
Last Updated: April 20, 2026
Introduction to Iron and Copper
Iron serves numerous important functions in the body with the most significant function being related to the overall metabolism of oxygen, particularly its role in hemoglobin transport of oxygen. Indeed, approximately 70% of iron in the human body is found in hemoglobin in red blood cells.
Within the body iron exist in two oxidation states: ferrous [Fe2+, Fe(II)] or ferric [Fe3+, Fe(III)]. Under conditions of neutral or alkaline pH, iron is found in the Fe3+ state and at acidic pH the Fe2+ state is favored.
Aside from its role in oxygen transport, iron is critical to the overall process of oxidative phosphorylation where it is also found in the heme of cytochromes and in the Fe-S (iron-sulfur) centers of proteins of the various complexes of oxidative phosphorylation. Because iron has an affinity for electronegative atoms such as oxygen, nitrogen, and sulfur, these atoms are found at the heart of the iron-binding centers of the proteins that require iron for function.
Ferrous iron is also a critical component in the activity of a large family of dioxygenases identified as the 2-oxoglutarate (α-ketoglutarate) and Fe2+-dependent dioxygenases, 2-OGDD.
Iron in the human body is toxic if allowed to remain free in the plasma or the fluid compartments of cells. When in the Fe3+ state, iron will form large complexes with anions, water, and peroxides. These large complexes have poor solubility and upon their aggregation lead to pathological consequences. In addition, iron can bind to, and interfere with the structure and function of various macromolecules.
The severe toxicity of free iron is related to the ability of ferrous iron to rapidly generate the highly toxic hydroxyl free radical (HO•) from hydrogen peroxide (H2O2) via the Fenton reaction. For these reasons there are extremely tight controls on overall iron homeostasis.
Fe2+ + H2O2 → Fe3+ + OH– + OH•
Iron is associated with proteins either by incorporation into protoporphyrin IX or by binding to other ligands such as the sulfur of the cysteine R-group. When the ferrous form (Fe2+) of iron and protoporphyrin IX are complexed the structure is referred to as heme. There are a number of heme containing proteins involved in the transport of oxygen (hemoglobin), oxygen storage (myoglobin) and enzyme catalysis such as nitric oxide synthase (NOS) and prostaglandin synthase (cyclooxygenase). A number of non-heme iron containing proteins are also known such as the iron-sulfur proteins of oxidative phosphorylation and the iron transport and storage proteins, transferrin and ferritin, respectively.
Like iron, copper is an essential trace element that serves numerous vital functions in the body. Within the human body copper exists in two oxidation states: cuprous [Cu+, Cu(I)] and cupric [Cu2+, Cu(II)]. The ability of this metal to readily shift between these two oxidation states, by virtue of either donating or accepting electrons, explains its essential role in numerous oxidation and reduction reactions within cells.
In addition, to its role in redox reactions of the oxidative phosphorylation pathway, copper is essential for the function of numerous metalloenzymes referred to as cuproenzymes. Among many numerous pathways, these copper-dependent enzymes function in connective tissue formation, nerve transmission, iron homeostasis, and angiogenesis.
Another critical role for copper is its participation in free radical detoxification, principally via the function of super oxide dismutase 1 (SOD1). Copper in the free state in the human body is, like free iron, highly toxic.
The toxicity of free copper is due to its high capacity to participate in redox reactions. For this reason intracellular copper homeostasis is critical and is carried out by a number of copper binding proteins and copper chaperones..
Iron Metabolism
Dietary Iron
Iron consumed in the diet is either free iron or heme iron. Free iron in the intestines is reduced from the ferric (Fe3+) to the ferrous (Fe2+) state primarily on the luminal surface (apical membrane) of intestinal enterocytes and then transported into the cell (see Figure below). The reduction reaction is catalyzed by the transmembrane ferrireductase called duodenal cytochrome b (DCYTB), a reaction facilitated by intracellular ascorbate. In the section below the role of vitamin C in iron homeostasis is described in detail.
The DCYTB protein is encoded by the cytochrome b reductase 1 (CYBRD1) gene. The CYBRD1 gene is located on chromosome 2q31.1 and is composed of 5 exons that generate three alternatively spliced mRNAs encoding three protein isoforms. The CYBRD1 gene is a member of the cytochrome b561 gene family.
Ferrous iron is then transported into the intestinal enterocyte via the action of the divalent metal transporter, DMT1. The DMT1 transporter is a member of the SLC family of transporters and is encoded by the SLC11A2 gene. The SLC11A2 gene is located on chromosome 12q13.12 and is composed of 26 exons that generate eleven alternatively spliced mRNAs that collectively encode six isoforms of the transporter.
Intestinal uptake of heme iron occurs through the interaction of dietary heme with the heme carrier protein (HCP1) encoded by the SLC family member transporter gene, SLC46A1. The SLC46A1 gene is located on chromosome 17q11.2 and is composed of 6 exons generate two alternatively spliced mRNAs encoding two distinct isoforms (isoform 1 is 459 amino acids; isoform 2 is 431 amino acids).
The iron in the heme is released within the enterocytes via the action the heme catabolizing enzyme heme oxygenase (see below). Within tissues outside the small intestine the SLC46A1 transporter functions as a proton-coupled folate transporter. Mutations in the SLC46A1 gene are, as might be expected, associated with an autosomal recessive folate malabsorption disorder.
Intestinal Iron Transport to Blood
The absorbed dietary iron can be stored within intestinal enterocytes or transported to the blood. Iron is transported across the basolateral membrane of intestinal enterocytes into the circulation, through the action of the transport protein ferroportin 1 (also called IREG1: iron-regulated transporter 1).
Ferroportin 1 is a member of the SLC family of membrane transporters and as such is encoded by the SLC40A1 gene. The SLC40A1 gene is located on chromosome 2q32.2 and is composed of 9 exons that encode a 571 amino acid protein. Ferroportin is the only identified iron export transporter in humans.
Associated with ferroportin 1 is the intestine-specific enzyme hephaestin (a copper-containing ferroxidase with homology to ceruloplasmin) which oxidizes the ferrous iron back to the ferric state. Hephaestin is encoded by the HEPH gene. The HEPH gene is located on the X chromosome (Xq12) and is composed of 28 exons that generate fourteen alternatively spliced mRNAs that collectively encode ten distinct protein isoforms.
Once dietary iron is exported to the circulation, the ferric ion is bound to transferrin and passes through the portal circulation of the liver. The liver is the major storage site for iron. The major site of iron utilization is the bone marrow where it is used in heme synthesis for incorporation into hemoglobin.
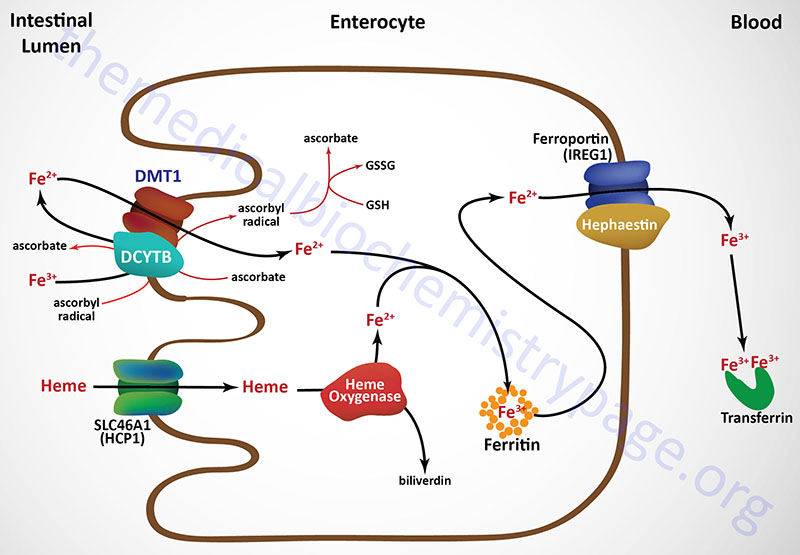
Ferritin and Iron Storage
Absorbed dietary iron can be stored within intestinal enterocytes or transported to the blood. When stored intracellularly in intestinal enterocytes, as well as all other cells (particularly hepatocytes of the liver), iron is bound inside a protein complex called ferritin. Iron-free ferritin is called apo-ferritin. Each functional ferritin complex is composed of 24 subunits that forms a shell into which the iron atoms are bound. Each ferritin complex can bind from 2,000-4,500 atoms of iron.
The ferritin complex is composed of heavy ferritin (H-ferritin) and light ferritin (L-ferritin) subunits. The H-ferritin subunits are encoded by the FTH1 gene. The FTH1 gene is located on chromosome 11q12.3 and is composed of 4 exons that encode a protein of 183 amino acids.
The L-ferritin subunits are encoded by the FTL gene. The FTL gene is located on chromosome 19q13.33 and is composed of 4 exons that encode a protein of 175 amino acids.
Ferritin complexes possess ferroxidase activity which is associated with the H-ferritin subunits. The L-ferritin subunits lack ferroxidase activity. The H-ferritin subunits thus, oxidize ferrous iron to ferric which is the form bound by ferritin.
An additional H-type ferritin gene (symbol: FTMT) encodes a ferritin protein that is localized to the mitochondria. This form of ferritin possesses ferroxidase activity as for the cytosolic H-ferritin protein and is, therefore, also involved in the sequestration of free iron. Expression of the FTMT gene is nearly exclusive to the testes with very low levels of expression detectable in other iron storage tissues.
Inside the ferritin shell, iron ions form a crystalline structure with phosphate and hydroxide ions [FeO(OH)]8[FeO(H2PO4)] that is similar to the mineral called ferrihydrite.
When iron is released from ferritin it is reduced back to the ferrous state prior to transport into the blood. The major intracellular ferrireductase is identified as STEAP3 (six-transmembrane epithelial antigen of prostate protein family, member 3). Humans express five genes in the STEAP family (STEAP1, STEAP1B, STEAP2, STEAP3, and STEAP4) with the proteins encoded by the STEAP2, STEAP3, and STEAP4 genes being metalloreductases. The STEAP1, STEAP2, STEAP3, and STEAP4 encoded proteins also belong to the large family of flavoproteins that also includes the acyl-CoA dehydrogenases and acyl-CoA oxidases that are involved in lipid oxidation.
The STEAP3 gene is located on chromosome 2q14.2 and is composed of 9 exons that generate four alternatively spliced mRNAs that collectively encode three distinct protein isoforms. The STEAP3 protein is embedded in membranes and in addition to its ability to function as a ferrireductase, it is also an iron transporter and a cupric reductase (Cu2+ to Cu+ reaction). The redox state of the STEAP3 reaction involves intracellular ascorbic acid, vitamin C (see also the Role of Ascorbate in Iron Homeostasis section).
Iron Transport, Cellular Uptake, and Storage
Transferrin (Tf), made in the liver, is the serum protein responsible for the transport of iron. Although several metals can bind to transferrin, the highest affinity is for the ferric form (Fe3+) of iron. The ferrous form (Fe2+) of iron does not bind to transferrin. Transferrin can bind two moles of ferric iron due to the protein having homologous N-and C-terminal domains, each of which can bind a ferric iron.
The transferrin is encoded by the TF gene. The TF gene is located on chromosome 3q22.1 and is composed of 24 exons that generate three alternatively spliced mRNAs encoding a 698 amino acid isoform 1 precursor protein, a 654 amino acid isoform 2 protein, and a 571 amino acid isoform 3 protein. Expression of the TF gene is almost exclusively found in the liver.
Cells take up the transported iron through interaction of iron-bound transferrin with cell-surface transferrin receptors. The major transferrin receptor is derived from the TFRC gene. The TFRC gene is located on chromosome 3q29 and is composed of 19 exons that generate four alternatively spliced mRNAs that collectively encode three distinct protein isoforms. Expression of the TFRC gene is ubiquitous with highest levels seen in the bone marrow.
Another transferrin receptor-like protein is identified as the transferrin receptor 2 encoded by the TFR2 gene. The TFR2 gene is located on chromosome 7q22.1 and is composed of 18 exons that generate two alternatively spliced mRNAs encoding two protein isoforms. Expression of the TFR2 gene is almost exclusively found in the liver.
Internalization of the iron-transferrin-receptor complexes is initiated following receptor phosphorylation by PKC. Following internalization, the iron is released from the transferrin protein due to the acidic nature of the endosomes. The transferrin receptor, along with iron-free transferrin (apotransferrin), is then recycled back to the cell surface and the apotransferrin is released back into the circulation.
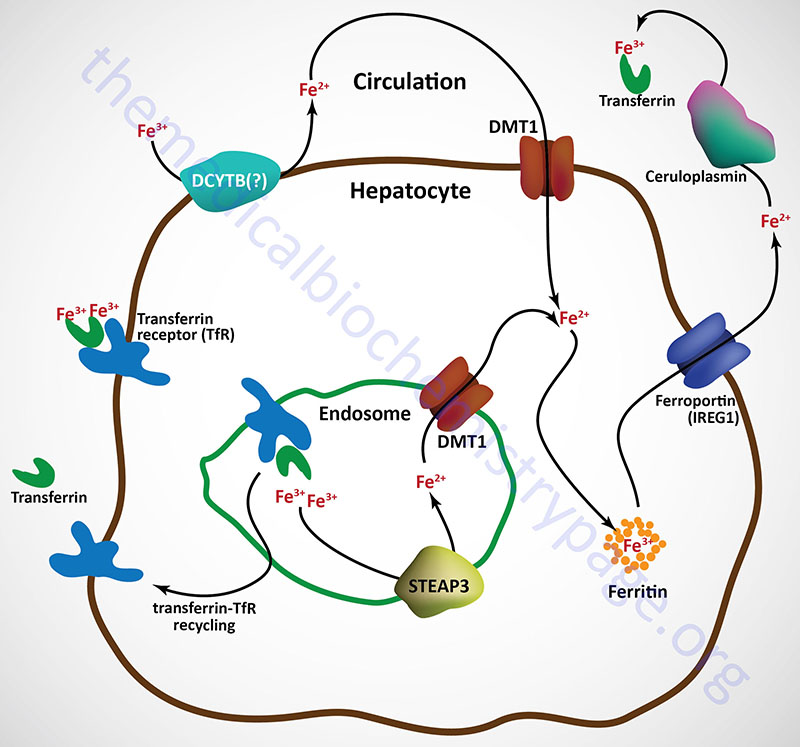
Free iron (called non-transferrin bound iron, NTBI) in the plasma can also be absorbed by hepatocytes via the action of DMT1. However, the ferric form predominates in the blood and must first be reduced by ferrireductases prior to DMT1 transport. Although not yet definitively demonstrated, the CYBRD encoded protein (DCYTB) may be the ferrireductase in the plasma membrane of the hepatocyte. NTBI can also be taken up by cells via the actions of the zinc transporters encoded SLC39A8 (ZIP8) and SLC39A14 (ZIP14) genes.
Upon binding transferrin, the transferrin receptor is internalized via receptor-mediated endocytosis. The acidic environment of the endosome results in the release of ferric iron from transferrin. The ferric iron is reduced in the endosome to the ferrous form via the action of an endosomal ferrireductase, identified as STEAP3. The ferrous iron is transported out of the endosome via DMT1 action and can then be stored in the hepatocyte bound to ferritin as in intestinal enterocytes.
The transferrin-transferrin receptor complexes are recycled back to the surface of the hepatocyte and the transferrin is released to the blood where it can bind more ferric iron in the circulation.
Ferrous iron is released from hepatocytes to the circulation through the action of ferroportin 1. When in the circulation ferrous iron is oxidized to the ferric form by the plasma ferroxidase known as ceruloplasmin. Ferrous iron release to the blood from other tissues involves a membrane-bound form of ceruloplasmin that is directly associated with ferroportin 1 as in the case of hephaestin and ferroportin 1 in intestinal enterocytes.
Ceruloplasmin is a copper-dependent ferroxidase and as such is a major Cu2+-containing protein in the blood. Loss of copper uptake from the intestines, as in the case of Menkes disease, is therefore, associated with defective iron homeostasis via the effects of copper deficiency on ceruloplasmin function. The ferric iron produced via the action of ceruloplasmin can then be bound by transferrin and delivered to other tissues of the body.
The majority of intracellularly stored iron is found in the liver, skeletal muscle, and reticuloendothelial cells. If the storage capacity of the ferritin is exceeded, iron will deposit adjacent to the ferritin-iron complexes in the cell and these complexes are partially digested by the lysosomes. Histologically these amorphous iron deposits are referred to as hemosiderin (“hE mO syd er in”). Hemosiderin is composed of ferritin, denatured ferritin, and other materials and its molecular structure is poorly defined. The iron present in hemosiderin is not readily available to the cell and thus, cannot supply iron to the cell when it is needed. Hemosiderin is found most frequently in macrophages and is most abundant following hemorrhagic events.
Hepcidin in Iron Homeostasis
In humans approximately 70% of total body iron is found in hemoglobin. Because of storage and recycling very little (1-2mg) iron will need to be replaced from the diet on a daily basis. Any excess dietary iron is not absorbed or is stored in intestinal enterocytes bound in ferritin. Refinement in the understanding of the regulation of iron absorption, recycling and release from intracellular stores was expanded through the discovery of the actions of the hepatic iron regulatory protein hepcidin.
Hepcidin is encoded by the HAMP (hepcidin antimicrobial peptide) gene. The HAMP gene is located on chromosome 19q13.12 and is composed of 3 exons encoding a preproprotein of 84 amino acids. In addition to being expressed by hepatocytes, hepcidin is also expressed by astrocytes and microglial cells in the brain and in the heart.
Hepcidin was initially described as a 25 amino acid peptide resembling cysteine-rich antimicrobial peptides. In humans the preprohepcidin protein is post-translationally cleaved into three distinct proteins of 20, 22, and 25 amino acids. The 25 amino acid form, derived from the C-terminus of the proprotein, is the functional hepcidin protein. Functional hepcidin is folded into a hairpin-like structure that is stabilized by four disulfide bonds.
Hepcidin functions by binding to the basolateral membrane-associated iron efflux transporter, ferroportin 1, in intestinal enterocytes and reticuloendothelial macrophages. Upon interaction with hepcidin, ferroportin 1 is internalized and degraded.
Regulation of Hepcidin Expression
An increased level of HAMP gene expression occurs as a consequence of iron status, erythropoiesis, inflammation, hypoxia, and endoplasmic reticulum (ER) stress (also referred to as the unfolded protein response, UPR). Several signaling processes are involved in the activation of HAMP gene expression including p53, Toll-like receptor 4 (TLR4), the IL-6/JAK2/STAT3 (interleukin-6/Janus kinase 2/signal transducer and activator of transcription 3), and the BMP/Smad pathways.
When there is excess iron in the body, or under conditions of inflammation, the HAMP gene is turned on resulting in decreased iron absorption and decreased iron release from macrophages. The decreased release of iron from macrophages during infections is believed to result in iron deprivation from invading bacteria which require the metal for their growth, specifically siderophilic bacteria. Conversely, when iron levels are deficient or during erythropoiesis or hypoxic events, expression of the HAMP gene decreases. When hepcidin expression is decreased there is increased iron absorption and increased iron release from macrophages.
The iron-mediated activation of hepatic HAMP gene expression is regulated by the level of hepatic iron, and in part, by the level of transferrin-iron saturation in the blood. The iron-dependent transcriptional activation of the hepcidin gene involves the BMP-SMAD signaling pathway. Within the liver iron triggers activation of the BMP6 gene in sinusoidal endothelial cells and the resultant BMP6 protein is released to the circulation. The released BMP6 binds to the BMP receptor on the surface of hepatocytes along with the co-receptor protein hemojuvelin (encoded by the HJV gene). The activation of the BMP receptor triggers the phosphorylation of several SMAD proteins (SMAD1, SMAD5, and SMAD8). Phosphorylation of the SMAD proteins then leads to activation of SMAD4 and/or SMAD7 and migration into the nucleus where it activates transcription of the HAMP gene.
As transferrin-iron saturation increases, more transferrin-bound iron is taken into hepatocytes which activates a signaling cascade involving the primary hemochromatosis protein (HFE; also known as HFE1), transferrin receptor 2 protein (TFR2), and the hemojuvelin protein which ultimately triggers increased HAMP gene transcription. For more information on the genes encoding these three proteins go to the Hemochromatosis page.
Additionally, inflammatory pathways are known to activate expression of the HAMP gene. The pro-inflammatory cytokine, IL-6, induces the HAMP gene through activation of the signal transducer and activator of transcription 3 (STAT3) pathway. IL-6 induces the phosphorylation of STAT3 which leads to its translocation to the nucleus and subsequent HAMP gene activation. Another pro-inflammatory cytokine, IL-1β, activates the HAMP gene via the C/EBPα and BMP/SMAD signaling pathways. The ER stress pathway leads to activation of the HAMP gene via the cAMP-responsive element-binding protein 3-like protein 3 (CREB3L3; also known as CREBH) and/or the C/EBP homologous protein (CHOP) pathways. The CHOP protein is encoded by the DNA damage inducible transcript 3 (DDIT3) gene.
The inflammation-mediated activation of the HAMP gene plays a critical role in the development of iron deficiency in chronic infections. This syndrome is commonly, although erroneously, referred to as anemia of chronic disease. It is more correctly an anemia of chronic infections or inflammation. The inflammation results in elevated expression of IL-6 which then leads to inappropriate levels of hepcidin being produced by the liver. The excess hepcidin prevents adequate intestinal iron absorption leading to deficiency-associated anemia.
Conversely, during erythropoiesis the expression of the HAMP gene is repressed to allow for adequate dietary iron uptake. This erythropoiesis-mediated inhibition of HAMP expression involves the protein erythroferrone that is released from erythroblasts. Erythroferrone is a member of the the C1q/TNF-related protein (CTRP) family and as such was designated CTRP15. Erythroferrone is also produced by skeletal muscle and was identified as myonectin, a nutrient-responsive metabolic regulator (a myokine) secreted from this tissue. Erythroferrone suppresses the BMP/SMAD pathway in hepatocytes by inhibiting the phosphorylation of SMAD1, SMAD5, and SMAD8 which in turn prevents activation of expression of the HAMP gene.
Hepcidin Effects in Intestines
The hepcidin that is produced in the liver, in response to increased serum iron, is released to the circulation. Once in the circulation, hepcidin interacts with the iron transporter, ferroportin 1, in the membranes of intestinal enterocytes and macrophages promoting the phosphorylation of the transporter leading to its internalization. Once internalized in response to hepcidin interaction ferroportin 1 is ubiquitylated by the E3 ubiquitin ligase encoded by the RNF217 (ring finger protein 217) gene. The function of RNF217 requires the E1 protein encoded by the UBA6 (ubiquitin like modifier activating enzyme 6) gene.
Downregulation of basolateral ferroportin 1 results in increased intracellular iron levels in enterocytes. The increased iron levels promotes iron-dependent degradation of the the HIF2α subunit of the HIF2 transcription factor complex. HIF2 is involved in the transcription of the genes encoding the enterocyte apical membrane ferrous iron transporter, DMT1, and the gene encoding the enterocyte ferrireductase, DCYTB.
The net effect of hepcidin, at the level of intestinal iron homeostasis is reduced uptake from the intestinal lumen and reduced transport from the enterocyte to the blood.
Hepcidin Imbalance and Associated Pathology
Given that too little or too much iron can have serious pathological consequences, hemochromatosis in iron excess, and anemia in iron deprivation, the regulation of iron homeostasis by hepcidin is of critical importance. Numerous proteins that function in overall iron homeostasis directly, or indirectly, interact with and modify the functions of hepcidin. Details on the various forms of inherited hemochromatosis (hereditary hemochromatosis) can be found in the Hemochromatosis page so the details will not be covered here but simply reviewed in the context of hepcidin function.
The majority of patients suffering from hereditary hemochromatosis harbor mutations in the HFE gene and this disorder is referred to as type 1 hemochromatosis. The protein encoded by the HFE gene interacts with the transferrin receptor 1 (TFR1) to regulate its internalization following binding of transferrin-iron complexes.
A related protein, identified as transferrin receptor 2 (TFR2). is essentially exclusively expressed in hepatocytes where it senses iron levels and is involved in the regulation of hepcidin expression. Mutations in the TFR2 gene are associated with a form of hereditary hemochromatosis referred to as type 3 (often designated HFE3).
Mutations in the gene encoding hepcidin (the HAMP gene) are associated with a juvenile onset form of hemochromatosis termed type 2B.
Another juvenile onset hemochromatosis (identified as type 2A) results from mutations in the HJV gene which encodes the protein identified as hemojuvelin. The function of hemojuvelin is in the overall regulation of hepcidin expression. Hemojuvelin serves as a co-receptor for BMP6 (bone morphogenetic protein 6) at both type I and type II BMP receptors, thereby serving to participate in the regulation of transcriptional activation of the hepcidin gene. In response to increased serum iron there is increased BMP6 expression which then binds to and activates BMP receptors on hepatocytes resulting in increased hepcidin expression.
On the opposite side of the spectrum is iron deficiency and there are aspects of hepcidin regulation that contribute to these pathologies. A serine protease identified as matripase-2, which is encoded by the TMPRSS6 gene, functions to reduce the level of hemojuvelin that interacts with the BMP receptors. The actions of matripase-2, therefore, will result in reduced levels of hepcidin gene expression. Mutations in the TMPRSS6 gene are associated with an inherited form of iron deficiency known as iron-refractory iron deficiency anemia (IRIDA).
The loss of matripase-2 activity results in loss of negative regulation of hepcidin expression causing elevated hepcidin release from the liver and the consequent reduction in ferroportin 1 levels in the basolateral membranes of intestinal enterocytes and reticuloendothelial macrophages. The effect of this dysregulation is a deficiency in adequate iron delivery to the blood and an associated anemia. As described above, chronic infection can lead to increased IL-6 production which results in excessive hepcidin expression in hepatocytes and, due to the resultant reduction in basolateral membrane ferroportin, an iron deficiency associated anemia.
Iron Homeostasis via Regulated Translation
The regulation of iron utilization in the body is primarily controlled via iron-mediated regulation of mRNA translation. The description of this process can be found in the Protein Synthesis (Translation): Processes and Regulation page. Both the transferrin receptor and the ferritin mRNAs contain stem-loop structures termed iron responsive elements, IREs. These IREs are recognized by an iron-binding protein containing an iron-sulfur center similar to that of the TCA cycle enzyme aconitase. This IRE-binding aconitase is called aconitase 1 and is encoded by the ACO1 gene. The TCA cycle aconitase is called aconitase 2 and is encoded by the ACO2 gene. Other IRE containing mRNAs include those encoding ferritin, ferroportin 1, DMT1, the erythrocyte protoporphyrin synthesis enzyme, ALAS2, and mitochondrial aconitase 2 (ACO2).
Role of Ascorbate in Iron Homeostasis
Ascorbate (vitamin C) is an essential co-factor in the overall process of iron homeostasis. Ascorbate stimulates dietary iron absorption, contributes to plasma transferrin-bound iron uptake following binding of the transferrin-iron complexes to the plasma membrane transferrin receptor, stimulates the synthesis of the iron storage protein ferritin, inhibits lysosomal ferritin degradation, and inhibits cellular iron efflux.
The absorption of non-heme iron, from the lumen of the small intestine, requires that the dietary iron, which is predominantly in the oxidized state (Fe3+), be reduced to the ferrous (Fe2+) state. Ferrous iron is then taken up into intestinal enterocytes via the action of the divalent metal transporter 1 (DMT1) protein.
There are at least three mechanisms known to exist that contribute to the process of dietary iron reduction, all of which involve ascorbic acid. The major mechanism for intestinal iron reduction involves a ferrireductase that is a member of the cytochrome b561 class of transmembrane redox enzymes. This intestinal ferrireductase is known as duodenal cytochrome b561 (DCYTB) which is encoded by the CYBRD1 (cytochrome b reductase 1) gene. During the process of DCYTB-mediated reduction of luminal ferric iron, intracellular ascorbate is oxidized to an ascorbyl radical and then to dehydroascorbate.
Ascorbic acid, as an ascorbyl radical within the lumen of the gut is also capable of reducing ferric iron to ferrous iron which can then be absorbed via DMT1. A third mechanism for intestinal ferric iron reduction involves ascorbic acid being oxidized to dehydroascorbate in the lumen of the intestine concomitant with reduction of ferric to ferrous iron. The resulting ferrous iron is absorbed by the enterocyte via DMT1 while the dehydroascorbate is transported into the enterocyte most likely via the action of GLUT1.
When stored within cells, iron is complexed with a heteromeric protein called ferritin. Ferritin, that stores iron is composed of 24 subunits, that include heavy ferritin (H-ferritin) and light ferritin (L-ferritin). When stored inside ferritin, the ferrous iron that was transported into the cell is oxidized back to the ferric state. The ferroxidase activity of ferritin is associated with H-ferritin. When ferritin stored iron is released it is reduced back to the ferrous state. This intracellular reduction process involves ascorbic acid.
Ferrous iron can be transported into the circulation from intestinal enterocytes and from hepatocytes which represent the major iron storage cells in the body. This transport involves the transmembrane transporter identified as ferroportin 1. Associated with ferroportin 1 in the membrane of intestinal enterocytes is the ferroxidase called hepheastin which oxidizes the ferrous iron to the ferric state. When iron is transported from hepatocytes to the blood via ferroportin 1 action, the ferrous iron is oxidized by the major plasma copper-dependent ferroxidase, ceruloplasmin.
The oxidation of ferrous iron to ferric is required for plasma transport as the major plasma iron transport protein, transferrin, only binds the ferric state iron. The iron bound to transferrin is taken up by most cells after binding of the transferrin-iron complexes to the plasma membrane transferrin receptor followed by endocytosis of the ligand-receptor complex. The internalization and release of iron and its reduction to the ferrous state within the cell involves intracellular ascorbic acid.
Role of Lactate in Iron Homeostasis
Elevated serum levels of lactate are common following exercise and are also observed to be associated with sepsis, hypoxia, and many cancers. Observations made in individuals with hyperlactic acidemia have found that many have associated anemia, decreased serum levels of iron, and increased levels of hepcidin. Indeed, experiments in humans and mice have shown a correlation between serum lactate levels and the levels of hepcidin. Most of the lactate produced by the body, predominantly red blood cells and skeletal muscle, is transported into the blood via the monocarboxylate transporter 4 (MCT4) and is then taken up by hepatocytes via MCT1 and MCT2. The uptake of lactate allows the liver to eliminate this acid, predominantly through the process of gluconeogenesis but also through oxidation to pyruvate which can then be completely oxidized within the TCA cycle.
Recent studies have shown that lactate plays a role in iron homeostasis through its ability to directly bind to soluble adenylate cyclase (sAC) thereby activating a signaling cascade resulting in increased expression of the HAMP gene that encodes hepcidin. The specificity of this effect for lactate was demonstrated by the lack of HAMP gene induction with pyruvate or citrate. As described in the section above on hepcidin, numerous stimuli result in the activation of hepcidin synthesis. Elevated levels of lactic acid result in increased levels of SMAD1, SMAD5, and SMAD8 phosphorylation resulting in increased expression of the SMAD7 gene and subsequently increased HAMP gene expression.
Plasma membrane receptors to which lactate is known to bind, such as HCA1 (formerly identified as GPR81) and GPR132, are not involved in the lactate-mediated increases in HAMP gene expression. Nonetheless, it has been shown that cAMP levels increase in response to increased lactate indicating that the cAMP, and subsequently activated PKA, are involved in the signaling pathway that leads to increased HAMP gene expression. Lactate treatment results in the activation of several genes that are downstream of PKA activation. In hepatocytes, the activation of PKA and increased expression of the HAMP gene requires both lactate uptake and the presence of an active soluble adenylate cyclase (sAC). It has been shown that lactate activates sAC through direct binding.
Major Iron-Dependent Proteins
Proteins that require iron for activity can be divided into two broad categories that are defined as the heme iron and the non-heme iron proteins. Well over 75 enzymes that require iron for activity are active in human tissues. The major non-enzymatic heme iron-dependent proteins are hemoglobin and myoglobin. The major heme iron-dependent enzymes are the cytochromes of the oxidative phosphorylation pathway, soluble guanylate cyclase involved in the function of nitric oxide (NO), the anti-oxidant enzyme catalase, and the multiple members of the cytochrome P450 family of xenobiotic metabolizing enzymes found in the liver.
The non-heme iron-dependent enzymes can be divided into two sub-groups dependent upon whether the protein utilizes ferrous (Fe2+) or ferric (Fe3+) iron. In addition, many of the non-heme iron-dependent enzymes function as a result of coordination of the iron to sulfur groups of cysteine residues in the protein. The resultant functional domains in these latter types of enzymes are referred to as iron-sulfur (Fe-S) centers. Probably the two most significant enzymes non-heme iron-dependent enzymes that possess Fe-S centers are the NADH:ubiquinone reductase of Complex I and succinate:ubiquinone reductase of Complex III, both components of the electron transport chain.
Table of Several Iron-Dependent Enzymes
| Enzyme Name | Gene Symbol | Form of Iron | Functions / Comments |
| Aconitases | ACO1, ACO2 | Fe4S4 | the protein encoded by ACO1 functions in the iron-mediated control of translation of the H-ferritin, L-ferritin, transferrin receptor, DMT1, ferroportin 1, ALAS2, and ACO2 mRNAs; the ACO2 encoded protein is involved in the TCA cycle |
| Alcohol dehydrogenases | 7 different genes | belong to medium-chain dehydrogenase/reductase (MDR) superfamily; catalyze the oxidation of various alcohols to their corresponding aldehydes; important in the detoxification/metabolism of ethanol | |
| Catalase | CAT | Fe3+ protoporphyrin IX | primary reaction is to detoxify the reactive oxygen species (ROS) hydrogen peroxide (H2O2) to water; can also oxidize certain alcohols to corresponding aldehydes |
| Cytochrome c reductase | multiple subunits including the Fe-S protein encoded by the UQCRFS1 gene | Fe2S2 | multisubunit component of oxidative phosphorylation Complex III; contains two cytochromes b (b-562 and b-566), cytochrome c1, and the Fe-S protein which is called the Rieske Fe-S protein after its discoverer J.S. Rieske; official name of this enzyme complex is ubiquinol-cytochrome c reductase |
| Lipoxygenases | ALOX5, ALOX12, ALOX15 | Fe3+ | all three lipoxygenases (5-LOX, 12-LOX, and 15-LOX) are involved in arachidonic acid oxidation during the synthesis of the leukotrienes and the lipoxins |
| Lysyl hydroxylases | PLOD1, PLOD2, PLOD3 | Fe2+ | official name for these enzymes is procollagen-lysine, 2-oxoglutarate 5-dioxygenase; PLOD1 is the major procollagen lysine hydroxylating enzyme; all 3 enzymes function as homodimers; PLOD2 and PLOD3 carry out hydroxylations in collagen-like proteins; mutations in PLOD1 are associated with Ehlers-Danlos syndrome (EDS) type VI, mutations in PLOD2 or PLOD3 are associated with EDS type VIB |
| NADH-ubiquinone reductase | multiple Fe-S subunit genes | three Fe4S4 clusters and three Fe2S2 clusters | multisubunit component of oxidative phosphorylation Complex I |
| Phenylalanine hydroxylase | PAH | Fe2+ | catalyzes the conversion of phenylalanine to tyrosine; mutations in the PAH gene result in phenylketonuria, PKU |
| Prolyl 4-hydroxylase (two α, two β subunits) | three α subunit genes: P4HA1, P4HA2, P4HA3; β subunit gene: P4HB | Fe2+ | catalyzes the formation of 4-hydroxyproline residues in procollagen |
| Ribonucleotide reductase (contains 2 subunits) | RRM1, RRM2 | Fe2+ | catalyzes the conversion of ribonucleoside diphosphates to their corresponding deoxyribonucleotide diphosphates |
| Stearoyl-CoA desaturase | SCD | Fe2+ | one of three fatty acid desaturases in humans; stearoyl-CoA desaturase is the rate-limiting enzyme catalyzing the synthesis of monounsaturated fatty acids (MUFA), primarily oleate (18:1; a physiologically significant omega-9 fatty acid) and palmitoleate (16:1) |
| Succinate-ubiquinone reductase | multiple Fe-S subunit genes | three Fe-S centers: Fe2S2, Fe4S4, Fe3S4 | multisubunit component of oxidative phosphorylation Complex II |
| Thyroid peroxidase | TPO | Fe2+ protoporphyrin IX | exclusively expressed in the thyroid gland; within the thyroid colloid TPO oxidizes I– to I+; reaction requires H2O2 |
| Tryptophan hydroxylase | TPH2 | Fe2+ | initial enzyme in the conversion of tryptophan to the neurotransmitters, serotonin and melatonin |
| Tyrosine hydroxylase | TH | Fe2+ | initial enzyme in the conversion of tyrosine to the catecholamines, dopamine, norepinephrine and epinephrine |
| Xanthine oxidase (derived from xanthine dehydrogenase) | XDH | Fe2S2 | also requires molybdenum for function; xanthine dehydrogenase can be converted to xanthine oxidase by reversible sulfhydryl oxidation or by irreversible proteolytic modification; catalyzes the conversion of hypoxanthine to xanthine and xanthine to uric acid in the catabolism and salvage of purine nucleotides |
Clinical Aspects of Abnormal Iron Metabolism
Iron can bind to and form complexes with numerous macromolecules, the consequences of which can be a disruption in normal activities of the affected complexes. Excess intracellular iron results in formation and deposition of hemosiderin which can lead to cellular dysfunction and damage. Thus, the consequences of excess iron intake and storage can have profound consequences. However, one must also consider that a reduction in iron intake can also lead to untoward consequences. Most notably, a reduced iron level negatively affects the function of oxygen transport in red blood cells. Defects in iron metabolism can result from impaired intestinal absorption, excess loss of heme iron due to bleeding as well as to mutations in the iron response elements (IRE) of iron regulated mRNAs.
Hemochromatosis is defined as a disorder in iron metabolism that is characterized by excess iron absorption, saturation of iron-binding proteins and deposition of hemosiderin in the tissues. The primary affected tissues are the liver pancreas and skin. Iron deposition in the liver leads to cirrhosis and in the pancreas causes diabetes. The excess iron deposition leads to bronze pigmentation of the organs and skin. In fact, the bronze skin pigmentation seen in hemochromatosis, coupled with the resultant diabetes lead to the designation of this condition as bronze diabetes.
The primary cause of hemochromatosis is the inheritance of an autosomal recessive allele. The locus causing hemochromatosis has been designated the HFE1 and is a major histocompatibility complex (MHC) class-1 gene located on chromosome 6. The gene encodes an α chain protein with three immunoglobulin-like domains. This α chain protein associates with β2-microglobulin. Normal HFE1 has been shown to form a complex with the transferrin receptor and in so doing is thought to regulate the rate of iron transfer into cells. A mutation in HFE1 will therefore, lead to increased iron uptake and storage.
The majority of hereditary hemochromatosis patients have inherited a mutation in HFE1 that results in the substitution of Cys 282 for a Tyr (C282Y). This mutation causes loss of conformation of one of the immunoglobulin domains in HFE1. Another mutation found in HFE1 causes a change of His 63 to Asp (H63D). There are several additional causes of iron overload, although none are as common as classic hemochromatosis. There are at least four additional genetic loci, that when defective, lead to hemochromatosis. Two of these additional forms of inherited iron overload are juvenile forms, identified as type 2A and 2B hemochromatosis. The other two forms are referred to type 3 and type 4 hemochromatosis. See the Hemochromatosis page for more information.
Iron deficiency anemia is characterized by microcytic (small) and hypochromic (low pigment) red blood cells. Reduced iron intake and/or excess iron excretion results in a decreased globin protein content in red blood cells as a consequence of the heme control of globin synthesis [go to the Protein Synthesis (Translation): Processes and Regulation page for details]. The most common causes of iron deficient anemia are excess menstrual flow or gastrointestinal (GI) bleeding. Causes of GI bleeding can include the use of medications that lead to ulceration or erosion of the gastric mucosa, peptic ulcer disease, gastric tumors, hiatal hernia or the gastritis associated with chronic alcohol consumption. Treatment of iron deficiency anemia is to first determine the cause and source of the excess bleeding. Oral administration of ferrous sulfate is commonly used to supplement the iron loss, however, intravenous iron therapy may be called for in some cases. Severe iron deficiency anemia may necessitate transfusion with packed red blood cells.
GRACILE syndrome (GRACILE = growth retardation, aminoacidurina, cholestasis, iron overload, lactic acidosis, early death) is a very rare neonatal lethal disorder caused by defects in the BCS1L gene located on chromosome 2q33. BCS1L stands for BCS1-like which is the human homolog of a yeast gene identified as BCS1. BCS1L encodes a member of the AAA family of ATPases that is necessary for the assembly of complex III of oxidative phosphorylation. Defects in BCSL1 are also responsible for Björnstad syndrome.
Ferroptosis
Ferroptosis is the term used to describe an iron-dependent form of regulated cell death (RCD) that is distinct from the classical pathway of caspase-mediated apoptosis, autophagy, and necrosis. Ferroptosis is characterized by iron-dependent accumulation of lipid peroxides. There are two major pathways via which ferroptosis can occur, the extrinsic and the intrinsic pathways.
The extrinsic pathway is the transporter dependent pathway of ferroptosis that involves inhibition of the heterodimeric cystine and glutamate antiporters that transport cystine in the opposite direction to that of glutamate. This transporter system is a member of the acidic amino acid transporter family and is commonly designated as the Xc– transporter system. The Xc– transporter is composed of a heavy chain (4F2hc) and a light chain (xCT). The 4F2hc protein is encoded by the SLC3A1 gene. The xCT protein is encoded by the SLC7A11 gene.
The intrinsic pathway of ferroptosis is the more well characterized and is also referred to as the enzyme regulated
pathway. Inhibition of glutathione peroxidase 4 (GPX4), starvation of cysteine, and the induction of the peroxidation of arachidonic acid all trigger the intrinsic pathway of ferroptosis. Inhibition of glutathione peroxidase activity results in a decrease in antioxidant capacity and an accumulation of lipid reactive oxygen species (ROS), ultimately leading to oxidative cell death.
For details on the processes of ferroptosis go to the Protein, Organelle, and Cell Turnover page.
Copper Metabolism
Dietary Copper Uptake
The absorption of dietary copper occurs almost exclusively within the small intestine following consumption of the free metal or after its release from proteins during gastric and duodenal digestive processes. Absorption of copper from the lumen of the intestines is very high ranging from 55% to 75% of the amount in the chyme.
In order for copper to be absorbed it first needs to be reduced from the cupric (Cu2+) to the cuprous (Cu+) state. This reduction occurs via the action of one or more cupric reductases. Several enzymes on the apical membrane of intestinal enterocytes are capable of reducing cupric copper including the duodenal cytochrome b (DCYTB) that is also capable of reducing ferric iron. An additional plasma membrane-associated enzyme shown to reduce cupric copper is the six-transmembrane epithelial antigen of prostate protein family member 2 (STEAP2) protein. As indicated above in the discussion of iron homeostasis, humans express five genes that encode proteins of the STEAP family with the proteins encoded by the STEAP2, STEAP3, and STEAP4 genes being metalloreductases.
Once in the cuprous state, copper is transported into the intestinal enterocyte primarily by the transporter identified as CTR1 (copper transporter 1). The CTR1 transporter functions as a homotrimer. Some cupric copper (Cu2+) uptake from the lumen of the intestines has also been shown to occur through the activity of the divalent metal transporter 1 (DMT1) that functions principally in intestinal iron uptake. Within cells, CTR1 is localized both to the plasma membrane and to membranes of intracellular vesicles. The intracellular localization of CTR1 allows it to function in the maintenance of copper storage and intracellular transport homeostasis. One of the intracellular functions of CTR1, in enterocytes, is to make the absorbed dietary copper available for further utilization by facilitating its release from intracellular vesicles.
In addition to CTR1, another copper transport protein has been characterized and is identified as CTR2. In comparison to CTR1, CTR2 is a low affinity copper transporter. Both copper transporters are members of the solute carrier family of transporters. The CTR1 protein is encoded by the SLC31A1 gene while the CTR2 protein is encoded by the SLC31A2 gene. Both genes are located on chromosome 9 in close proximity to each other. Interestingly, although both proteins participate in some pathway of copper transport, there is no amino acid sequence identity between the two proteins. Both the SLC31A1 and SLC31A2 genes are located at 9q32. The SLC31A1 gene is composed of 5 exons that encode a protein of 190 amino acids. The SLC31A2 gene is composed of 4 exons that encode a protein of 143 amino acids.
Once copper is picked up by intestinal CTR1 it is transferred directly to the intracellular chaperone identified as antioxidant 1 copper chaperone (ATOX1) which is encoded by the ATOX1 gene. Copper is then transported from the apical membrane to either intracellular vesicles (where CTR1 and/or CTR2 promote its uptake into the vesicles) or to the copper transporting ATPase (Cu-ATPase), ATP7A, within the trans-Golgi network (TGN) for ultimate export from the enterocyte to the blood. The significance of ATP7A function to overall copper homeostasis is evidenced by the fact that loss of its function due to inherited mutations in the ATP7A gene result in the lethal disorder identified as Menkes disease.
Copper Transport and Storage
Once in the blood, copper is transported throughout the body bound primarily to albumin and to a much lesser extent by α2-macroglobulin. Although up to 95% of copper in the blood is bound to the ferroxidase called ceruloplasmin, ceruloplasmin copper is not part of the exchangeable plasma copper pool. Indeed, ceruloplasmin does not directly bind nor take up copper ions. Copper is incorporated into soluble ceruloplasmin only during the synthesis of the protein by the liver. Most cells also synthesize ceruloplasmin but the form of the enzyme synthesized by non-hepatic tissues remains bound to the plasma membrane via a GPI linkage.
The major function of soluble ceruloplasmin is as the primary ferroxidase in the blood responsible for the oxidation of iron from the ferrous (Fe2+) to the ferric (Fe3+) state. This oxidation of iron is critical to prevent the formation of the hydroxyl and hydroxide free radicals that occurs via the interaction of Fe2+ with hydrogen peroxide (H2O2) in what is termed the Fenton reaction. In addition, transport of iron throughout the body in the blood is the function of transferrin which only binds ferric (Fe3+) iron. The role of the GPI-linked form of ceruloplasmin is to facilitate iron efflux from cells. Indeed, the role of copper in the ferroxidase function of ceruloplasmin is the reason copper is so very critical for overall iron homeostasis.
Copper (Cu+) uptake into cells from the blood occurs predominantly via the action of CTR1 as in the case of dietary copper uptake by intestinal enterocytes. Recent (2024) evidence has shown that the zinc export transporter identified as ZnT1 is also involved in the uptake of copper, but in the case of ZnT1 it is cupric (Cu2+) copper transport. The gene encoding ZnT1 is a member of the solute carrier (SLC) family of transporters, SLC30A1. Both Cu2+ and Zn2+ bind to the same sites on ZnT1 but their transport is not coupled. The identification of the role of ZnT1 in copper transport into cells explained previous observations that zinc blocks copper uptake and provided the mechanism of therapeutic use of zinc in the treatment of Wilson disease.
As in the case of intestinal enterocytes, within other cells of the body copper is transferred from CTR1 to the ATOX1 protein. Another intracellular copper chaperone important in overall copper homeostasis is copper chaperone for superoxide dismutase, CCS. The function of CCS is to activate cytosolic Cu, Zn-dependent superoxide dismutase (SOD1) by inserting copper ion and by introducing a disulfide bond in the enzyme.
Although the majority of copper is stored within hepatocytes, most cells have the capacity to store some copper. This stored copper can be accessed so as to incorporate it into proteins that require bound copper for their functions, several of which are listed in the Table below.
While stored inside the cell, copper is primarily localized to lysosome-like compartments. These storage sites are oxidizing compared with the cytosol and, therefore, specific cupric reductases are required to maintain copper in the cuprous state. The STEAP family of reductases are the most likely enzymes carrying out this function since STEAP3 and STEAP4 are located in intracellular vesicles. Intracellular copper is also stored bound to copper-binding proteins, with the major class of proteins being the metallothioneins.
Metallothioneins are so named because of their ability to bind heavy metals and their high cysteine content. The primary metals bound by the metallothioneins are zinc and copper. Humans express four distinct metallothioneins identified as MT1, MT2, MT3, and MT4. The MT1 and MT2 proteins are the most widely expressed while MT3 and MT4 expression is primarily within the central nervous system. Both the MT1 and MT2 genes are inducible by numerous stimuli, such as heavy metals, whereas the MT3 and MT4 genes are not.
The function of ATOX1 in liver, and other non-intestinal cells, is the same as that of its function in intestinal enterocytes, to carry copper from the plasma membrane to intracellular vesicles via the trans-Golgi network (TGN). Within the TGN ATOX1 can transfer copper to the primary copper-transporting ATPase (Cu-ATPase) of the secretory pathway. This ATPase is identified as ATP7B. The significance of ATP7B function to overall copper homeostasis is evidenced by the fact that loss of its function due to inherited mutations in the ATP7B gene result in the disorder identified as Wilson disease.
The distribution of CTR1 between the plasma membrane and intracellular membranes is a dynamic process regulated by the overall level of copper ions in the body. When copper levels rise in the extracellular compartment, CTR1 is endocytosed from the plasma membrane to intracellular vesicles, thereby, decreasing copper uptake. Even in the absence of CTR1 some copper still enters cells, most likely as a result of CTR2 function, however, the amount of copper entry is insufficient to support the activity of all of the copper-dependent enzymes.
In addition to ATOX1 and CCs there are several other important copper chaperones, one of which is critical for overall mitochondrial function. This chaperone is identified as cytochrome c oxidase copper chaperone 17 (COX17). Cytochrome c oxidase is the terminal enzyme of the mitochondrial respiratory chain where it is a component of complex IV and catalyzes electron transfer from reduced cytochrome c to molecular oxygen generating H2O. COX17 carries copper ions from the cytoplasm into the mitochondrial intermembrane space. Cytochrome c oxidase assembly factor 6 (COA6) functions as a thiol-disulfide oxidoreductase that reduces the formation of disulfide bonds between cysteine residues in two proteins involved in cytochrome c oxidase assembly, synthesis of cytochrome c oxidase1 (SCO1) and SCO2. The function of COA6 allows copper transfer from COX17 to SCO1 and SCO2 and subsequently from SCO1 and SCO2 to cytochrome c oxidase. Thus, mitochondrial oxidative phosphorylation is dependent on overall copper homeostasis. In addition to cytochrome c oxidase assembly, SCO1 and SCO2 are involved in overall intracellular copper homeostasis as is evidenced by the decrease in cellular copper levels when either protein is non-functional.
Table of Major Copper-Dependent Proteins
Numerous proteins are dependent upon copper for function. It is not the intention of this discussion to cover them all
| Enzyme Name | Gene | Functions |
| Amine oxidase | AOC1, AOC3 | official names are amine oxidase copper containing 1 and 3; AOC1 encoded enzyme is most commonly identified as diamine oxidase, DAO; AOC1 oxidatively deaminates amine species into aldehydes and ammonia; major substrates for AOC1 include putrescine and histamine |
| Ceruloplasmin | CP | major ferroxidase in the blood; each enzyme binds 6–7 Cu2+ (cupric) ions; plays a major role in ensuring no free iron in the circulation; oxidizes Fe2+ (ferrous) iron to Fe3+ (ferric) iron which can then be bound to transferrin, the major iron transporting protein in the blood; ceruloplasmin is often misrepresented as the major copper transporting protein of the blood due to the fact that up to 95% of copper in the blood is found in this enzyme, however, the major function of ceruloplasmin is as a ferroxidase not as a copper transporter; two CP isoforms generated via alternative mRNA splicing, one form is secreted the other is attached to the plasma membrane via a GPI linkage; secreted CP synthesized exclusively by the liver, the GPI-linked CP is expressed by numerous organs including the brain, liver, kidneys, and lungs; the GPI-linked CP is primarily responsible for iron efflux from tissues; aceruloplasminemia, due to defects in the CP gene, doesn’t affect copper homeostasis but manifests with iron overload of a form referred to as hemosiderosis; the CP gene is located on chromosome 3q24-q25.1 and is composed of 21 exons that 1065 amino acid precursor protein; although the CP gene is expressed in tissues other than the liver, the level of expression in the liver is on the order of 400 times that of any of these other tissues |
| Cytochrome c oxidase | 13 genes | composed of 13 subunits that comprise the mitochondrial oxidative phosphorylation complex IV; mitochondrial genome harbors MT-CO1, MT-CO2, and MT-CO3 genes; nuclear genome harbors the other ten genes: COX4, COX5A, COX5B, COX6A, COX6B, COX6C, COX7A, COX7B, COX7C, COX8; functions to re-oxidized reduced cytochrome c while subsequently reducing molecular oxygen to water; the ferric (Fe3+) iron in complex IV is the site of cyanide (CN– binding) |
| Dopamine β-hydroxylase (dopamine β-monooxygenase) | DBH | involved in catecholamine synthesis, catalyzes hydroxylation of dopamine to norepinephrine; expression limited to adrenal medulla and post-ganglionic sympathetic neurons |
| Hephaestin | HEPH | functions as a ferroxidase (similar to ceruloplasmin); expression is limited to intestinal villi cells; required for iron transport from intestinal enterocytes to the blood; dietary iron is transported from enterocytes to the blood via the action of ferroportin 1 with simultaneous oxidation of Fe2+ (ferrous) iron to Fe3+ (ferric) iron by hephaestin; ensures the iron can be bound to transferrin for delivery to the tissues |
| Lysyl oxidase | LOX | catalyzes the oxidative deamination of the ε-amino group of lysine and hydroxylysine residues in collagens and lysine residues of elastin; results in cross-linking of protein forming fibrils |
| Methionine synthase (homocysteine methyltransferase) | MTR | official name is 5-methyltetrahydrofolate-homocysteine S-methyltransferase; catalyzes the conversion of homocysteine to methionine; is one of only two enzymes that require vitamin B12 (as methylcobalamin); as the name implies the enzyme also requires N5-methyl-THF for activity; defects in the MTR gene, or deficiency in either folate or B12 (or both), result in homocysteinemia/homocystinuria and macrocytic anemia |
| Cu-Zn Superoxide dismutase | SOD1 | major cytoplasmic anti-oxidant enzyme; catalyzes conversion of superoxide free radicals to molecular oxygen (O2) and hydrogen peroxide (H2O2); the major mitochondrial superoxide dismutase (SOD2) is a manganese-dependent enzyme |
Clinical Aspects of Copper Homeostasis
As discussed above, copper plays a crucial role in the overall state of iron homeostasis, in addition to its role in numerous other critical metabolic processes. For these reasons a loss of copper absorption from the gut can result in profound clinical consequences. Like iron in the free state, copper in the free state is toxic to the body when at high levels. This consequence of copper is due to its ability to disturb the redox state of the cell. For this latter reason excess intracellular copper, in amounts that swamp out the intracellular copper chaperone and copper-binding protein capacity can lead to serious clinical outcomes. With respect to copper, two proteins, encoded by two different yet functionally related genes are associated with the two highest impact inherited disorders in copper homeostasis. These two genes encode the Cu-transporting ATPases, ATP7A and ATP7B.
As indicated above, mutations in the ATP7A gene result in the lethal disorder, Menkes disease. The details of Menkes disease are described in the Menkes disease page. Briefly, the consequences of defective ATP7A function are the result of loss of copper absorption from the diet and, therefore, to global loss of copper-dependent enzyme function. Three important copper-requiring enzymes reflect most of the clinical consequences of Menkes disease. These enzymes are lysyl oxidase, ceruloplasmin, and dopamine β-hydroxylase. The neurological complications of Menkes disease are due not only to loss of important copper-dependent enzymes, such as dopamine β-hydroxylase, but also to loss of iron efflux from the brain, as a result of the loss of ceruloplasmin function. The inability to undertake iron efflux leads to iron overload in neurons and glial cells. The increased iron results in redox-active iron and an associated increase in the Fenton catalysis of ferrous (Fe2+) iron which involves the generation of the highly cytotoxic hydroxyl free radical from hydrogen peroxide (H2O2).
Mutations in the ATP7B gene result in Wilson disease, the details of which are discussed in the Wilson disease page. Loss of the Cu-transporting ATPase encoded by the ATP7B gene results in excess copper storage intracellularly, most significantly within hepatocytes. This is due to the fact that ATP7B is responsible for copper export from the liver via the secretory pathway of the bile canaliculi. Excess intracellular copper deposition most affects the liver as this is the major tissue for overall whole body copper homeostasis.
Cuproptosis
intracellular copper concentration is tightly regulated given the fact that even moderate increases can cause cytotoxicity and even lead to cell death. Copper excess is associated with the induction of a novel type of cell death that has been termed cuproptosis.
Cuproptosis represents a form of regulated cell death that is characterized by the aggregation of lipoylated mitochondrial enzymes and a loss of iron-sulfur (Fe–S) proteins. The distinction of the cell death pathway identified as cuproptosis has been demonstrated through the use of specific pharmacological inhibition of other known cell death pathways including ferroptosis (inhibited with ferrostatin-1) and apoptosis (inhibited with Z-VAD-FMK). The inhibition of these other cell death pathways failed to inhibit copper mediated cell death initiated with the use of a copper-specific ionophore. In contrast, the use of a copper chelator was shown to rescue cells from excess Cu2+-induced cell death.
For details on the processes of cuproptosis go to the Protein, Organelle, and Cell Turnover page.