% of the coding capacity of the APOB encoded mRNA |
| apoC-I | APOC1: 19q13.32; five exons; 83 amino acid precursor protein | Chylomicrons, HDL, VLDL, IDL | clustered with APOE, APOC4, and APOC2 genes on chromosome 19; expression exclusive to the liver and macrophages; may also activate LCAT |
| apoC-II | APOC2: 19q13.32; four exons; 101 amino acid precursor protein; mature protein is 79 amino acids | Chylomicrons, HDL, VLDL, IDL | clustered with APOE, APOC1, and APOC4 on chromosome 19; expression exclusive to the liver and macrophages; expression regulated by bile acid-mediated activation of the FXR family of nuclear receptors as well as by hepatocyte nuclear factor-4α (HNF4α) which is a master regulator of hepatocyte lipid metabolism; activates lipoprotein lipase; mutations in the APOC2 gene are associated with familial chylomicronemia syndrome, FCS |
| apoC-III | APOC3: 11q23.3; four exons; 99 amino acid precursor protein | Chylomicrons, HDL, VLDL, IDL | APOC3, APOA1, APOA4, and APOA5 genes are clustered at 11q23.3; predominantly expressed in liver and to a lessor extent in the intestines; hepatic expression is regulated by glucose through activation of ChREBP and HNF4α; hepatic expression is induced by saturated fatty acids via activation of PGC-1β; insulin and polyunsaturated fatty acids (PUFA) inhibit expression through inhibition of the FOXO1 transcription factor; inhibits lipoprotein lipase, endothelial lipase, and hepatic lipase; interferes with hepatic uptake and catabolism of apoB-containing lipoproteins; appears to enhance the catabolism of HDL particles; enhances monocyte adhesion to vascular endothelial cells; activates inflammatory signaling pathways |
| apoC-IV | APOC4: 19q13.32; four exons; 127 amino acid precursor protein | Chylomicrons, HDL, VLDL, IDL | clustered with APOE, APOC1, and APOC2 gene on chromosome 19; expression exclusive to the liver and macrophages; precise function is yet to be elucidated |
| apoD | APOD: 3q29; five exons; 189 amino acid precursor | HDL | produced by numerous tissues with highest levels in the testes and brain; closely associated with LCAT; amino acid homology to α2-microglobulin super family of proteins that are also known as the lipocalins; elevated levels in the brain associated with Parkinson and Alzheimer diseases |
| cholesterol ester transfer protein, CETP | CETP: 16q13; 17 exons; two alternatively spliced mRNAs; two protein isoforms: 493 and 433 amino acid precursor proteins | HDL | not considered a apolipoprotein although it is associated with HDL; plasma glycoprotein secreted primarily from the liver and is associated with cholesteryl ester transfer from HDL to LDL and VLDL in exchange for triglycerides |
| apoE | APOE: 19q13.32; six exons; five alternatively spliced mRNAs, four of which have alternate 5′-terminal exons compared to the longest protein coding mRNA | Chylomicron remnants, HDL, VLDL, IDL | clustered with APOC1, APOC2, and APOC4 genes on chromosome 19; expression exclusive to liver and macrophages; at least 3 alleles E2, E3, E4; the apoE2 allele has Cys at amino acids 112(TGC) and 158(TGC); apoE3 has Cys(TGC) and Arg(CGC) at these two positions, respectively; apoE4 has Arg(both codons CGC) at both positions; binds to LDL receptor; the apoE3 allele is the most common; apoE4 allele amplification associated with an inherited form of late-onset Alzheimer disease |
| apoF | APOF: 12q13.3; two exons; 326 amino acid precursor protein | primarily HDL, some LDL | initially called lipid transfer inhibitor protein (LITP) due to its ability to inhibit the activity of CETP; accelerates cholesterol clearance from the blood |
| apoH | APOH: 17q24.2; eight exons; 345 amino acid precursor protein | negatively charged surfaces | was originally identified as β2-glycoprotein 1; binds to phospholipids, primarily cardiolipins; inhibits serotonin release from platelets; alters ADP-mediated platelet aggregation |
| apoO | APOO: Xp22.11; 11 exons; 198 amino acid precursor protein | primarily HDL; also intracellular | expression is increased in heart of obese individuals; in addition to being secreted it is associated with mitochondria where it promotes mitochondrial uncoupling and enhancement of fatty acid oxidation; only apolipoprotein to have a chondroitin sulfate glycosylation |
| apoM | APOM: 6p21.33; seven exons; two alternatively spliced mRNAs; 188 and 116 amino acid proteins | nearly exclusive to HDL; only very small amounts in VLDL, LDL, and triglyceride-rich lipoproteins | exclusively expressed in liver and kidneys; membrane-bound; involved in lipid transport; exhibits antioxidant and anti-atherosclerotic activity through cholesterol efflux from cells; plasma form only from liver |
| apo(a) | LPA: 6q25.3-q26; 39 exons; the 2040 amino acid precursor represents the reference genome sequence | LDL | protein ranges in size from 300,000–800,000 as a result of from 2–43 copies of the Kringle-type domain; Kringle domains contain around 80 amino acids which form the domain via three intrachain disulfide bonds; disulfide bonded to apoB-100, forms a complex with LDL identified as lipoprotein(a), Lp(a); functions as a serine protease that inhibits tissue-type plasminogen activator 1 (tPA); strongly resembles plasminogen; may deliver cholesterol to sites of vascular injury, high risk association with premature coronary artery disease and stroke |
*apoC-IV and apoL1-apoL5 not included; apoJ not included as it is not an apolipoprotein but actually the chaperone clusterin
Apolipoprotein B
Chylomicrons and VLDL contain one molecule each of the protein (apoB) derived from the APOB gene. The apoB protein in chylomicrons is different from that found in VLDL as a result of editing of the primary APOB derived mRNA within intestinal enterocytes. Editing of the APOB mRNA changes a CAA codon (at amino acid 2180) to a UAA translational stop codon leading to premature termination of protein synthesis and the generation of a smaller protein called apoB-48 due to the fact that it comprises 48% of the possible coding capacity of the full-length apoB mRNA. This apolipoprotein (apoB-48) is found exclusively associated with chylomicrons.
When the APOB gene is transcribed within hepatocytes the mRNA is not edited and a full-length apoB protein is generated called apoB-100. This apolipoprotein (apoB-100) is found exclusively with the VLDL particles produced and secreted by the liver.
The C-to-U editing of the APOB mRNA requires a single-stranded RNA template with well defined characteristics in the immediate vicinity of the edited base, as well as protein cofactors that assemble into a functional complex referred to as a holoenzyme or editosome. This functional complex includes a minimal core composed of apolipoprotein B mRNA editing enzyme, catalytic polypeptide 1 (APOBEC-1; the catalytic deaminase) and a competence factor, APOBEC-1 complementation factor (A1CF). The function of A1CF is to act as an adaptor protein by binding both the APOBEC-1 enzyme and the mRNA substrate.
The APOBEC-1 protein is encoded by the APOBEC1 gene. The APOBEC1 gene is located on chromosome 12p13.31 and is composed of 6 exons that generate three alternatively spliced mRNAs that collectively encode two protein isoforms, the 236 amino acid isoform a and the 191 amino acid isoform b. Expression of the APOBEC1 gene is exclusive to the gastrointestinal system with the highest levels of expression in the duodenum.
Lipoprotein Lipase, LPL
In the capillaries of adipose tissue, skeletal muscle, and the heart, the fatty acids of chylomicrons and VLDL/IDL/LDL are removed from the triglycerides by the action of lipoprotein lipase (LPL), which is found on the surface of the endothelial cells of the capillaries.
Lipoprotein lipase is encoded by the LPL gene. The LPL gene is located on chromosome 8p21.3 and is composed of 10 exons that encode a 475 amino acid precursor protein.
Synthesis of LPL occurs on the rough ER within parenchymal cells of adipose tissue (adipocytes) and of cardiac and skeletal muscle (cardiomyocytes and myocytes, respectively). As LPL is being synthesized, the activity of lipase maturation factor 1 (LMF1) is required for maturation of LPL and its migration through the secretory pathway. Mutations in the LMF1 gene result in a form of familial chylomicronemia syndrome (FCS). Lipase maturation factor 1 is also responsible for the maturation of hepatic lipase (encoded by LIPC gene) and endothelial lipase (encoded by the LIPG gene).
The significance of LMF1 in overall blood lipid homeostasis was demonstrated when a mutation in the gene was identified in mice. Mice homozygous for the mutation died shortly after birth with massive hypertriglyceridemia. These mice exhibited essentially no LPL nor hepatic lipase activity. In 2007 a nonsense mutation in the LMF1 gene was identified in a patient suffering from severe hypertriglyceridemia, repeated episodes of pancreatitis, tuberous xanthomas, and acquired partial lipodystrophy in conjunction with type 2 diabetes. This mutation was at tyrosine 439 and is identified as Y439X. Subsequent to the identification of the Y439X mutation an additional nonsense mutation was found in the tryptophan codon at position 464 (W464X).
LPL is then subsequently exocytosed from the parenchymal cell where it binds to heparin sulfated proteoglycans (HSPG) present on the surface of the cell facing the subendothelial spaces. The enzyme is then picked up and transported from the subendothelial spaces of the capillaries to the capillary endothelial cells via the action of glycosylphosphatidylinositol-anchored HDL binding protein 1 which is encoded by the GPIHBP1 gene.
The GPIHBP1 gene is located on chromosome 8q24.3 and is composed of 4 exons that generate two alternatively spliced mRNAs, both of which encode distinct protein isoforms. Mutations in the GPIHBP1 gene result in a form of familial chylomicronemia syndrome (FCS) that was originally identified as hyperlipoproteinemia type 1D.
Functional LPL is a homodimer which remains associated with the apical (lumen) surface of the endothelial cells through its interactions with GPIHBP1. The role of the angiopoietin-like proteins in the processing of LPL are detailed below.
The apoC-II and apoA-V in the chylomicrons and VLDL activates LPL in the presence of phospholipid. In addition to apoC-II and apoA-V, several angiopoietin-like proteins (discussed in the next section) are critical to the regulation of LPL activity.
The free fatty acids released by the action of LPL are then absorbed by the tissues and the glycerol backbone of the triglycerides is returned, via the blood, to the liver and kidneys. Within the liver the glycerol is phosphorylated by glycerol kinase. The resultant glycerol-3-phosphate is then converted to the glycolytic intermediate, dihydroxyacetone phosphate (DHAP), via the action of cytosolic glycerol-3-phosphate dehydrogenase (GPD1), and is utilized for glucose synthesis via the gluconeogenesis pathway.
During the removal of fatty acids, a substantial portion of phospholipid, apoA-V and apoC-II is transferred to HDL. The loss of apoC-II and apoA-V prevents LPL from further degrading the chylomicron remnants.
Angiopoietin-Like Proteins and Regulation of LPL Activity
As described in the previous section, lipoprotein lipase (LPL) is a rate-limiting enzyme for the removal of fatty acids present in the triglycerides in circulating lipoproteins, particularly chylomicrons. The major sites of LPL activity are the endothelial cells of the capillaries of the heart, skeletal muscle, and adipose tissue. Given that the heart and skeletal muscle depend on fatty acid oxidation for energy production, it is not surprising that these tissues abundantly express this enzyme. Since adipose tissue is the major site of fatty acid storage, through fatty acid esterification into triglycerides, LPL is abundantly expressed in white adipose tissue (WAT) as well. High levels of LPL in adipose tissue allows adipocytes access to the fatty acids in circulating triglycerides.
Although found tethered to the apical (luminal) membranes of capillary endothelial cells, the endothelial cells themselves do not synthesize the enzyme. LPL is synthesized in the parenchymal cells of adipose tissue, heart muscle and skeletal muscle and is secreted into the subendothelial spaces. Endothelial cell surface GPIHBP1 then picks up LPL and transfers it from the basolateral side of the endothelial cell to the apical side. In the lumen of the capillaries LPL remains tethered to GPIHBP1 as a functional homodimer.
Because LPL plays such a central role in triglyceride metabolism specifically, and in the regulation of overall lipoprotein metabolism in general, its activity is carefully regulated in a tissue-specific manner in relation to the daily feed-fast cycles. Following the intake of food, the level of LPL in WAT is increased whereas its levels in heart and skeletal muscle declines. Conversely, during periods of fasting LPL activity in WAT declines, whereas in heart and skeletal muscle LPL levels rise. These rapid changes in LPL activity are determined by post-translational mechanisms that involve interactions with proteins of the angiopoietin-like protein family as well as with apolipoproteins in the circulating chylomicrons and VLDL. The predominant lipoprotein-associated regulator of LPL activity is apoC-II (apoC-2) which chylomicrons and VLDL acquire from HDL. Another minor apolipoprotein that can activate LPL is apoA-V (apoA-5).
The activity of LPL is also regulated by proteins of the angiopoietin-like protein (ANGPTL) family. Humans express eight angiopoietin-like protein encoding genes identified as ANGPTL1–ANGPTL8. The angiopoietin-like proteins share structural, but not functional, similarity to angiopoietins which are members of the vascular endothelial growth factor family. Humans express four angiopoietin family member proteins (ANGPT1–ANGPT4) that exert their effects by binding to two related receptors called tyrosine kinase with immunoglobulin-like and EGF-like domains 1 (TIE1; encoded by the TIE1 gene) and TIE2. The TIE2 protein is encoded by the TEK gene (TEK refers to Tyrosine kinase, Endothelial, Kinase). The angiopoietin-like proteins do not bind to these receptors. Three of the angiopoietin-like proteins (ANGPTL3, ANGPTL4, and ANGPTL8) play crucial roles in the post-translational regulation of LPL activity.
ANGPTL3
Angiopoietin-like protein 3 (also known as angiopoietin 5) is produced exclusively by the liver and secreted into the circulation. Prior to its release from hepatocytes, a portion of the ANGPTL3 proprotein is cleaved into N-terminal and C-terminal fragments via the action of the proprotein convertase subtilisin/kexin type (PCSK) family member enzyme, furin (encoded by the FURIN gene). Furin is also known as PCSK3.
Both full-length and the cleaved fragments of ANGPTL3 are secreted into the circulation. The N-terminal portion of ANGPTL3 is involved in the regulation of overall lipoprotein metabolism through its ability to inhibit the activity of LPL, as well as to inhibit the activity of endothelial lipase, EL. Endothelial lipase is so-called because it is expressed exclusively by endothelial cells. Endothelial lipase functions almost exclusively as a phospholipase with highest affinity for HDL. The C-terminal fragment of ANGPTL3 is involved in angiogenesis.
The LPL inhibitory activity of ANGPTL3 on its own is minimal but is greatly enhanced upon its interaction with ANGPTL8.
The ANGPTL3 gene is located on chromosome 1p31.3 and is composed of 7 exons that encode a preproprotein of 460 amino acids. Expression of the ANGPTL3 gene is restricted to the liver and kidney. Mutation in the ANGPTL3 gene are the cause of familial combined hypolipidemia which is associated with reduced circulating levels of VLDL, LDL, and HDL due to increased activities of both LPL and endothelial lipase.
ANGPTL4
The ANGPTL4 gene was initially identified as a gene induced during periods of fasting and also as a result of its induction by peroxisome proliferator-activated receptor-γ (PPARγ) in white adipose tissue (WAT). As a result of the observation of its induction during fasting, ANGPTL4 was originally called fasting-induced adipose factor (FIAF). Conversely, food intake results in reduced expression of ANGPTL4.
Like the ANGPTL3 proprotein, the ANGPTL4 proprotein is cleaved by furin and it is the N-terminal domain that is the potent LPL inhibitor. ANGPTL4 inhibits LPL activity by preventing the active homodimeric form of LPL from forming and by reducing the affinity of the monomers for binding to GPIHBP1 on the surface of capillary endothelial cells. In addition, ANGPTL4 promotes intracellular furin-mediated cleavage and inactivation of LPL.
Inhibition of adipose tissue LPL by ANGPTL4 during fasting is critical to the ensuring the delivery of fatty acids to cardiac muscle and skeletal muscle from circulating lipoproteins. Unlike ANGPTL3, ANGPTL4 is a potent LPL inhibitor in the absence of ANGPTL8. Interaction of ANGPTL4 with ANGPTL8 in WAT results in marked reduction in its ability to inhibit LPL.
The ANGPTL4 gene is located on chromosome 19p13.2 and is composed of 7 exons that generate two alternatively spliced mRNAs, both of which encode distinct protein isoforms. Expression of the ANGPTL4 gene is ubiquitous with the highest levels of expression being in the liver and adipose tissue.
ANGPTL8
Expression of the ANGPTL8 gene is enhanced in both the liver and white adipose tissue (WAT) in response to food intake, while fasting results in reduced levels of expression. Expression of ANGPTL8 is also induced in brown adipose tissue (BAT) in response to prolonged exposure to cold. Although the ANGPTL8 protein possesses sequence homology to both ANGPTL3 and ANGPTL4, it lacks the C-terminal fibrinogen-like domain that is present in both the ANGPTL3 and ANGPTL4 proteins.
ANGPTL8 forms complexes with both ANGPTL3 and ANGPTL4. ANGPTL8 functions in an endocrine manner with ANGPTL3 in the heart and skeletal muscle following food intake. The ANGPTL8-mediated activation of ANGPTL3 in these two tissues following feeding allows more fatty acids from circulating chylomicrons to be taken up by adipose tissue. Conversely, ANGPTL8 inhibits the activity of ANGPTL4 which normally inhibits LPL, thus resulting in increased LPL activity.
Loss-of-function (LOF) mutations in the ANGPTL8 gene have been identified and are associated with decreased levels of circulating triglycerides, decreased circulating levels of LDL-cholesterol (LDL-C), and increased circulating levels of HDL-cholesterol (HDL-C). These observations clearly point to a critical role for ANGPTL8 in the overall regulation of lipid metabolism.
Through differences in tissue expression and feed-fast cycle regulation of both ANGPTL4 and ANGPTL8, thereby resulting in regulation of ANGPTL3 activity, the activity of LPL is tightly controlled. During periods of feeding LPL activity is highest in adipose tissue and lowest in heart and skeletal muscle and during fasting LPL activity is highest in heart and skeletal muscle and lowest in adipose tissue.
The ANGPTL8 gene is also located on chromosome 19p13.2 and is composed of 4 exons that encode a precursor protein of 198 amino acids. Expression of the ANGPTL8 gene is restricted to liver and adipose tissue.
Role of CREBH in the Regulation of LPL Activity
The major transcription factor regulating the expression of the LPL gene in the liver is cAMP response element-binding protein, hepatocyte specific, CREBH. CREBH is critically involved in overall hepatic lipid homeostasis, particularly during periods of fasting and stress, such that there is increased triglyceride breakdown and fatty acid oxidation.
The gene encoding CREBH is CREB3L3 (cAMP responsive element binding protein 3 like 3). Expression of the CREB3L3 gene is restricted to the liver and the intestines.
The CREBH protein is localized the to endoplasmic reticulum (ER) which regulates its overall activity. When the CREBH protein is proteolyzed the N-terminal fragment is released and migrates to the nucleus where it functions as a transcription factor. With respect to the regulation of overall LPL activity, CREBH activates the expression of both the APOC2 (encoding apoC-II) and APOA5 (encoding apoA-V) genes allowing for increased LPL activity. ApoC-II directly activates LPL whereas apoA-V inhibits the ANGPTL3/ANGPTL8 complex that inhibits LPL activity.
In addition, following proteolytic activation the C-terminal fragment of CREBH functions as a hepatokine by being released. Following its release the C-terminal fragment interacts with ANGPTL3/ANGPTL8 complexes resulting in the blocking of the ability of these complexes to inhibit LPL activity.
Microsomal Triglyceride Transfer Protein: MTP
Apolipoproteins generated from the APOB gene are large hydrophobic proteins that exists in plasma as apoB-48 or apoB-100 that, as outlined in Apolipoprotein B section above, are associated with intestinally derived chylomicrons or the liver derived VLDL, respectively.
Newly synthesized apoB containing lipoproteins, referred to as ‘nascent’ particles, undergo apolipoprotein exchange and enzymatic lipolysis as soon as they reach the lymph or the plasma. The incorporation of apoB-48 into chylomicrons, and of apoB-100 into VLDL, requires the activity of the endoplasmic reticulum (ER) chaperone identified as microsomal triglyceride transfer protein (MTP; also identified as MTTP) which is encoded by the MTTP gene.
The MTTP gene is located on chromosome 4q23 and is composed of 19 exons that generate three alternatively spliced mRNAs that collectively encode two precursor proteins of 894 amino acids (isoform 1) and 921 amino acids (isoform 2). Expression of the MTTP gene is essentially exclusive to the liver and the small intestines with very low level of expression seen in the kidney and testes.
There are three essential functions associated with MTP including lipid transfer, apoB binding, and membrane association. Functional MTP is composed of a large catalytic α-subunit encoded by the MTTP gene and a small β-subunit which is a member of the protein disulfide isomerase (PDI) family of proteins that are involved in the processes of protein folding in the ER. The PDI family member gene that encodes the MTP β-subunit is P4HB (prolyl 4-hydroxylase subunit beta) and is also known as PDIA1.
Microsomal triglyceride transfer protein functions in the assembly and secretion of primordial apoB-containing lipoproteins in a process that includes two major steps. In the first step, the so-called primordial lipoprotein particle is synthesized while in the second step there is core expansion of the primordial lipoproteins and the synthesis of what is referred to as nascent lipoproteins.
The first step of primordial lipoprotein assembly involves both a nucleation and a desorption. When the newly synthesized apoB protein is translocated into the endoplasmic reticulum, the N-terminus of the protein associates with the inner membrane and acquires neutral lipids forming nucleation sites. These nucleation sites most likely arise from endoplasmic reticulum microdomains harboring triglyceride-synthesizing enzymes. The formation of these nucleation sites is enhanced in the presence of MTP due to its neutral lipid transfer activity.
In addition to its effect on the composition of ER membrane lipids, MTP can also transfer triglycerides to nascent apoB proteins either during or following their translation. The phospholipid transfer activity of MTP is critical for desorption of primordial apoB particles along with neutral lipids from the ER-associated nucleation sites. This latter process renders the apoB containing particles able to be secreted.
A form of abetalipoproteinemia is associated with mutations in the MTTP gene. Abetalipoproteinemia is characterized by fat malabsorption, steatorrhea, acanthocytosis (thorny looking cells), and hypocholesterolemia in infancy. Later in life, deficiency of fat-soluble vitamins is associated with development of atypical retinitis pigmentosa, coagulopathy, posterior column neuropathy, and myopathy.
Phospholipids of Lipoprotein Particles
Lipoprotein particles possess a monolayer of phospholipids that cover the surface of the particles. The major phospholipids of lipoprotein particles are phosphatidylcholines, PC. The majority of phospholipid synthesis occurs on the cytoplasmic surface of the membranes of the endoplasmic reticulum, ER. Therefore, in order for their incorporation into lipoprotein particles, these phospholipids need to be shuttled across the membrane into the ER lumen.
Recent studies have demonstrated that two proteins that reside in the ER membrane likely serve as phospholipid scramblases to facilitate phospholipid transfer into the ER lumen. These proteins are encoded by the TMEM41B (transmembrane protein 41B) and VMP1 (vacuole membrane protein 1) genes. Prior experiments demonstrated that the TMEM41B encoded protein functions in intracellular lipid transport and in the assembly of autophagosomes. The VMP1 protein is also involved in autophagosome biogenesis.
The phospholipid transfer protein (encoded by the PLTP gene) that is involved in the transfer of phospholipids from the two triglyceride-rich lipoproteins (TRL), chylomicrons and VLDL, to circulating HDL particles is also involved in the overall process of phospholipid incorporation into lipoproteins.
Another important enzyme involved in overall lipoprotein maturation related to phospholipid incorporation is the lysophospholipid acyltranstransferase (LPLAT) family member, LPLAT12 which is encoded by the LPCAT3 gene. LPLAT12 also belongs to the membrane bound O-acyltransferase domain containing (MBOAT) family and as such is identified as MBOAT5. Expression of the LPCAT3 gene is highest in the intestines and the liver, as would be expected from a gene encoding a protein playing an important role in lipoprotein particle assembly.
Chylomicrons
Chylomicrons are assembled in intestinal enterocyte as a means to transport dietary fatty acids and cholesterol to the rest of the body. Following the digestion of dietary triglycerides and phospholipids into free fatty acids, monoglycerides, and lysophosphatidic acids and the uptake of these molecules into intestinal enterocytes, triglycerides and phospholipids are re-synthesized and free cholesterol is esterified. These lipids are then packaged into the lipoprotein particles termed chylomicrons. Chylomicrons are, therefore, the molecules formed to mobilize dietary (exogenous) lipids. The predominant lipids of chylomicrons are triglycerides (see Table above). The apolipoproteins that predominate before the chylomicrons enter the circulation include apoB-48, apoA-I, apoA-II, and apoA-IV.
ApoB-48 combines only with chylomicrons. ApoB-48 incorporation into forming chylomicrons involves the function of the endoplasmic reticulum (ER) associated heterodimeric complex called microsomal triglyceride transfer protein, MTTP (also identified as MTP). Chylomicrons leave the intestine via the lymphatic system and enter the circulation at the left subclavian vein. In the bloodstream, chylomicrons acquire apoC-II and apoE from plasma HDL.
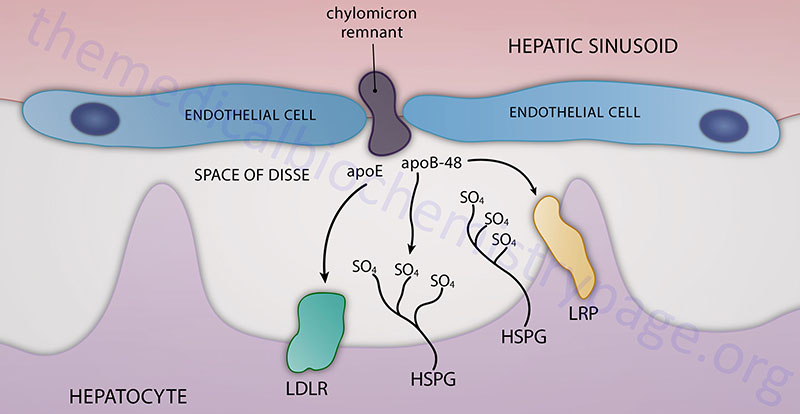
Hepatic Chylomicron Remnant Uptake
Chylomicron remnants, containing primarily cholesteryl esters, apoE, and apoB-48, are then delivered to, and taken up by, the liver. The remnant particle must be of a sufficiently small size such that can pass through the fenestrated endothelial cells lining the hepatic sinusoids and enter into the space of Disse. Chylomicron remnants can then be taken up by hepatocytes via interaction with the LDL receptor which is facilitated by the presence of apoE. In addition, while in the space of Disse chylomicron remnants can accumulate additional apoE that is secreted free into the space. This latter process allows the remnant to be taken up via the chylomicron remnant receptor, which is a member of the LDL receptor-related protein (LRP) family. The recognition of chylomicron remnants by the hepatic remnant receptor also requires apoE. Chylomicron remnants can also remain sequestered in the space of Disse by binding of apoE to heparan sulfate proteoglycans and/or binding of apoB-48 to hepatic lipase. While sequestered, chylomicron remnants may be further metabolized which increases apoE and lysophospholipid content allowing for transfer to LDL receptors or LRP for hepatic uptake.
VLDL, IDL, and LDL
The dietary intake of both fat and carbohydrate, in excess of the needs of the body, leads to the conversion, in the liver, of the excess carbons into fatty acids which are then incorporated into triglycerides. These triglycerides are packaged into VLDL and released into the circulation for delivery to the various tissues (primarily muscle and adipose tissue) for storage or production of energy through oxidation. VLDL are, therefore, the molecules formed to transport endogenously derived triglycerides to extra-hepatic tissues. A critical contributor to the hepatic synthesis of VLDL is phosphatidylcholine which is necessary for the packaging of triglycerides into VLDL.
In addition to triglycerides, VLDL contain some cholesterol and cholesteryl esters and the apoproteins, apoB-100 (a single copy). ApoB-100 incorporation into forming VLDL particles, similar to the incorporation of apoB-48 into chylomicrons, involves the function of the endoplasmic reticulum (ER) associated microsomal triglyceride transfer protein, MTTP. Upon secretion into the blood, VLDL accumulates apoC-I, apoC-II, apoC-III, and apoE from circulating HDL similarly to the incorporation of these apolipoproteins into nascent chylomicrons.
The fatty acid portion of VLDL is released to adipose tissue and muscle in the same way as for chylomicrons, through the action of lipoprotein lipase. The action of lipoprotein lipase coupled to a loss of certain apoproteins (e.g. apoC-II) converts VLDL to intermediate density lipoproteins (IDL), also termed VLDL remnants. IDL contain multiple copies of apoE and a single copy of apoB-100. The presence of the multiple copies of apoE enable these lipoprotein particles to have very high affinity for the LDL receptor on cells such as hepatocytes and adrenal cortex cells. Conversion of VLDL to IDL is also associated with loss of apoCs by transfer back to HDL. Further loss of fatty acids from triglycerides, as well as transfer of apoE back to HDL converts IDL to LDL. The presence of the apoB-100 protein allows LDL to be recognized by the LDL receptor but the lack of apoE makes the affinity much lower compared to that of IDL.
The liver takes up IDL and LDL after they have interacted with the LDL receptor to form a complex, which is then endocytosed by the cell. For LDL receptors in the liver to recognize IDL and LDL requires the presence of apoB-100 and is enhanced (in the case of IDL) by the presence of apoE. The LDL receptor is also sometimes referred to as the apoB-100/apoE receptor. The importance of apoE in cholesterol uptake by LDL receptors has been demonstrated in transgenic mice lacking functional apoE genes. These mice develop severe atherosclerotic lesions at 10 weeks of age even when fed a low-fat diet.
Of significance to overall serum total cholesterol levels is the fact that one of the events that results in the conversion of IDL to LDL is the loss of apoE which is returned to HDL. Although LDL particles still possess apoB-100 which is required for LDL receptor binding, the loss of apoE makes their affinity for the receptor reduced. Any perturbation in serum total cholesterol regulation will result in increased circulating LDL. The longer that LDL remains in the blood the greater the likelihood the protein and lipid components will become oxidized (forming oxLDL). The significance of oxLDL is that these particles are bound to the oxLDL receptors, primarily on macrophages, leading to enhanced intravascular inflammation. The significances of increased serum oxLDL is discussed below in the section on lipoprotein receptors, specifically the scavenger receptors.
The cellular requirement for cholesterol as a membrane component is satisfied in one of two ways: either it is synthesized de novo within the cell, or it is supplied from extra-cellular sources, namely, chylomicrons and IDL/LDL. As indicated above, the dietary cholesterol that goes into chylomicrons is supplied to the liver by the interaction of chylomicron remnants with the remnant receptor. In addition, cholesterol synthesized by the liver can be transported to extra-hepatic tissues if packaged in VLDL. In the circulation VLDL are converted to IDL and LDL through the action of lipoprotein lipase. IDL and LDL are the primary plasma carriers of cholesterol for delivery to all tissues via their LDL receptor mediated uptake.
The exclusive apolipoprotein of LDL is apoB-100. LDL are taken up by cells via LDL receptor-mediated endocytosis, as described above for IDL uptake. The uptake of LDL occurs predominantly in liver (75%), adrenals and adipose tissue. As with IDL, the interaction of LDL with LDL receptors requires the presence of apoB-100. The endocytosed membrane vesicles (endosomes) fuse with lysosomes, in which the apoproteins are degraded and the cholesterol esters are hydrolyzed to yield free cholesterol. The cholesterol is then incorporated into the plasma membranes as necessary.
Excess intracellular cholesterol is re-esterified, by sterol O-acyltransferase 2 (SOAT2), for intracellular storage. The activity of SOAT2 is enhanced by the presence of intracellular cholesterol. The original name given to SOAT2 was acyl-CoA: cholesterol acyltransferase 2 (ACAT2). This designation conflicts with that for the official ACAT2 enzyme (a member of thiolase family of enzymes), acetyl-CoA acetyltransferase 2.
The SOAT2 gene is located on chromosome 12q13.13 and is composed of 16 exons that encode a 522 amino acid protein.
Another SOAT gene, SOAT1, encodes a protein that is also involved in the regulation of intracellular cholesterol concentrations. The SOAT1 gene is located on chromosome 1q25 and is composed of 17 exons that generate three alternatively spliced mRNAs.
Insulin and triiodothyronine (T3) increase the binding of LDL to liver cells, whereas glucocorticoids (e.g., dexamethasone) have the opposite effect. The precise mechanism for these effects is unclear but may be mediated through the regulation of apoB degradation. The effects of insulin and T3 on hepatic LDL binding may explain the hypercholesterolemia and increased risk of atherosclerosis that have been shown to be associated with uncontrolled diabetes or hypothyroidism.
The consumption of alcohol is associated with either a protective or a negative effect on the level of circulating LDL. Low level alcohol consumption, particularly red wines which contain the antioxidant resveratrol, appear to be beneficial with respect to cardiovascular health. Resveratrol consumption is associated with a reduced risk of cardiovascular, cerebrovascular, and peripheral vascular disease. One major effect of resveratrol in the blood is the prevention of oxidation of LDL, (forming oxLDL). Oxidized LDL contribute significantly to the development of atherosclerosis. Conversely excess alcohol consumption is associated with the development of fatty liver which in turn impairs the ability of the liver to take up LDL via the LDL receptor resulting in increased LDL in the circulation. Clearly a reduction in alcohol consumption will have a significant impact on overall cardiovascular and hepatic function.
High Density Lipoproteins, HDL
HDL represent a heterogeneous population of lipoproteins in that they exist as functionally distinct particles possessing different sizes, protein content, and lipid composition. One of the major functions of HDL is to acquire cholesterol from peripheral tissues and transport this cholesterol back to the liver where it can ultimately be excreted following conversion to bile acids. This function is referred to as reverse cholesterol transport (RCT). The role of HDL in RCT represents the major atheroprotective (prevention of the development of atherosclerotic lesions in the vasculature) function of this class of lipoprotein. In addition to RCT, HDL exert anti-inflammatory, antioxidant, and vasodilatory effects that together represent additional atheroprotective functions of HDL. Evidence has also been generated that demonstrates that HDL possess anti-apoptotic, anti-thrombotic, and anti-infectious properties. With respect to these various atheroprotective functions of HDL, it is the small dense particles (referred to as HDL3) that are the most beneficial.
HDL begin as the single protein, apolipoprotein A-I (apoA-I), which is synthesized de novo in the liver and small intestine. These newly formed HDL are devoid of any cholesterol, cholesteryl esters, lipids, and any other proteins. ApoA-I, as well as more complex HDL containing apoA-I, acquire cholesterol as described in the following paragraphs and outlined in the Figure below. As apoA-I picks up cholesterol, the resultant nascent HDL particle begins to accumulate numerous proteins from the blood. The primary apolipoproteins of HDL are apoA-I, apoC-I, apoC-II, apoD, apoE, apoF, apoM, and apoO. In fact, a major function of HDL is to act as a circulating store of apoC-I, apoC-II, and apoE. ApoA-I is the most abundant protein in HDL constituting over 70% of the total protein mass. In addition to apolipoproteins, HDL carry numerous enzymes that participate in the anti-oxidant activities of HDL.
Proteomics studies have demonstrated that as many as 300 different proteins can be found associated with HDL, many of which have no known role in lipid transport. Some of the critical enzymes in HDL include glutathione peroxidase 1 (GPx), paraoxonase 1 (PON1), and platelet activating factor acetylhydrolase (PAF-AH, also called lipoprotein-associated phospholipase A2, Lp-PLA2). Two additional functionally important enzymes found associated with HDL are lecithin:cholesterol acyltransferase (LCAT) and cholesterol ester transfer protein (CETP). Another important HDL component is the compound sphingosine-1-phosphate (S1P; details of S1P activities can be found in the Sphingolipid Metabolism and the Ceramides page).
The primary mechanism by which HDL acquire peripheral tissue cholesterol is via an interaction with monocyte-derived macrophages in the subendothelial spaces of the tissues. Macrophages bind nascent HDL, that contain primarily apoA-I, through interaction with the ATP-binding cassette transport protein A1 (ABCA1). The transfer of cholesterol from macrophages, via the action of ABCA1, involves apoA-I and results in the formation of nascent discoidal lipoprotein particles termed pre-β HDL. The free cholesterol transferred in this way is esterified by HDL-associated LCAT.
LCAT is synthesized in the liver and so named because it transfers a fatty acid from the C-2 position of a lecithin (phosphatidylcholine, PC) to the C3-OH of cholesterol, generating a cholesteryl ester and a lysolecithin. The activity of LCAT requires interaction with apoA-I, which remains on the surface of HDL as they get larger through protein, cholesterol, and triglyceride acquisition. The cholesteryl esters formed via LCAT activity are internalized into the hydrophobic core of the pre-β HDL particle. As pre-β HDL particles progressively take up cholesterol and proteins they become larger and spherical generating the mature HDL particles.
The importance of ABCA1 in reverse cholesterol transport is evident in individuals harboring defects in ABCA1 gene. These individuals suffer from a disorder called Tangier disease which is characterized by two clinical hallmarks; enlarged lipid-laden tonsils and low serum HDL.
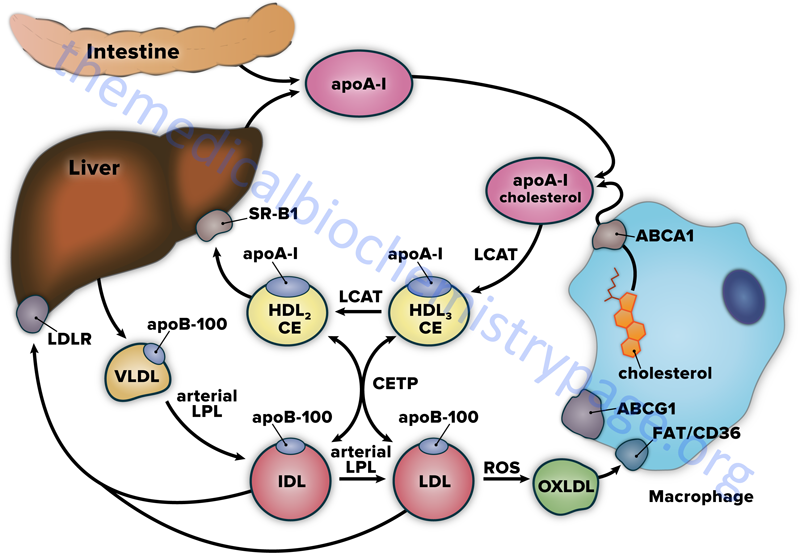
HDL also acquire cholesterol by extracting it from cell surface membranes. This process has the effect of lowering the level of intracellular cholesterol, since the cholesterol stored within cells as cholesteryl esters will be mobilized to replace the cholesterol removed from the plasma membrane. The transfer of cholesterol from peripheral tissue cells to HDL in this way involves the action of the ATP-binding cassette protein G1 (ABCG1). Approximately 20% of HDL uptake of peripheral tissue cholesterol occurs via the ABCG1-mediated pathway.
Cholesterol-rich HDL return to the liver, where they bind to a receptor that is a member of the scavenger receptor family, specifically the scavenger receptor BI: SR-BI (see below). When HDL binds to SR-BI it is not internalized as is the case for IDL or LDL following their binding to the LDL receptor. Following HDL binding to SR-BI the cholesteryl esters are taken up by the hepatocytes through caveolae while the HDL and SR-BI remain on the plasma membrane. Caveolae (Latin for little caves) are specialized “lipid rafts” present in flask-shaped indentations in the plasma membranes of many cells types that perform a number of signaling functions.
HDL particles exhibit complex, and sometimes contradictory rolls in vascular biology. Depending upon the vascular context, as well as the make-up of HDL particle, these lipoproteins can serve antiatherogenic or proatherogenic functions. In the absence of systemic inflammation many of the enzymes and apolipoproteins associated with HDL play important roles in reducing the amount of oxidized lipid to which peripheral tissues are exposed. Some of these important proteins are apoA-I, PON1, GPx (an important anti-oxidant enzyme), and PAF-AH (see section below for the discussion of this important activity). However, when an individual has an ongoing systemic inflammatory state, these anti-oxidant proteins can be dissociated from the HDL or become inactivated resulting in the increased generation of oxidized and peroxidized lipids which are proatherogenic. Atherosclerotic plaques also produce myeloperoxidase which chemically modifies HDL-associated apoA-I rendering it less capable of interacting with cell surfaces such as macrophages. This latter effect results in a reduced capacity for removal of cholesterol from lipid-laden macrophages (foam cells) leaving the foam cells in a more pro-inflammatory state.
Reverse cholesterol transport can also involve the transfer of cholesterol esters from HDL to VLDL, IDL, and LDL. This transfer requires the activity of the plasma glycoprotein cholesterol ester transfer protein (CETP). The transfer of cholesteryl esters from HDL via CETP activity also involves an exchange of triglycerides to the HDL. This action of HDL associated CETP has the added effect of allowing the excess cellular cholesterol to be returned to the liver through the LDL receptor. However, some of the LDL is oxidized in the periphery (generating oxLDL) where it can participate in atherogenesis.
Additionally, when HDL particles become enriched with triglycerides they are better targets for the action of hepatic lipase. As hepatic lipase acts on the triglyceride-rich HDL they become progressively smaller and unstable which results in the release of apoA-I. The loss of apoA-I renders the HDL particle unable to participate in reverse cholesterol transport. Blocking the activity of CETP keeps HDL particles less triglyceride-enriched while also reducing cholesterol transfer to VLDL, IDL, and LDL ultimately resulting in reduced circulating levels of proatherogenic oxLDL. This latter observation suggests that CETP inhibition may be a viable therapeutic approach for elevating the circulating levels of HDL. This is discussed below.
Anti-oxidant & Anti-inflammatory Activities of HDL
Using a range of both in vitro and in vivo assays it has been possible to quantify the anti- and pro-inflammatory properties, as well as the anti-oxidant functions of HDL. Cell-free assays have been used to measure the ability of HDL to prevent the formation of oxidized phospholipids in LDL as well as to determine the ability of HDL to degrade oxidized phospholipids that are already formed. In cell culture assays HDL have been shown to inhibit monocyte chemotaxis in response to oxidized LDL or to prevent the upregulation of cell adhesion molecules on endothelial cells. Both of these latter effects are strongly anti-inflammatory since monocytes need to migrate to a site of inflammation via a chemotactic gradient and then adhere to the endothelium at the site of injury or inflammatory event. The role of HDL in promoting cholesterol efflux from cells, especially from macrophages, (the process of reverse cholesterol transport) reduces the activation of inflammatory responses in these cells. The analysis of HDL functions in oxidative and inflammatory events has identified the role of various apolipoproteins associated with HDL in these processes which are outlined in the following sections.
Apolipoprotein A-I
Numerous lines of evidence demonstrate that apoA-I is a major anti-atherogenic and anti-oxidant factor in HDL due to its critical role in the HDL-mediated process of reverse cholesterol transport. In addition to reverse cholesterol transport, apoA-I can remove oxidized phospholipids from oxidized LDL (oxLDL) and from cells. Specific methionine residues (Met112 and Met148) of apoA-I have been shown to directly reduce cholesterol ester hydroperoxides and phosphatidylcholine hydroperoxides.
Apolipoprotein A-II
Experiments in transgenic mice have demonstrated that human apoA-II-enriched HDL served to protect VLDL from oxidation more efficiently than HDL from control animals. The human apoA-II-enriched HDL support highly effective reverse cholesterol transport from macrophages. Although there is a demonstrated benefit of apoA-II in reverse cholesterol transport and in reduced LDL oxidation, these transgenic mice exhibited increased displacement of PON1 and PAF-AH from HDL. The displacement of these two beneficial HDL-associated proteins (see below) likely explains the increased atherosclerosis seen in dyslipidemic mice that overexpress either human or murine apoA-II. However, recent clinical studies in human patients show that the higher the plasma apoA-II concentration the lower is the risk of developing coronary artery disease (CAD).
Apolipoprotein A-IV
Apolipoprotein A-IV has multiple activities related to lipid and lipoprotein metabolism as well as the control of feeding behaviors. ApoA-IV participates in reverse cholesterol transport by promoting cholesterol efflux as well as through by activation of LCAT. ApoA-IV has also been shown to have anti-oxidant, anti-inflammatory and anti-atherosclerotic actions. ApoA-IV is secreted only by the small intestine in humans (although it is expressed in the hypothalamus) and its synthesis in the gut is stimulated by active lipid absorption. Intestinal apoA-IV synthesis is enhanced by protein tyrosine-tyrosine (PYY) secreted from the ileum. Intestinal apoA-IV, present in the circulation following ingestion of fat, as well as hypothalamic apoA-IV is an anorexigenic peptide which mediates, in part, the appetite suppressing effects of a lipid-rich meal.
Apolipoprotein E
The anti-atherosclerotic activity associated with apoE is well known. This beneficial effect of apoE is due primarily to its role in the process of receptor-mediated uptake of LDL by the liver. Although apoE-mediated hepatic uptake of LDL results in a reduction in hypercholesterolemia, apoE has also been shown to inhibit atherosclerosis without any significant effect on hypercholesterolemia. In addition, different apoE alleles have demonstrated activities. For example apoE2 stimulates endothelial nitric oxide (NO) release and has anti-inflammatory activities, whereas, apoE4 is pro-inflammatory.
Paroxonases 1 and 3
Paraoxonases are a family of enzymes that hydrolyze organophosphates. Paraoxonase 1 (PON1) is synthesized in the liver and is carried in the serum by HDL. PON1 possesses anti-oxidant properties, in particular it prevents the oxidation of LDL. Evidence suggests that the direct anti-oxidant effect of HDL, on LDL oxidation, is mediated by PON1. PON1 has been shown to enhance cholesterol efflux from macrophages by promoting HDL binding mediated by ABCA1, which in turn results in a reduction of pro-inflammatory signaling. This anti-inflammatory action of PON1 serves an anti-atherosclerotic function of the protein. That PON1 is indeed important in preventing atherosclerosis has been demonstrated in mice deficient in the protein. Atherosclerotic lesions that develop in these mice when fed a high-fat diet are twice the size that develop in similarly fed control mice. In human clinical studies, a higher level of PON1 activity is associated with a lower incidence of major cardiovascular events. Other pathological conditions in humans that are associated with oxidative stress, such as rheumatoid arthritis and Alzheimer disease, are frequently associated with reduced activity of PON1.
PON3, which is another HDL-associated paraoxonase, has also been shown to prevent the oxidation of LDL. Transgenic mice expressing human PON3 have been shown to be protected from the development of atherosclerosis, without any significant changes in plasma lipoprotein cholesterol, triglyceride or glucose levels.
Platelet-Activating Factor Acetylhydrolase (PAF-AH)
There are two major forms of PAF-AH, cytosolic and plasma lipoprotein-associated. The plasma form of PAF-AH circulates bound to HDL. Given that PAF-AH is a member of the PLA2 family and that it also circulates bound to lipoprotein it is more commonly referred to as the lipoprotein-associated PLA2 (Lp-PLA2). Experimental data suggests that Lp-PLA2, rather than PON1, is the major HDL-associated hydrolase that is responsible for the hydrolysis of oxidized phospholipids. Lipoproteins that are isolated from transgenic mice expressing human Lp-PLA2 are more resistant to oxidative stress. In addition, these mice have been shown to have reduced levels of foam cell (lipid-rich macrophages) formation and enhanced rates of cholesterol efflux from macrophages. In experimental atherosclerosis models, gene transfer of LP-PLA2 inhibits atherosclerotic lesion formation in apoE-deficient mice. In humans, Lp-PLA2 deficiency is associated with increases in cardiovascular disease, while conversely circulating levels of Lp-PLA2 serve as an independent marker of the risk for developing coronary artery disease.
Glutathione Peroxidase 1
Glutathione peroxidase 1 (GPx1) functions primarily to reduce hydrogen peroxide to water, but it has been shown to also reduce lipid hydroperoxides to corresponding hydroxides effectively detoxifying these types of abnormally modified lipids. Numerous human clinical studies indicated that GPx1 provides a protective role against the development of atherosclerosis. These effects of GPx1 have also been shown in mice deficient in apoE where concomitant loss of the peroxidase results in increased rates of atherosclerotic plaque formation. The role of GPx1 in the protection from development of atherosclerosis is most pronounced under conditions of significant oxidative stress.
Sphingosine-1-Phosphate (S1P)
S1P is a bioactive lysophospholipid involved in a number of physiologically important pathways. For more detailed information of S1P activities visit the Sphingolipid Metabolism and the Ceramides page as well as the Bioactive Lipids and Lipid Sensing Receptors page. Within the blood, HDL are known to be the most prominent carriers of S1P. Indeed, many of the biological effects of HDL are mediated, in part, via S1P binding to its cell surface receptors. Effects of HDL on endothelial cells, such as migration, proliferation, and angiogenesis, are mediated, in part, by S1P associated with HDL. HDL-associated S1P inhibits pro-inflammatory responses, such as the generation of reactive oxygen species, activation of NAD(P)H oxidase and the production of monocyte chemoattractant protein-1. While the HDL-associated forms of S1P exhibit these anti-inflammatory effects, free plasma S1P can activate inflammatory events dependent upon the receptor sub-type to which it binds.
Therapeutic Benefits of Elevating HDL
Numerous epidemiological and clinical studies over the past 10 years have demonstrated a direct correlation between the circulating levels of HDL cholesterol (most often abbreviated HDL-c) and a reduction in the potential for atherosclerosis and coronary heart disease (CHD). Individuals with levels of HDL above 50mg/dL are several time less likely to experience CHD than individuals with levels below 40mg/dL. In addition, clinical studies in which apoA-I, (the predominant protein component of HDL-c) or reconstituted HDL are infused into patients, raises circulating HDL levels and reduces the incidence of CHD. Thus, there is precedence for therapies aimed at raising HDL levels in the treatment and prevention of atherosclerosis and CHD. Unfortunately current therapies only modestly elevate HDL levels. Both the statins and the fibrates have only been shown to increase HDL levels between 5%–20% and niacin is poorly tolerated in many patients. Therefore, alternative strategies aimed at increasing HDL levels are being tested.
Cholesterol ester transfer protein (CETP) is plasma glycoprotein secreted primarily from the liver and plays a critical role in HDL metabolism by facilitating the exchange of cholesteryl esters (CE) from HDL for triglycerides (TG) in apoB containing lipoproteins, such as LDL and VLDL. The activity of CETP directly lowers the cholesterol levels of HDL and enhances HDL catabolism by providing HDL with the TG substrate of hepatic lipase. Thus, CETP plays a critical role in the regulation of circulating levels of HDL, LDL, and apoA-I. It has also been shown that in mice naturally lacking CETP most of their cholesterol is found in HDL and these mice are relatively resistant to atherosclerosis.
The potential for the therapeutic use of CETP inhibitors in humans was first suggested when it was discovered in 1985 that a small population of Japanese had an inborn error in the CETP gene leading to hyperalphalipoproteinemia and very high HDL levels. To date three CETP inhibitors have been used in clinical trials. These compounds are anacetrapib, torcetrapib, evacetrapib, and dalcetrapib. Although torcetrapib is a potent inhibitor of CETP, its’ use has been discontinued due to increased negative cardiovascular events and death rates in test subjects. Clinical trials with dalcetrapib resulted in increases in HDL (19–37%) and a modest decrease (≈6%) in LDL levels. Clinical trials with evacetrapib raised HDL by more 125% and lowered LDL by more than 30%. Clinical trials with anacetrapib resulted in a significant increase in HDL (≈130%) and lowered LDL (≈40%).
Although the outcomes of large clinical trials with CETP inhibitors have not yet led to approved therapies they have found that one potential impact of CETP inhibitors is on the subsequent development of type 2 diabetes. Regardless of the ultimate impact on cardiovascular events, administration of CETP inhibitors was found to be associated with a reduced rate of development of type 2 diabetes and also with improved plasma glucose control in test subjects who already had diabetes. Whereas the specific mechanism underlying these observations remains unclear, there is evidence that HDL exert favorable effects on pancreatic β-cell function.
As described in the section below on therapeutic intervention in hyperlipidemias/hypercholesterolemias, the fibrates (e.g. fenofibrate) are a class of drugs that has been shown to result in small increases in HDL levels. The fibrates function by activation of the peroxisome proliferator-activated receptor-α (PPARα) class of transcription co-activators. However, the level of HDL increase with the current PPARα agonists is minimal at best primarily due to lack of specificity for PPARα. Therefore, current research is focused on subtype-specific PPARα agonists that have increased potency. One compound currently being tested, GFT505, is a selective PPARα agonist with a potency 100-fold greater than fenofibrate.
The liver X receptors (LXRα and LXRβ) are transcription co-activators that are involved in the regulation of lipid metabolism and have also been associated with regulation of inflammation. LXR agonists have been shown to inhibit the progression of atherosclerosis in mouse models of the disorder. Although the precise mechanism by which these LXR agonists effect a reduction in the progression of atherosclerosis is not clear, it is known that the genes encoding ABCA1 and ABCG1 contain LXR-binding sites. In fact, LXR agonists up-regulate the expression of both ABCA1 and ABCG1 in macrophages which leads to increased reverse cholesterol transport. Less cholesterol in macrophages leads to a reduced inflammatory activity of the macrophage which in turn likely contributes to the reduced atherosclerosis.
However, there is a limitation to the utility of LXR agonists as shown by the first generation synthetic LXR ligands which activate both LXRs and lead to marked increases in hepatic lipogenesis and plasma triglyceride levels. These effects are due to the role of LXRs in activation of hepatic SREBP-1c and the resultant activation of each of its target genes as described above. Although it could be theoretically possible to enhance the reverse cholesterol effects of LXRs without targeting hepatic lipogenesis with the use of LXRβ-specific ligands since most of the hepatic responses are due to activation of LXRα, this will be a difficult challenge as the ligand binding pocket in both isoforms has been shown to be nearly identical. In addition, there are species-specific differences in overall LXR responses that need to be carefully considered meaning the use of animal models that more closely resemble humans in their metabolic pathways.
Lipoprotein Receptors
LDL Receptors
LDL are the principal plasma carriers of cholesterol delivering cholesterol from the liver (via hepatic synthesis of VLDL) to peripheral tissues, primarily the adrenals, the gonads, and adipose tissue. LDL also return cholesterol to the liver. The cellular uptake of cholesterol from both IDL and LDL occurs following the interaction of the lipoprotein particles with the LDL receptor (also called the apoB-100/apoE receptor). IDL possess both apoB-100 and apoE, where the presence of apoE enhances the binding of IDL to the LDL receptor. On the other hand, the sole apoprotein present in LDL is apoB-100, which is required for interaction with the LDL receptor, but the lack of apoE reduces the overall affinity of LDL for the LDL receptor.
The LDL receptor is encoded by the LDLR gene. The LDLR gene is located on chromosome 19p13.2 and is composed of 18 exons that generate six alternatively spliced mRNAs which encode six distinct isoforms of the LDL receptor. The longest LDL receptor isoform is a 860 amino acid precursor protein. The LDL receptor spans the plasma membrane and it is the extracellular domain that is responsible for apoB-100/apoE binding. The intracellular domain is responsible for the clustering of LDL receptors into regions of the plasma membrane termed coated pits. Associated with the extracellular domain of the LDL receptor is the enzyme called proprotein convertase subtilisin/kexin type 9 (PCSK9).
Once LDL binds the receptor, the LDL-PCSK9-LDLR complexes are rapidly internalized (endocytosed). ATP-dependent proton pumps lower the pH in the endosomes, which results in dissociation of the LDL from the receptor. The portion of the endosomal membranes harboring the LDL receptor are then recycled to the plasma membrane and the LDL-containing endosomes fuse with lysosomes. However, within the endosome some of the LDL receptor protein is degraded via the action of PCSK9 resulting in less than 100% recycling of the LDL receptor to the plasma membrane. Acid hydrolases of the lysosomes degrade the apoproteins and release free fatty acids and cholesterol. As indicated above, the free cholesterol is either incorporated into plasma membranes or esterified (by SOAT2; formerly called ACAT) and stored within the cell.
The level of intracellular cholesterol is regulated through cholesterol-induced suppression of LDL receptor synthesis and cholesterol-induced inhibition of cholesterol synthesis. The increased level of intracellular cholesterol that results from LDL uptake has the additional effect of activating SOAT2, thereby allowing the storage of excess cholesterol within cells. However, the effect of cholesterol-induced suppression of LDL receptor synthesis is a decrease in the rate at which LDL and IDL are removed from the serum. This can lead to excess circulating levels of cholesterol and cholesteryl esters when the dietary intake of fat and cholesterol exceeds the needs of the body. The excess cholesterol tends to be deposited in the skin, tendons and (more gravely) within the arteries, leading to atherosclerosis.
LDL Receptor-Related Proteins (LRP)
The LDL receptor-related protein family represents a group of structurally related transmembrane proteins involved in a diverse range of biological activities including lipid metabolism, nutrient transport, protection against atherosclerosis, as well as numerous developmental processes. The LDL receptor (LDLR) described above represents the founding member of this family of proteins. The additional LRP include LRP1, LRP1B, LRP2 (also called megalin), LRP3, LRP4 (also called MEGF7 for multiple epidermal growth factor-like domains protein 7), LRP5 (also assigned the designation LRP7), LRP6, LRP8 (also called apoE receptor 2, APOER2), LRP10, the VLDL receptor (VLDLR), and SORL1 (sortilin related receptor 1; also called sorting protein related receptor containing LDLR class A repeats).
LRP1 is also known as the apoE receptor (APOER), CD91, or α2-macroglobulin receptor (A2MR). This receptor is expressed in numerous tissues and is known to be involved in diverse activities that include lipoprotein transport, modulation of platelet derived growth factor receptor-β (PDGFRβ) signaling, regulation of cell-surface protease activity, and the control of cellular entry of bacteria and viruses. Regulation of PDFGRβ activity mediates the protective effects of LRP1 in development of atherosclerosis. LRP1 is synthesized as a 600kDa precursor that is proteolytically processed into a 85kDa transmembrane protein and a 515kDa extracellular protein. The extracellular protein non-covalently associates with the transmembrane protein. LRP1 has been shown to bind more than 40 different ligands that include lipoproteins, extracellular matrix proteins, cytokines and growth factors, protease and protease inhibitor complexes, and viruses. This diverse array of ligands clearly demonstrates that LRP1 is involved in numerous biological and physiological processes.
LRP2 was originally identified as an autoantigen in a rat model of autoimmune kidney disease called Heymann nephritis. LRP2 is expressed in numerous tissues and is found in the apical surfaces of epithelial borders as well as intracellularly in endosomes. In the proximal convoluted tubule of the kidney LRP2 is involved in the reabsorption of numerous molecules. LRP2 binds lipoproteins, hormones, vitamins, vitamin-binding proteins, proteases and, protease inhibitor complexes.
The LRP5 and LRP6 proteins serve as co-receptors in Wnt signaling (see the Signal Transduction by Wnts, TGFs, and BMPs page for more details).
Scavenger Receptors
The founding member of the scavenger receptor family was identified in studies that were attempting to determine the mechanism by which LDL accumulated in macrophages in atherosclerotic plaques. Macrophages ingest a variety of negatively charged macromolecules that includes modified LDL such as oxidized LDL (oxLDL). These studies led to the characterization of two types of macrophage scavenger receptors identified as type I and type II.
Subsequent research determined that the scavenger receptor gene family in humans consists of 27 genes. Of these 27 genes 19 encode proteins that have been classified into several subfamilies identified as the class A (SCARA) through class J (SCARJ) receptors, although the class C receptors are only expressed in insects.
After binding ligand the scavenger receptors can either be internalized, similar to the process of internalization of LDL receptors, or they can remain on the cell surface and transfer lipid, or other ligands, into the cell through caveolae or they can mediate adhesion.
Class A Scavenger Receptors
The class A receptors include the SCARA1, SCARA2, SCARA3, SCARA4, and SCARA5 receptors. The SCARA1 protein is encoded by the MSR1 (macrophage scavenger receptor 1) gene. The SCARA2 protein is encoded by the MARCO gene (macrophage receptor with collagenous structure). The SCARA4 protein is encoded by the COLEC12 (collectin subfamily member 12) gene.
The SCARA1 receptor is expressed by macrophages, mast cells, dendritic cells, vascular endothelial cells, and vascular smooth muscle cells. SCARA1 has been shown to be a receptor for oxidized LDL (oxLDL) as well as for β-amyloid, apoptotic cells, and bacteria.
Class B Scavenger Receptors
The class B receptors include the SCARB1 (more commonly called SR-B1), SCARB2, and SCARB3 receptors. The SCARB3 protein is encoded by the CD36 gene. The CD36 receptor is also known as fatty acid translocase (FAT; thus often designated CD36/FAT or FAT/CD36) and it is one of the receptors responsible for the cellular uptake of fatty acids as well as for the uptake of oxidized LDL (oxLDL) by macrophages.
The CD36 and SCARB1 genes encode closely related multi-ligand receptors that are most recognized for their roles in lipid and lipoprotein metabolism. The role of these receptors in platelet function has recently been the focus of numerous studies. Several of the identified ligands for FAT/CD36 include the gut hormone ghrelin, phosphatidylserine (PS), β-amyloid, serum amyloid A, bacterial lipopeptides, and specific forms of oxidized phospholipids (oxPL) either associated with LDL (referred to as oxLDL) or free that contain an oxidized polyunsaturated fatty acid at the sn-2 position. These latter oxPL are referred to as oxPCCD36 because they are predominantly phosphatidylcholine PL and they bind FAT/CD36.
Class D Scavenger Receptors
Other members of the human scavenger receptor superfamily include the CD68 gene encoded protein (also identified as SCARD1 or SR-D1). Expression of the CD68 gene predominates in immune cells such as monocytes, macrophages, and dendritic cells. Like the CD36/FAT receptor, CD68 binds oxLDL.
Class E Scavenger Receptors
The endothelial receptors that bind oxLDL are members of the SR-E family of scavenger receptors. The human SR-E1 (SCARE1) protein is commonly called the LOX-1 receptor (lectin-like oxidized LDL receptor-1). LOX-1 is also a member of the C-type lectin superfamily of carbohydrate recognition proteins. The receptor is also called the oxidized LDL receptor 1 (OLR1) and as such the LOX-1 protein is encoded by the OLR1 gene. The human SCARE2 protein is encoded by the CLEC7A (C-type lectin domain containing 7A) gene.
Class F Scavenger Receptors
The human SCARF class receptors include the SCARF1 and SCARF2 gene encoded proteins. The SCARF1 protein is also known as SR-F1 or SREC1. The SCARF2 protein is also known as SR-F2 or SREC2.
Class G Scavenger Receptors
The human class G receptor is more commonly called C-X-C motif chemokine ligand 16 (encoded by the CXCL16 gene). CXCL16 is expressed in vascular smooth muscle cells, monocytes, macrophages, and endothelial cells and mediates the binding of phosphatidylserine and oxLDL.
Class H Scavenger Receptors
The human SCARH class receptors include SR-H1 and SR-H2, both of which are fasciclin, EGF-like, laminin-type EGF-like and link (FEEL) domain-containing scavenger receptors. The accepted designation for the human SR-H class genes are STAB1 and STAB2 which encode the proteins called stabilin 1 (FEEL-1) and stabilin 2 (FEEL-2), respectively.
Class I Scavenger Receptors
The human class I receptors include two members commonly identified as SCARI1 and SCARI2. The SCARI1 protein is more commonly called CD163 and as such is encoded by the CD163 gene. Expression of CD163 predominates in monocytes and macrophages where it is responsible for hemoglobin recognition and clearance. As a result of this activity the protein is often referred to as the hemoglobin scavenger receptor. The SCARI2 protein is CD163 molecule like 1 which is encoded by the CD163L1 gene.
Class J Scavenger Receptors
The human class J receptor (commonly called SCARJ1) is also referred to as the Receptor for Advanced Glycation End-products (RAGE). The SCARJ1 protein is encoded by the AGER (advanced glycosylation end-product specific receptor) gene.
Significance of Macrophage Lipoprotein Receptors
The significance of the numerous receptors on the surface of macrophages that specifically bind oxLDL is that the consequences are an enhanced intravascular inflammatory state. Intravascular inflammation is a major contributor to the generation of atherosclerotic lesions. Macrophages bind oxLDL, preferentially via the FAT/CD36 receptor, but also through oxLDL binding to LOX-1, SR-AI, SR-AII, and proteins of the toll-like receptor (TLR) family. Internalization of oxLDL by macrophages induces these cells to release pro-inflammatory cytokines (predominantly IL-1β and TNFα) and smooth muscle cell growth factors as well as reactive oxygen species (ROS) and metalloproteases. All of the substances released from macrophages, following uptake of oxLDL, strongly promote the intravascular inflammatory state.
Unlike where LDL internalization (mediated via the LDL receptor) results in a reduction in LDL receptor expression, through the effects of intracellular cholesterol on the activity of SREBP, macrophage internalization of oxLDL results in increased synthesis of FAT/CD36. Within the macrophage the oxLDL serves as a ligand for PPARγ which alters the gene expression profile in these cells, where the FAT/CD36 gene is one of the major targets. The net effect is that macrophage internalization of oxLDL leads to enhanced FAT/CD36 receptor levels on the macrophage allowing enhanced uptake of oxLDL.
In addition, the release of pro-inflammatory cytokines by the macrophage results in upregulation of LOX-1 on the surface of the macrophage. The combined effects of oxLDL internalization by the macrophage are an ever progressing intravascular proinflammatory state leading to progressive atherogenesis.
The SR-BI protein has been shown to be the endogenous receptor for HDL in the liver. Additionally, the HDL-SR-BI interaction in the adrenal glands is the mechanism for the delivery of cholesterol to the steroid hormone synthesizing cells of this tissue. HDL first bind to SR-BI and then the cholesteryl esters present in the HDL are transferred to the membrane for uptake via caveolae. The importance of the fact that the HDL-SR-BI complex remains at the cell surface is evident from the observation that this ligand-receptor interaction is also involved in the removal of cholesterol from cells by HDL in the process of reverse cholesterol transport.
Lipoprotein-Associated Phospholipase A2: Lp-PLA2
Platelet activating factor (PAF) is a lipid compound of the plasmalogen family of phospholipids (ether-linked glycerophospholipid) that is involved in numerous proinflammatory activities. Inactivation of PAF was originally ascribed to an activity called PAF-acetylhydrolase (PAF-AH). Subsequent to its initial characterization, PAF-AH was shown to be a member of a large family of enzymes that all hydrolyze the sn-2 position of glycerophospholipids. This family of enzymes is the PLA2 family. A detailed discussion of the PLA2 family of enzymes can be found on the Bioactive Lipid and Lipid Sensing Receptors page.
There are two major forms PAF-AH, one that is cytosolic and one that is secreted and found in the plasma. The plasma form of PAF-AH circulates bound to lipoproteins. Given that PAF-AH is a member of the PLA2 family and that it also circulates bound to lipoprotein it is more commonly referred to as the lipoprotein-associated PLA2 (Lp-PLA2). Lp-PLA2 is found in the plasma bound primarily to LDL but is also found associated with HDL and lipoprotein(a) [Lp(a)]. Of clinical significance is the fact that Lp-PLA2 has been implicated in atherosclerosis and cardiovascular disease but its precise role in these pathophysiological processes is not completely understood.
The human Lp-PLA2 protein is encoded by the PLA2G7 gene. The PLA2G7 gene is located on chromosome 6p12.3 and is composed of 13 exons that generate two alternatively spliced mRNAs, both of which encode the same 441 amino acids precursor protein. The Lp-PLA2 protein contains two sites of N-glycosylation. The enzymatic activity of Lp-PLA2 is specific for short chain acyl groups (up to 9 methylene groups) at the sn-2 position of phospholipids. When PAF is the substrate for Lp-PLA2 the products are lyso-PAF and acetate. When phospholipids of the phosphatidylcholine (PC) family are oxidized by free radical activity (referred to as oxPL) they can be a substrate for Lp-PLA2 even if the unsaturated fatty acid at the sn-2 position is longer than 9 carbon atoms.
The ability of Lp-PLA2 to recognize oxPL as substrates is due to the presence of aldehydic or carboxylic moieties at the omega (ω) end of the sn-2 peroxidized fatty acyl residues. The products of Lp-PLA2 activity on oxPL are oxidized free fatty acids (oxFFA) and lyso-PC. Numerous types of oxPL have been identified in oxidized LDL (oxLDL) particles and many of them exhibit biological activity and exert key effects in the development of atherosclerotic lesions in the vasculature. Lp-PLA2 can also hydrolyze long chain fatty acyl phospholipid hydroperoxides, phospholipids containing isoprostanes esterified at the sn-2 position and other lipid esters such as short-chain diglycerides, triglycerides, and acetylated alkanols.
In addition to its hydrolytic activity Lp-PLA2 exhibits transacetylase activity. The transacetylase function transfers acetate and short-chain fatty acids from PAF to ether- and ester-linked lysophospholipids. The transacetylase function is evident when Lp-PLA2 is associated with LDL.
In humans with normal levels of circulating lipids and no detectable Lp(a), essentially all of the Lp-PLA2 in the plasma is bound to LDL. The interaction of Lp-PLA2 with LDL occurs through apolipoprotein B-100 (apoB-100). When plasma levels of Lp(a) rise in excess of 30mg/dL there is an enrichment in the association of Lp-PLA2 with this abnormal lipoprotein particle. When expressed as enzyme mass, Lp(a) carries 1.5–2 times more Lp-PLA2 than does LDL. As in its association with LDL, Lp-PLA2 interacts with apoB-100 in Lp(a) particles. Abnormalities in lipoprotein metabolism, such as those resulting in Lp(a) production, significantly affect the plasma levels of Lp-PLA2. For example in familial hypercholesterolemia the level of LDL-Lp-PLA2 activity increases in parallel with the severity of the hypercholesterolemia. The level of plasma Lp-PLA2 can be positively affected by low-calorie diets associated with weight loss or after drug treatment with the various classes of hypolipidemic drugs discussed below in Pharmacologic Intervention in Hyperlipidemias.
In the context of atherosclerosis and cardiovascular disease numerous epidemiological studies have shown that increased levels of plasma Lp-PLA2 approximately doubles the risk for primary and secondary cardiovascular events. In fact it is suggested that measurement of Lp-PLA2 levels is useful as a cardiovascular risk marker independent of and additive to traditional risk factors. However, whether Lp-PLA2 is a novel biomarker or is causal in the development of atherosclerotic diseases remains controversial. This is because there are both anti- or proatherogenic activities associated with Lp-PLA2.
The antiatherogenic functions of Lp-PLA2 are attributed to its role in hydrolyzing and inactivating the powerful proinflammatory lipid, PAF. Additionally, by hydrolyzing oxPL, Lp-PLA2 effectively lowers the circulating levels of this class of inflammatory mediators. On the other hand the proatherogenic and proinflammatory actions associated with Lp-PLA2 are in fact due to its hydrolysis of oxPL. The hydrolysis of oxPL releases both lyso-PC and oxFFA both of which have been shown to have proatherogenic effects.
Clinical Significances of Lipoprotein Metabolism
Fortunately, few individuals carry the inherited defects in lipoprotein metabolism that lead to hyper- or hypolipoproteinemias (see Tables below for brief descriptions). Persons suffering from type 2 diabetes, hypothyroidism, cholestatic liver disease, and kidney disease (not meant to represent all associated disorders) often exhibit abnormal lipoprotein metabolism as a result of secondary effects of their disorders.
For example, because lipoprotein lipase (LPL) synthesis is regulated by insulin, LPL deficiencies, resulting in classic familial chylomicronemia syndrome (hyperlipoproteinemia, type 1A), may occur as a secondary outcome of type 2 diabetes. Additionally, insulin and thyroid hormones positively affect hepatic LDL-receptor interactions; therefore, the hypercholesterolemia and increased risk of atherosclerosis associated with uncontrolled diabetes or hypothyroidism is likely due to decreased hepatic LDL uptake and metabolism. The cardiovascular risks associated with the level of lipoprotein(a) [Lp(a)] is discussed in the next section.
An abnormal lipoprotein, identified as lipoprotein-X (Lp-X), predominates in the circulation of patients suffering from cholestatic liver disease and is also observed in familial LCAT deficiency (FLD). Lp-X is characterized by the very low protein content of the particle (around 5%) and its predominant phospholipid (>65%) content. The protein in Lp-X is almost exclusively albumin and apoC.
Of the many disorders of lipoprotein metabolism, the familial hypercholesterolemias (FH) may be the most prevalent in the general population. Classic FH (type 2a hyperlipidemia) is an autosomal dominant disorder that results from mutations affecting the structure and function of the cell-surface receptor that binds plasma LDL removing them from the circulation. The defects in LDL-receptor (LDLR) interaction result in lifelong elevation of LDL-cholesterol (LDL-C) in the blood. The resultant hypercholesterolemia leads to premature coronary artery disease and atherosclerotic plaque formation in the coronary arteries and the aorta.
Familial hypercholesterolemia was the first inherited disorder that was recognized as being a cause of myocardial infarction (heart attack). Although the primary causes of FH are mutations in the LDLR gene (representing 60%–80% of FH patients), mutations in the apolipoprotein B (APOB) gene that cause familial ligand-defective apoB (often referred to as familial hypercholesterolemia type 2) and gain-of-function mutations in the proprotein convertase subtilisin/kexin type 9 (PCSK9) gene are also associated with autosomal dominant forms of familial hypercholesterolemia. Mutations in the APOB gene represent about 1%–5% of FH cases, whereas mutations in the PCSK9 gene represent no more than 3% of identified cases of FH.
Heterozygosity at the LDLR locus in classic FH occurs in approximately 1:500 individuals, whereas, homozygosity is observed in approximately 1:1,000,000 individuals. Familial hypercholesterolemia caused by mutations in the LDLR gene comprises six different classes of mutation. The class 1 defect (the most common) results in a complete loss of receptor synthesis. The class 2 defect results in the synthesis of a receptor protein that is not properly processed in the endoplasmic reticulum (ER) and Golgi apparatus and therefore is not transported to the plasma membrane. The class 3 defect results in an LDL receptor that is incapable of binding LDL. The class 4 defect results in receptors that bind LDL but do not cluster in coated pits and are, therefore, not endocytosed. The class 5 defects are in the EGF domains and as such affect LDL binding and therefore, this class of mutation exhibits a similar phenotype to the class 3 mutations. The class 6 defects result from mutations affecting the cytoplasmic tail of the receptor and fail to be targeted to the basolateral membrane.
FH sufferers may be either heterozygous or homozygous for a particular mutation in the LDLR, APOB, or PCSK9 genes. Homozygotes exhibit grossly elevated serum cholesterol (primarily in LDL). The elevated levels of LDL result in their phagocytosis by macrophages. These lipid-laden phagocytic cells tend to deposit within the skin and tendons, leading to xanthomas. A greater complication results from cholesterol deposition within the arteries, leading to atherosclerosis, the major contributing factor of nearly all cardiovascular diseases.
Lipoprotein(a) and Atherogenesis
Lipoprotein(a) [Lp(a)] was originally described as a new serum lipoprotein particle by Kare Berg in 1963. Lp(a) is composed of a common LDL nucleus linked to a molecule of apolipoprotein(a) [apo(a); encoded by the LPA gene] by disulfide bonds between a cysteine residue in a Kringle-IV (KIV) type 9 domain in apo(a) and a cysteine residue in apolipoprotein B-100 (apoB-100). When attached to apoB-100 the apo(a) protein surrounds the LDL molecule. Synthesis of Lp(a) occurs in the liver. The half-life of Lp(a) in the circulation is approximately 3–4 days. Although Lp(a) was described over 40 years ago its precise physiological function remains unclear. However, numerous epidemiological studies have demonstrated that elevated plasma levels of Lp(a) are a significant risk factor for the development of atherosclerotic disease.
The Kringle domains of apo(a) exhibit 75%-85% similarity to the KIV domains of plasminogen. The Kringle domain is a highly glycosylated domain found in numerous proteins and is so-called because the three dimensional structure resembles a looped Danish pastry. Each Kringle domain is composed of approximately 80 amino acid residues and the structure is stabilized by three internal disulfide bonds. There are 10 distinct sub-classes of KIV domains in apo(a) designated KIV1 through KIV10. The apo(a) KIV1 and KIV3 through KIV10 domains are present as single-copy domains. The KIV2 domain is present in a variable number of repeated copies (from 2–43) and constitutes the molecular basis for the highly variable size of Lp(a) in different individuals. Apo(a) also contains a Kringle V (KV) domain that resembles the catalytic domain of plasminogen. Indeed, the gene (LPA) encoding apo(a) gene is a member of the plasminogen superfamily and given the similarities between apo(a) and plasminogen it has been hypothesized that apo(a) influences the processes of hemostasis.
Apo(a) proteins exhibit a variability in size due to a polymorphism caused by a variable number of the KIV repeats. To date at least seven different isoforms of Lp(a) have been characterized based upon electrophoretic mobilities. These different isoforms are designated F, B, and S1 through S5. The different isoforms are grouped into low molecular weight (LMW) and high molecular weight (HMW) isoforms determined by the number of KIV repeats in the apo(a) protein found in the Lp(a). The level of Lp(a) found in healthy individuals depends upon whether their plasma contains the LMW or HMW isoforms. Individuals with the LMW isoforms have high plasma Lp(a) concentration while those with the HMW isoforms have low concentrations.
When in the circulation Lp(a) particles can be affected by oxidative modification similar to that of the other plasma lipoprotein particles. Lp(a) and oxidized Lp(a) [oxLp(a)] particles interact with macrophages via scavenger receptor uptake leading to cholesterol accumulation and foam cell formation. Indeed, oxLp(a) are phagocytosed more rapidly than other lipoprotein particles and therefore accumulate in the subendothelial space at high levels. This process can lead to progression of atherogenesis, thus accounting for the direct correlation between the plasma level of Lp(a) and coronary artery disease. In addition to oxidation of Lp(a) leading to increased foam cell production, glycation of the particle also may contribute to atherogenesis. In fact, there is a strong correlation in the level of glycated Lp(a) and the severity of hyperglycemia observed in poorly controlled type 2 diabetes.
Although the precise physiology of Lp(a) is poorly understood, as indicated above, there is a strong correlation between plasma concentration of Lp(a) and atherogenic events that lead to coronary artery disease. For a discussion of the processes of blood coagulation and the role of plasminogen visit the Hemostasis: Biochemistry of Blood Coagulation page.
Because of the high degree of similarity between apo(a) and plasminogen it is suggested that Lp(a) may contribute to the thrombotic aspects of ischemic heart disease. Lp(a) has been shown to competitively inhibit the binding of plasminogen to its receptor on endothelial cells as well as to its binding sites on fibrinogen and fibrin. This interference of plasminogen binding leads to reduced surface-dependent activation of plasminogen to plasmin.
The normal function of plasmin is to degrade the fibrin clot that forms as a result of vessel injury. Therefore, high plasma concentrations of Lp(a) may represent a source of antifibrinolytic activity. Of significance to the potential for atherogenesis, the antifibrinolytic potential of Lp(a) particles is related to their size. The LMW isoforms of Lp(a) have been shown to have a higher fibrin-binding capacity than the HMW isoforms. Lp(a) also interferes with other aspects of the normal processes of coagulation in addition to its effects on plasminogen function.
Lp(a) stimulates the production of plasminogen activator inhibitor-1 (PAI-1) leading to a reduced ability of t-PA to activate the process of clot dissolution. Increased production of PAI-1 also leads to enhanced proinflammatory events via activation of monocyte adhesion to the vessel wall.
Lp(a) has also been shown to modulate platelet activation interfering with the interaction of platelets with exposed collagen fibers in the injured vessel wall. In addition to the role of Lp(a) in inhibiting plasminogen binding, Lp(a) has been shown to inhibit the release of tissue plasminogen activator (t-PA) from endothelial cells. With reduced release of the enzyme (t-PA) that converts plasminogen to plasmin and interference with plasminogen binding to fibrin clots Lp(a) can exert a significant negative effect on the ability to dissolve blood clots.
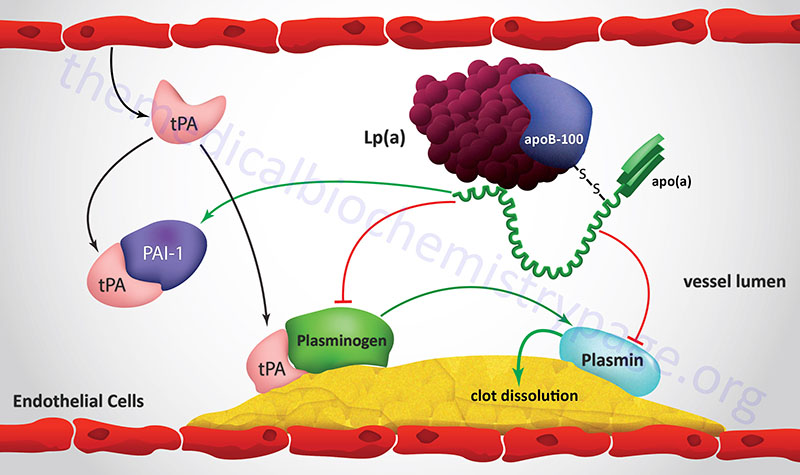
In addition to the interactions with plasminogen, leading to enhanced atherogenesis, Lp(a) has been shown to stimulate smooth muscle cell (SMC) growth. This effect of Lp(a) is exerted via an inactivation of transforming growth factor-β (TGF-β). Activated TGF-β inhibits the proliferation and migration of SMC, thus the inhibition of this regulatory effect of TGF-β leads to accelerated blood vessel stenosis with concomitant enhancement of the atherogenic process. oxLp(a) has also been shown to inhibit nitric oxide-dependent vasodilation which will tend to exacerbate the atherogenic process in hypertensive patients.
Lp(a) also interferes with other aspects of the normal processes of coagulation in addition to its effects on plasminogen function. Lp(a) stimulates the production of plasminogen activator inhibitor-1 (PAI-1) leading to a reduced ability of tissue plasminogen activator (t-PA) to activate the process of clot dissolution. Increased production of PAI-1 also leads to enhanced proinflammatory events via activation of monocyte adhesion to the vessel wall. Lp(a) has also been shown to modulate platelet activation by interfering with the interaction of platelets with exposed collagen fibers in the injured vessel wall. All of the observed effects of Lp(a) on hemostasis result in the persistence of clots which is a significant contributor to atherogenesis and increases the potential for abnormal thrombotic episodes.
Hyperlipoproteinemias and Hyperlipidemas
Numerous disorders have, at one time or another, been classified as a hyperlipoproteinemia, a hypertriglyceridemia, and/or as a chylomicronemia. Many of these disorders have been identified using each of these designations concurrently leading to confusions as to the cause and nature of a particular condition,
For example, there are several forms of inherited chylomicronemia syndromes, whose symptoms are highly similar, that are due to mutations in one of at least five genes that are involved in the removal of fatty acids from circulating lipoprotein particles. Since chylomicrons are the lipoprotein particles that are the most enriched in triglycerides, the loss of fatty acid removal results in increased circulating levels of chylomicrons and triglycerides. Hence, these disorders, which are inherited as monogenic disorders, are referred to as familial chylomicronemia syndromes, as hypertriglyceridemias, as hyperlipoproteinemias, and as hyperlipidemias.
Table of Hyperlipoproteinemias/Hyperlipidemias
| Disorder | Defect | Comments |
| Familial chylomicronemia syndrome, FCS; also known as familial LPL deficiency and as hyperlipoproteinemia, type 1A | mutations in lipoprotein lipase (LPL) gene | slow chylomicron clearance, reduced LDL and HDL levels; characterized by very severe hypertriglyceridemia, episodes of abdominal pain, recurrent acute pancreatitis, eruptive xanthomas, hepatosplenomegaly; treated by low fat/complex carbohydrate diet; no increased risk of coronary artery disease |
| Familial chylomicronemia syndrome, FCS; also known as familial apoC-II deficiency and as hyperlipoproteinemia, type 1B | mutations in APOC2 gene | loss of apoC-II function leads to reduced lipoprotein lipase activity; results in slow chylomicron clearance, reduced LDL and HDL levels; characterized by very severe hypertriglyceridemia, episodes of abdominal pain, recurrent acute pancreatitis, eruptive xanthomas, hepatosplenomegaly; treated by low fat/complex carbohydrate diet; no increased risk of coronary artery disease |
| Familial chylomicronemia syndrome, FCS; also known as hyperlipoproteinemia type 1D | mutations in GPIHBP1 gene | GPIHBP1 encoded protein required for movement of LPL from cells of adipose tissue, heart, and skeletal muscle to surface of endothelial cells of vasculature in these tissues; loss of LPL activity results in severe chylomicronemia and hypertriglyceridemia |
| Familial chylomicronemia syndrome, FCS; also known as hyperlipoproteinemia, type 5 | mutations in APOA5 gene | characterized by very severe hypertriglyceridemia, episodes of abdominal pain, recurrent acute pancreatitis, eruptive xanthomas, hepatosplenomegaly |
| Familial hypercholesterolemia, FH hyperlipidemia, type 2A | six classes of LDL receptor defect mutations in the APOB gene gain-of-function mutations in the PCSK9 gene | reduced LDL clearance leads to hypercholesterolemia (dramatic elevation in LDL cholesterol), resulting in early onset atherosclerosis and coronary artery disease |
| Familial ligand-defective apoB | four different mutations in APOB gene in the LDL receptor-binding domain: two most common are Gln for Arg at amino acid 3500 (R3500Q) or Cys for Arg at amino acid 3531 (R3531C) | is a form of autosomal dominant familial hypercholesterolemia; significant reduction in affinity of apoB-100 for the LDL receptor; dramatic increase in LDL levels; no affect on HDL, VLDL or plasma triglyceride levels; significant cause of hypercholesterolemia and premature coronary artery disease; symptoms very similar to classic familial hypercholesterolemia and as such often considered as a familial hypercholesterolemia |
| Familial dysbetalipoproteinemia hyperlipidemia, type 3; remnant removal disease, broad beta disease, apolipoprotein E deficiency | APOE gene mutations; patients only express the apoE2 isoform | apoE binds to several lipoprotein receptors including the LDL receptor, VLDL receptor, lipoprotein receptor-related protein 1 (LRP1; also called APOER), and LRP8 (also called apoE receptor 2, APOER2); apoE2 isoform interacts poorly with these receptors; results in severely reduced hepatic remnant clearance; causes tendon xanthomas, hypercholesterolemia and atherosclerosis in peripheral and coronary arteries due to elevated levels of chylomicrons and VLDL |
| Familial hypertriglyceridemia hyperlipidemia, type 4 | elevated production of VLDL associated with glucose intolerance and hyperinsulinemia | frequently associated with non-insulin dependent diabetes mellitus (type 2 diabetes), obesity, chronic alcohol consumption or administration of progesterone and related hormones; elevated total serum cholesterol as a result of increased VLDL |
| primary (familial) hyperalphalipoproteinemia 1: primary HALP1 (hyperlipidemia, type 2B) | mutations in the CETP gene | increased level of HDL; a rare condition that is beneficial for health and longevity; a related disorder, HALP2, results from mutations in the apoC-III (APOC3) gene |
| Familial LCAT deficiency (Norum disease) Fish-eye disease | complete (familial LCAT deficiency) or partial (Fish-eye disease) loss of LCAT | complete or partial absence of LCAT leads to inability of HDL to take cholesterol from cells due to loss of esterification capability; decreased levels of plasma cholesteryl esters and lysolecithin; abnormal LDL (Lp-X) and VLDL; diffuse corneal opacities, swelling of optic nerve, accumulation of fat under the skin around the eyes (xanthelasma), target cell hemolytic anemia, and proteinuria with renal failure |
| Wolman disease and cholesteryl ester storage disease (CESD) | mutations in lysosomal acid lipase (LIPA) gene | lysosomal acid lipase is also called cholesteryl ester hydrolase; Wolman disease is severe form of LIPA deficiency (no enzyme activity), CESD mild form (some residual enzyme activity); Wolman disease associated with massive infiltration of organs with macrophages filled with triglycerides and cholesteryl esters, death occurs early in life; CESD is slowly progressing disease that primarily affects the liver |
| hepatic lipase deficiency | results from mutations in the LIPC gene | deficiency of the lipase leads to accumulation of triglyceride-rich HDL and VLDL; premature atherosclerosis, tendon xanthomas, and coronary artery disease |
Table of Hypolipoproteinemias
| Disorder | Defect | Comments |
| Abetalipoproteinemia: ABL (acanthocytosis, Bassen-Kornzweig syndrome) | mutations in the MTTP gene | MTTP encodes microsomal triglyceride transfer protein; loss of function results similar pathology to familial hypobetalipoproteinemia (FHBL); intestine and liver accumulate VLDL and chylomicrons, respectively; results in malabsorption of fat, steatorrhea, deficiency in fat soluble vitamins, retinitis pigmentosa, acanthocytosis (erythrocytes with a thorny appearance), and ataxic neuropathic disease |
| Familial hypobetalipoproteinemia syndromes: FHBL | truncating mutations in the APOB gene also due to loss-of-function mutations in PCSK9 gene | both forms of FHBL inherited as autosomal dominant conditions; serum LDL concentrations 10-20% of normal; hepatic steatosis |
| Familial combined hypolipidemia | mutations in ANGPTL3 gene | angiopoietin-like protein 3 inhibits lipoprotein lipase and endothelial lipase activities; therefore loss of activity results in enhanced fatty acid removal from circulating triglycerides; associated with reduced levels of both apoB (VLDL/LDL) and apoA-I (HDL) containing lipoproteins; primary cause in most patients thus far characterized is a double mutation (TCC → TGA) converting the Ser at codon 17 to a stop codon (designated the S17X mutation) |
| Tangier disease | mutations in the ABCA1 gene | reduced HDL concentrations, no effect on chylomicron or VLDL production; tendency to hypertriglycerideemia; some elevation in VLDL; hypertrophic tonsils with orange appearance |
Pharmacologic Intervention in Hyperlipidemias
Drug treatment to lower plasma lipoproteins and/or cholesterol is primarily aimed at reducing the risk of atherosclerosis and subsequent coronary artery disease that exists in patients with elevated circulating blood lipids. Drug therapy usually is considered as an option only if non-pharmacologic interventions (altered diet and exercise) have failed to lower plasma lipids.
PCSK9 Inhibition: Alirocumab (Praluent®), Evolocumab (Repatha®)
These drugs are the newest types of anti-hyperlipidemia/hypercholesterolemia drugs recently approved by the FDA for use in the US. Both drugs are injectable antibodies that block the function of proprotein convertase subtilisin/kexin type 9, PCSK9.
PCSK9 is secreted by the liver and it binds to LDL-LDL receptor complexes on hepatocytes which, following endocytosis, stimulates the lysosomal degradation of the LDL receptor (LDLR), thereby reducing the recycling of the LDLR to the plasma membrane. This effect of PCSK9 leads to a reduced ability of the liver to remove IDL and LDL from the blood contributing to the potential for hypercholesterolemia.
The potential for the pharmaceutical benefits of the interference in the activity PCSK9 was recognized by a confluence of several studies. Patients with a specific form of familial hypercholesterolemia not due to mutations in the LDLR gene were shown to have severe hypercholesterolemia due to mutations in the PCSK9 gene resulting in hyperactivity (gain-of-function mutations) of the enzyme. In addition, it was found that in certain individuals with low serum LDL levels there was an association with the inheritance of nonsense mutations in the PCSK9 gene which result in loss of PCSK9 activity.
Hypercholesterolemic patients taking another cholesterol-lowering drug while simultaneously utilizing either of these new PCSK9 inhibitors saw further reductions in serum LDL levels of between 55% and 77%. Currently these PCSK9 antibodies can be used in conjunction with statin drugs or they can be used alone (monotherapy). Patients inject the drug subcutaneously every two weeks and even with monotherapy achieve reductions of circulating LDL in the range of 40%–60%.
A newer class of PCSK9 inhibitor that received US FDA approval in 2021, for the treatment of patients with heterozygous familial hypercholesterolemia, is based upon the RNA interference (RNAi) pathway. The drug, inclisiran (trade name Leqvio) is a small interfering RNA (siRNA) targeting the mRNA encoding the PCSK9 protein. Inclisiran is a synthetic double-stranded siRNA that has been modified with phosphorothioate substitutions and with the addition of 2′-deoxy, 2′-fluoro-RNA, 2′-O-methyl-RNA nucleotides which increase the stability and thus, the longevity and potency of the construct. The double stranded siRNA consists of a “passenger” ( the sense) strand and a “guide” (the antisense) strand. The antisense strand contains complementary sequences to PCSK9 mRNA. The “passenger” strand is conjugated to a triantennary N-acetylgalactosamine (GalNAc) carbohydrate. The use of the GalNAc carbohydrate guarantees that the siRNA molecule will gain entry into hepatocytes due to the asialoglycoprotein receptor which is abundantly expressed on hepatocytes. Once inside the hepatocyte, the “passenger” strand is degraded and the antisense strand is loaded onto the RNA-induced silencing complex (RISC), which will then detect and degrade PCSK9 mRNA. By inducing the degradation of the PCSK9 mRNA the result is a reduction in the level of functional PCSK9 enzyme associated with the LDL receptor and increased LDL receptor recycling to the plasma membrane of hepatocytes.
Statin Drugs: Atorvastatin (Lipotor®), Simvastatin (Zocor®), Lovastatin (Mevacor®)
These drugs are fungal HMG-CoA reductase (HMGR) inhibitors and are members of the family of drugs referred to as the statins. The net result of treatment is an increased cellular uptake of LDL, since the intracellular synthesis of cholesterol is inhibited and cells are therefore dependent on extracellular sources of cholesterol. However, since mevalonate (the product of the HMG-CoA reductase reaction) is required for the synthesis of other important isoprenoid compounds besides cholesterol, long-term treatments carry some risk of toxicity. A component of the natural cholesterol lowering supplement, red yeast rice, is in fact a statin-like compound.
The statins have become recognized as a class of drugs capable of more pharmacologic benefits than just lowering blood cholesterol levels via their actions on HMGR. Part of the cardiac benefit of the statins relates to their ability to regulate the production of S-nitrosylated cycloxygenase 2 (COX-2 or PGS-2). COX-2 is an inducible enzyme involved in the synthesis of the prostaglandins and thromboxanes as well as the lipoxins and resolvins. The latter two classes of compounds are anti-inflammatory lipids discussed in the Bioactive Lipid Mediators of Inflammation page. Evidence has shown that statins activate inducible nitric oxide synthase (iNOS) leading to nitrosylation of COX-2. The S-nitrosylated COX-2 enzyme produces the lipid compound 15R-hydroxyeicosatetraenoic acid (15R-HETE) which is then converted via the action of 5-lipoxygenase (5-LOX) to the epimeric lipoxin, 15-epi-LXA4. This latter compound is the same as the aspirin-triggered lipoxin (ATL) that results from the aspirin-induced acetylation of COX-2. Therefore, part of the beneficial effects of the statins are exerted via the actions of the lipoxin family of anti-inflammatory lipids.
Additional anti-inflammatory actions of the statins results from a reduction in the prenylation of numerous pro-inflammatory modulators. Prenylation refers to the addition of the 15 carbon farnesyl group or the 20 carbon geranylgeranyl group to acceptor proteins. The isoprenoid groups are attached to cysteine residues at the carboxy terminus of proteins in a thioether linkage (C-S-C). A common consensus sequence at the C-terminus of prenylated proteins has been identified and is composed of CAAX, where C is cysteine, A is any aliphatic amino acid (except alanine) and X is the C-terminal amino acid.
In addition to numerous prenylated proteins that contain the CAAX consensus, prenylation is known to occur on proteins of the RAB family of RAS-related G-proteins. There are at least 60 proteins in this family that are prenylated at either a CC or CXC element in their C-termini. The RAB family of proteins are involved in signaling pathways that control intracellular membrane trafficking. The prenylation of proteins allows them to be anchored to cell membranes. In addition to cell membrane attachment, prenylation is known to be important for protein-protein interactions. Thus, inhibition of this post-translational modification by the statins interferes with the important functions of many signaling proteins which is manifest by inhibition of inflammatory responses.
Some of the effects on immune function that have been attributed to the statins are attenuation of autoimmune disease, inhibition of T-cell proliferation, inhibition of inflammatory co-stimulatory molecule expression, decreases in leukocyte infiltration, and promotion of a shift in cytokine profiles of helper T-cell types from Th1 to Th2. Th1 cells are involved in cell-mediated immunity processes, whereas, Th2 cells are involved in humoral immunity process. The cytokines produced by Th2 cells include IL-4, IL-5, IL-10 and IL-13 and these trigger B cells to switch to IgE production and to activate eosinophils.
Complications of Statin Therapies
Although the use of statin class drugs have proven effective at preventing cardiovascular disease, these drugs are not without potentially severe negative outcomes. The most common negative effects of the statins are rhabdomyolysis and impaired liver function. As the use of statins has become quite broad, nearly 1 in 4 adults over 40 years of age are taking these drugs, additional negative outcomes have become apparent including the onset of diabetes and glucose intolerance.
Recent evidence has found that statin-induced dysregulation of insulin and glucose homeostasis is the result of the inhibition of GLP-1 synthesis and release by intestinal enteroendocrine L-cells. A major mechanism, contributing to reduced intestinal GLP-1 production with statin use, is a reduction in the level of gut bacteria of the Clostridium genus.
The role of Clostridium in intestinal GLP-1 production is related to the conversion of the bile acid, chenodeoxycholic acid (CDCA) to ursodeoxycholic acid (UDCA). The Clostridium enzymes of the 7α,β-hydroxysteroid dehydrogenase (HSDH) family are responsible for CDCA conversion to UDCA. Within the gut UDCA bids to and activates the bile acid receptor, G-protein coupled bile acid receptor 1, GPBAR1 (originally identified as TGR5 and also known as GPR131) to enhance the expression of the proglucagon (GCG) gene in enteroendocrine L-cells resulting in increased production and release of GLP-1.
The significance of gut microbiota production of UDCA to the synthesis of GLP-1 and the consequent positive effects on insulin secretion and glucose homeostasis has been demonstrated with the use of dietary UDCA supplementation. Administration of UDCA results in increased GLP-1 levels, enhanced hepatic expression of the gene (CYP7A1) encoding the rate-limiting enzyme in bile acid synthesis, improved insulin sensitivity, and improved glucose tolerance.
Nicotinic acid (Niacor® and Niaspan®)
Nicotinic acid reduces the plasma levels of both VLDL and LDL by inhibiting hepatic VLDL secretion, as well as suppressing the flux of FFA release from adipose tissue by inhibiting lipolysis. In addition, nicotinic administration strongly increases the circulating levels of HDL. Patient compliance with nicotinic acid administration is sometimes compromised because of the unpleasant side-effect of flushing (strong cutaneous vasodilation).
Recent evidence has shown that nicotinic acid binds to and activates the G-protein coupled receptor identified as hydroxycarboxylic acid receptor 2, HCA2 (originally identified as GPR109A, and also called HM74A or PUMA-G). For more detailed information on the normal biological function of HCA2 go to the Bioactive Lipids and Lipid Sensing Receptors page. The identity of a receptor to which nicotinic acid binds allows for the development of new drug therapies that activate the same receptor but that may lack the negative side-effect of flushing associated with nicotinic acid. Because of its ability to cause large reductions in circulating levels of cholesterol, nicotinic acid is used to treat Type II, III, IV and V hyperlipoproteinemias.
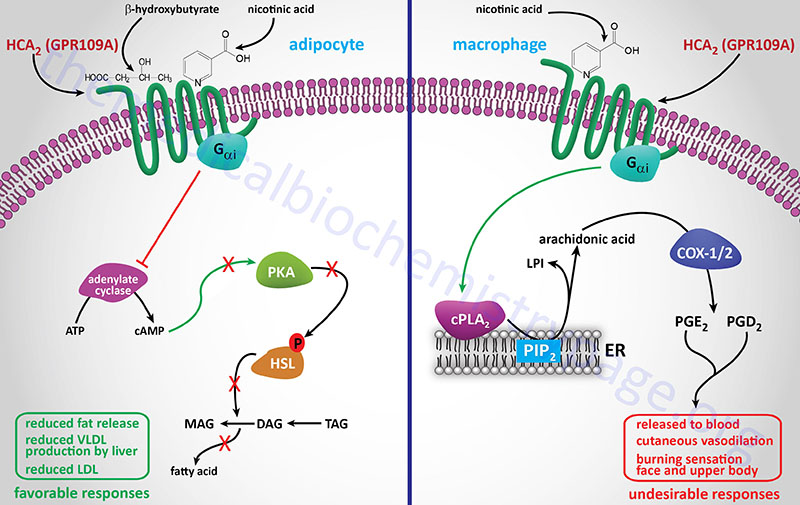
Gemfibrozil (Lopid®), Fenofibrate (TriCor®)
These compounds (called fibrates) are derivatives of fibric acid and although used clinically since the 1930’s were only recently discovered to exert some of their lipid-lowering effects via the activation of peroxisome proliferation. Specifically, the fibrates were found to be activators of the peroxisome proliferator-activated receptor-α (PPAR-α) class of proteins that are classified as nuclear receptor co-activators. The naturally occurring ligands for PPAR-α are leukotriene B4 (LTB4, see the Eicosanoid Metabolism: Prostaglandins, Thromboxanes, Leukotrienes, and Lipoxins page), unsaturated fatty acids and oxidized components of VLDL and LDL.
The PPAR interact with another receptor family called the retinoid X receptors (RXR) that bind 9-cis-retinoic acid. Activation of PPAR results in modulation of the expression of genes involved in lipid metabolism. In addition the PPAR modulate carbohydrate metabolism and adipose tissue differentiation.
Fibrates result in the activation of PPAR-α in liver and muscle. In the liver this leads to increased β-oxidation of fatty acids, thereby decreasing the liver’s secretion of triglyceride- and cholesterol-rich VLDL, as well as increased clearance of chylomicron remnants, increased levels of HDL, and increased lipoprotein lipase activity which in turn promotes rapid VLDL turnover. PPARα also activates expression of the apolipoprotein A-II () gene which contributes to the increased levels of HDL in patients administered fibrates.
Bile-Binding Resins: Cholestyramine, Colestipol
These compounds are nonabsorbable resins that bind bile acids which are then not reabsorbed by the liver but excreted. The drop in hepatic reabsorption of bile acids releases a feedback inhibitory mechanism that had been inhibiting bile acid synthesis. As a result, a greater amount of cholesterol is converted to bile acids to maintain a steady level in circulation. Additionally, the synthesis of LDL receptors increases to allow increased cholesterol uptake for bile acid synthesis, and the overall effect is a reduction in plasma cholesterol. This treatment is ineffective in homozygous FH patients, since they are completely deficient in LDL receptors.
Ezetimibe
This drug is sold under the trade names Zetia® or Ezetrol® and is also combined with the statin drug simvastatin and sold as Vytorin® or Inegy®. Ezetimibe functions to reduce intestinal absorption of cholesterol, thus effecting a reduction in circulating cholesterol. The drug functions by inhibiting the intestinal brush border transporter involved in absorption of cholesterol. This transporter is known as Niemann-Pick type C1-like 1 (NPC1L1). NPC1L1 is also highly expressed in human liver. The hepatic function of NPC1L1 is presumed to limit excessive biliary cholesterol loss. NPC1L1-dependent sterol uptake is regulated by cellular cholesterol content. In addition to the cholesterol lowering effects that result from inhibition of NPC1L1, its inhibition has been shown to have beneficial effects on components of the metabolic syndrome, such as obesity, insulin resistance, and fatty liver, in addition to atherosclerosis.
Ezetimibe is usually prescribed for patients who cannot tolerate a statin drug or a high dose statin regimen. There is some controversy as to the efficacy of ezetimibe at lowering serum cholesterol and reducing the production of fatty plaques on arterial walls. The combination drug of ezetimibe and simvastatin has shown efficacy equal to or slightly greater than atorvastatin (Lipitor®) alone at reducing circulating cholesterol levels.
Lomitapide
Certain patient populations, especially individuals that are homozygous for mutations in the LDL receptor are not effectively treated with drugs such as alirocumab and statins. Two recent drugs, that were designed to target liver lipoprotein homeostasis, have been approved for use in humans. One drug, lomitapide (Juxtapid®), is a small molecule inhibitor of the microsomal triglyceride transfer protein (MTTP; also referred to as MTP).
MTTP is a heterodimeric complex composed of a large subunit (encoded by the MTTP gene) and a small subunit which is a member of the protein disulfide isomerase (PDI) family of enzymes that are involved in protein folding. The MTTP complex is required for the incorporation of apoB-48 into chylomicrons in the intestines and apoB-100 into VLDL by the liver. Lomitapide has been shown to reduce circulating LDL in homozygous FH patients by up to 50%.
Mipomersen
The drug mipomersen (Kynamro®) is an anti-sense oligonucleotide (ASO) that targets the apoB mRNA in the liver, thereby resulting in reduced synthesis of the apoB-100 protein. Since apoB-100 is required for VLDL assembly in the liver there is reduced VLDL secretion by the liver. In homozygous LDL receptor gene mutation patients with familial hypercholesterolemia (FH) who take mipomersen there was an observed reduction of circulating LDL by approximately 25%.
Bempedoic Acid
Bempedoic acid is a dicarboxylic acid that was demonstrated to inhibit fatty acid and cholesterol synthesis in experimental animals and these effects were correlated to reductions in plasma triglyceride and lipoprotein levels. The ability of bempedoic acid to modulate lipid metabolism, in a manner reflective of the role of fatty acids in these processes, defines the molecule as a decoy fatty acid. Several other decoy fatty acids, such as gemcabene, are being tested for their efficacy at reducing hepatic lipid synthesis and thus improving health outcomes either alone or in combination with statins.
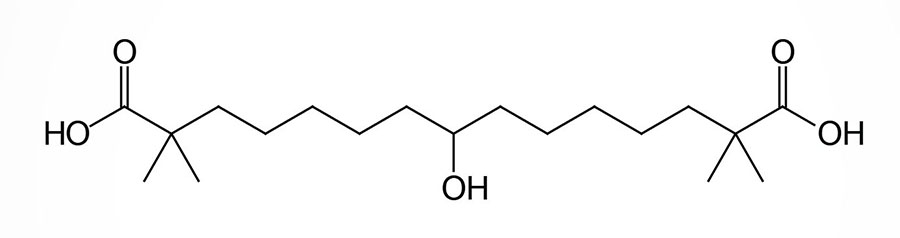
Bempedoic acid is a pro-drug that is converted exclusively in the liver to its active CoA-derivative, bempedoyl-CoA. The CoA addition to bempedoic acid is catalyzed by very long-chain acyl-CoA synthetase-1 (ACSVL1; also known as fatty acid transport protein 2: FATP2) which is encoded by the SLC27A2 gene (see the Lipolysis and the Oxidation of Fatty Acids page). The SLC27A2 gene is highly expressed in the liver but is not expressed in adipose tissue, the intestines, nor in skeletal muscle.
The conversion of bempedoic acid to its CoA derivative is believed to be required for its ability to suppress fatty acid and cholesterol synthesis and to also stimulate mitochondrial fatty acid β-oxidation. One of the targets of bempedoyl-CoA is the enzyme, ATP citrate lyase (ACLY) that hydrolyzes citrate to acetyl-CoA and oxaloacetic acid. This is a major part of the pathway of the conversion of glucose carbons into those of fatty acids and those of cholesterol. Within the liver, bempedoic acid is capable of reducing lipid levels in hepatocytes in the absence of the ACLY gene. These findings indicate that the mode of action of bempedoic acid on the lowering of hepatic lipids, and thus circulating LDL cholesterol levels, occurs through multiple distinct processes.
In addition to ATP-citrate lyase, bempedoic acid also modulates the activity of AMPK. In the case of AMPK, bempedoic acid exerts its effects in a manner similar to that of long-chain fatty acyl-CoAs (LCFA-CoAs). AMPK is a heterotrimeric enzyme composed of α-, β-, and γ-subunits. The effects of LCFA-CoAs is exerted by their activation of AMPK isoforms containing the β1-subunit. The AMPK isoform shown to be activated by bempedoic acid is the α1β1γ1 isoform. Although evidence has demonstrated that bempedoic acid does activate AMPK, this effect is dispensable for the effects of bempedoic acid on lowering plasma LDL cholesterol (LDL-C) levels.
Recent studies (2026) in mice have demonstrated that bempedoic acid can bind to an activate the nuclear receptor, peroxisome proliferator-activated receptor-alpha (PPARα). The ability of bempedoic acid to activate PPARα does not require that it first be activated to the CoA derivative, bempedoyl-CoA by the SLC27A2 encoded synthetase. The consequences of PPARα activation by bempedoic acid is enhanced hepatocyte fatty acid β-oxidation and a consequent reduction in lipid content in the hepatocytes.
One advantage of bempedoic acid over statins in the treatment of hypercholesterolemia is that the lack of SLC27A2 expression in skeletal muscle would prevent any adverse side effects in that tissue. The inhibition of muscle cholesterol synthesis by statins is a cause of the associated myotoxicity of that class of drug. Indeed, during clinical trials of bempedoic acid there was an absence of any muscle related symptoms. The US FDA approved the use of orally administered bempedoic acid alone (Nexletol™) or in combination with ezetimibe (Nexlizet™) in February of 2020.
Bempedoic acid given alone as a single daily dose of 180 mg reduces LDL cholesterol (LDL-C) by a mean of 24.5%. When administered with a statin drug the reduction in LDL-C is an additional 18% compared to a stain drug alone. The level of LDL-C reduction is between 38-40% when bempedoic acid is administered in a fixed-dose in combination with ezetimibe.
Angiopoietin-Like Protein Inhibitors
Humans express two additional LPL inhibitors encoded by the ANGPTL3 and ANGPTL4 genes. These genes encode proteins of the angiopoietin-like family that not only inhibit LPL but also inhibit endothelial lipase (encoded by the LIPG gene). Loss of function (LOF) mutations in the ANGPTL3 gene are associated with reduced levels of circulating triglycerides in a disorder called familial combined hypolipidemia. These genetic observations suggest that blocking the function of ANGPTL3 may be useful in treating hypertriglyceridemias.
However, given that ANGPTL3 activity is positively correlated to increased levels of phospholipid-rich HDL, which is highly active at cholesterol transport, there may be risks to blocking ANGPTL3 activity entirely. Indeed, humans with homozygous loss-of-function mutations in the ANGPTL3 gene have low serum levels of not only triglycerides and apo-B-containing lipoproteins (VLDL/LDL) but also low levels of apoA-I lipoproteins (HDL).
The use of a monoclonal antibody targeting circulating ANGPTL3 (evinacumab: trade name Evkeeza) demonstrated promise at lowering circulating levels of LDL in homozygous familial hypercholesterolemia (designated HoFH) patients. Patients were administered the monoclonal antibody over a period of 24 weeks and showed an average reduction in LDL cholesterol (LDL-c) of 49%. In 2021 the US FDA approved the use of Evkeeza as an adjunct to other lipid-lowering drugs for patients 12 years of age and older who are diagnosed with homozygous familial hypercholesterolemia (HoFH).
The use of RNA-targeting therapies to lower liver specific expression of ANGPTL3 has proved problematic. An antisense oligonucleotide (ASO) to the ANGPTL3 mRNA was developed that was conjugated to three molecules of N-acetylgalactosamine (GalNAc). This drug is called vupanorsen. The advantage of the GalNAc conjugation is that it specifically targets the conjugated molecule to the hepatocyte-specific asialoglycoprotein receptor 1 (encoded by the ASGR1 gene). However, clinical trials with vupanoresen were terminated due to adverse liver effects.
Nonetheless, another RNA-targeting approach has been developed utilizing an RNA interference methodology. A double stranded small-interfering RNA (siRNA) to the ANGPTL3 mRNA, that is conjugated to GalNAc (identified as ARO-ANG3), has shown promise at hepatocyte-specific reduction in ANGPTL3 mRNA levels. The results of early clinical trials with ARO-ANG3 demonstrated reductions in circulating levels of ANGPTL3 of up to 84%, reductions in circulating triglyceride levels of up to 58%, and reductions of non-HDL cholesterol of up to 26%.
Potential Therapies for Hyperlipidemias and Hyperlipoproteinemias
apoC-III Inhibitors
Several other potential targets have been identified that may prove useful for pharmacological intervention of hyperlipidemias and hypercholesterolemias. The apolipoprotein, apoC-III, (encoded by the APOC3 gene) inhibits the activity of lipoprotein lipase (LPL), endothelial lipoprotein lipase (LPL), as well as hepatic lipase. Individuals harboring loss-of-function mutations in the APOC3 gene have significantly reduced levels of circulating triglycerides suggesting that targeting this protein may be an effective treatment of hypertriglyceridemias.
The drug volanesorsen (Waylivra®), which is a 2’-O-methoxyl-modified single-stranded antisense oligonucleotide (ASO) to the mRNA encoding apoC-III, has been found to reduce triglyceride levels in patients with classic familial chylomicronemia syndrome (FCS) on the order of 50%–80%. ApoC-III is an inhibitor of LPL-dependent and LPL-independent pathways in the clearance of triglyceride-rich lipoproteins by the liver. Thus, its inhibition leads to enhanced hepatic triglyceride clearance. Although approved for use by the European Union, the US FDA has rejected approval of volanesoren due to a high correlation (25%) of thrombocytopenia in trial participants. The EU approval is exclusively for patients with documented FCS.
A next-generation version of an ASO to the apoC-III mRNA has been developed and identified as olezarsen. Olezarsen is an ASO conjugated with three molecules of N-acetylgalactosamine (GalNAc). The GalNAc conjugation allows for highly specific attachment to the hepatocyte specific asialoglycoprotein type 1 receptor which is encoded by the ASGR1 gene. The tissue specificity of this modified ASO allows for a lower dose with no loss of efficacy. The US FDA granted olezarsen an Orphan Drug designation in 2023 for the treatment of familial chylomicronemia syndrome (FCS).