
Last Updated: November 6, 2025
Introduction to Krabbe Disease
Krabbe (pronounced “crab A”) disease (also known as globoid cell leukodystrophy) is an autosomal recessive disorder that belongs to the family of disorders identified as lysosomal storage disorders. The frequency of Krabbe disease is 1 in 70,000 to 1 in 100,00 live births.
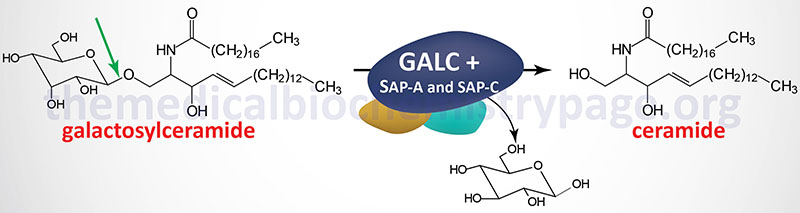
Krabbe disease is characterized by the lysosomal accumulation of galactosylceramides (galactocerebrosides) as a consequence of defects in the lysosomal hydrolase, galactosylceramidase (also called galactocerebrosidase). Galactosylceramides are almost exclusively found in the myelin sheaths of nerve cells. Thus, defects in their metabolism lead to the severe neurologic involvement of Krabbe disease.
Molecular Biology of Krabbe Disease
Galactosylceramidase is encoded by the GALC (galactocerebroside β-galactosidase) gene. The GALC gene is located on chromosome 14q31.3 spanning 56 kb and is composed of 21 exons that generate three alternatively spliced mRNAs encoding three distinct isoforms of the enzyme.
As of 2025 there have been a total of 205 pathogenic mutations identified in the GALC gene resulting in Krabbe disease.
The function of galactosylceramidase (galactocerebrosidase) requires cofactor proteins derived from the prosaposin protein, saposin A and saposin C. A more detailed description of the saposin proteins is presented in the Gaucher Disease page.
As of 2025 a total of 47 pathogenic mutations in the PSAP gene, that encodes prosaposin, have been identified, some of which affect the functions of saposin A and saposin C and thus, contribute to the delvelopment of Krabbe disease.
Clinical Features of Krabbe Disease
Because a characteristic feature of Krabbe disease is the accumulation of multinucleated globoid cells in the white matter of the brain, the disease is also known as infantile globoid cell leukodystrophy (GLD). This disease is a rapidly progressing disease that is invariably fatal before the end of the second year. There is a late-onset form of the disease that occurs in older children and adults of any age. The late-onset disease is characterized by blindness, dementia and spastic paraparesis with a longer time frame of progression but yet is still a fatal disorder.
Symptoms of the infantile form manifest around 3 to 6 months of age. Clinical manifestation of Krabbe disease is limited to the nervous system. In addition to the presence of the hematogenous globoid cells of macrophage lineage, there is near total loss of myelin and oligodendroglia as well as astrocytic gliosis. The earliest signs of Krabbe disease are hypersensitivity to external stimuli. The disease rapidly progresses to severe psycho-motor deterioration. Infants become decerebrate, are blind and usually deaf, and have no contact with their surroundings.
Treatment of Krabbe Disease
There is no treatment for the infantile form of Krabbe disease. Bone marrow transplantation (allogenic hematopoietic stem-cells) has been effective in patients with minimal neurologic involvement and the late-onset form of the disease. Bone marrow transplantation has not proven to be effective in the treatment of the infantile-onset form of Krabbe disease.
Although bone marrow transplantation does show promise for treating late-onset Krabbe disease there is a high incidence of morbidity and mortality associated with this form of therapy. In addition, the high doses of chemotherapy, with or without radiation, required to ablate the patients bone marrow prior to transplantation, the need for immunosuppressive therapy in conjunction with transplantation, and the immunological complications associated with graft-versus-host disease or graft rejection following bone marrow transplant markedly limit the effectiveness of this form of treatment.