
Last Updated: February 19, 2025
Keq, Kw and pH
As H2O is the medium of biological systems one must consider the role of this molecule in the dissociation of ions from biological molecules. Water is essentially a neutral molecule but will ionize to a small degree. This can be described by a simple equilibrium equation:
H2O ↔ H+ + OH–
This equilibrium can be calculated as for any reaction:
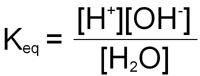
Since the concentration of H2O is very high (55.5M) relative to that of the [H+] and [OH–], consideration of it is generally removed from the equation by multiplying both sides by 55.5 yielding a new term, Kw:
Kw = [H+][OH–]
This term is referred to as the ion product. In pure water, to which no acids or bases have been added:
Kw = 1 x 10–14 M2
As Kw is constant, if one considers the case of pure water to which no acids or bases have been added:
[H+] = [OH–] = 1 x 10–7 M
This term can be reduced to reflect the hydrogen ion concentration of any solution. This is termed the pH, where:
pH = –log[H+]
pKa
Acids and bases can be classified as proton donors and proton acceptors, respectively. This means that the conjugate base of a given acid will carry a net charge that is more negative than the corresponding acid. In biologically relevant compounds various weak acids and bases are encountered, e.g. the acidic and basic amino acids, nucleotides, phospholipids etc.
Weak acids and bases in solution do not fully dissociate and, therefore, there is an equilibrium between the acid and its conjugate base. This equilibrium can be calculated and is termed the equilibrium constant = Ka. This is also sometimes referred to as the dissociation constant as it pertains to the dissociation of protons from acids and bases.
In the reaction of a weak acid:
HA ↔ A– + H+
the equilibrium constant can be calculated from the following equation:
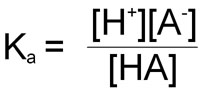
As in the case of the ion product:
pKa = –logKa
Therefore, in obtaining the –log of both sides of the equation describing the dissociation of a weak acid we arrive at the following equation:
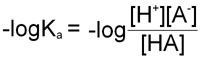
Since as indicated above –logKa = pKa and taking into account the laws of logarithms:
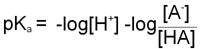
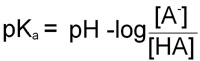
From this equation it can be seen that the smaller the pKa value the stronger is the acid. This is due to the fact that the stronger an acid the more readily it will give up H+ and, therefore, the value of [HA] in the above equation will be relatively small.
The Henderson-Hasselbalch Equation
By rearranging the last equation above we arrive at the Henderson-Hasselbalch equation:
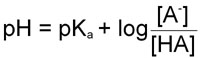
It should be obvious now that the pH of a solution of any acid (for which the equilibrium constant is known, and there are numerous tables with this information) can be calculated knowing the concentration of the acid, HA, and its conjugate base [A–].
At the point of the dissociation where the concentration of the conjugate base [A–] = to that of the acid [HA]:
pH = pKa + log[1]
The log of 1 = 0. Thus, at the mid-point of a titration of a weak acid:
pKa = pH
In other words, the term pKa is that pH at which an equivalent distribution of acid and conjugate base (or base and conjugate acid) exists in solution.
Buffering
It should be noted that around the pKa the pH of a solution does not change appreciably even when large amounts of acid or base are added. This phenomenon is known as buffering. In most biochemical studies it is important to perform experiments, that will consume H+ or OH– equivalents, in a solution of a buffering agent that has a pKa near the pH optimum for the experiment.
Clinical Significance of Blood Buffering
The pH of blood is maintained in a narrow range around 7.4. Even relatively small changes in this value of blood pH can lead to severe metabolic consequences. Therefore, blood buffering is extremely important in order to maintain homeostasis. Although the blood contains numerous cations (e.g., Na+, K+, Ca2+ and Mg2+) and anions (e.g., Cl–, PO43– and SO42–) that can, as a whole, play a role in buffering, the primary buffers in blood are hemoglobin in erythrocytes and bicarbonate ion (HCO3–) in the plasma. Buffering by hemoglobin is accomplished by ionization of the imidazole ring of histidines in the individual globin proteins.
The formation of bicarbonate ion in erythrocytes from CO2 and H2O allows the transfer of relatively insoluble CO2 from the tissues to the lungs, where it is expelled. The major source of CO2 in the tissues comes from the oxidation of ingested carbon compounds.
Carbonic acid is formed from the reaction of dissolved CO2 with H2O and is catalyzed by carbonic anhydrase. Humans express 12 functional isoforms of carbonic anhydrase (CA) including cytosolic, secreted, membrane-bound, and mitochondrial versions. Carbonic anhydrases I and II, both of which are cytosolic, are expressed within erythrocytes. Carbonic anhydrase II is also the major CA isoform expressed in the proximal convoluted tubule (PCT) of the kidneys. The relationship between carbonic acid and bicarbonate ion formation is shown in the two equations below:
CO2 + H2O ↔ H2CO3
H2CO3 ↔ H+ + HCO3–
These reactions occur predominately within erythrocytes, since nearly all of the CO2 leaving tissues via the capillary endothelium is taken up by these cells. Ionization of carbonic acid then occurs spontaneously yielding bicarbonate ion.
Carbonic acid is a relatively strong acid with a pKa of 3.8. However, carbonic acid is in equilibrium with dissolved CO2. Therefore, the equilibrium equation for the sum of the two previous equations requires a conversion factor, since CO2 is a dissolved gas. This factor has been shown to be approximately 0.03 times the partial pressure of CO2 (PCO2). When this is entered into the Henderson-Hasselbalch equation:
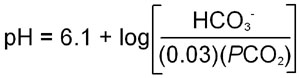
where the apparent pKa for bicarbonate formation, 6.1, has been introduced into the equation.
The PCO2 in the peripheral tissues is approximately 50mm Hg, whereas in the blood entering the peripheral tissues it is approximately 40mm Hg. This difference results in the diffusion of CO2 from the tissues into the blood in the capillaries of the periphery. When the CO2 is converted to H2CO3 within the erythrocytes and then ionizes, the hydrogen ions (H+) are buffered by hemoglobin. The production of H+ ions, within erythrocytes, and their subsequent buffering by hemoglobin results in a reduced affinity of hemoglobin for oxygen. This leads to a release of O2 to the peripheral tissues, a phenomenon is termed the Bohr effect.
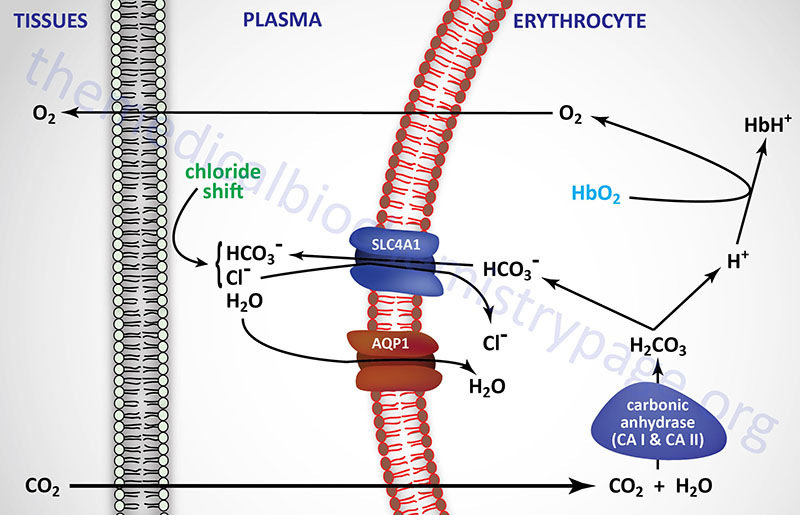
As CO2 passes from the tissues to the plasma a minor amount of carbonic acid takes form and ionizes. The H+ ions are then buffered predominantly by proteins and phosphate ions in the plasma. As the concentration of bicarbonate ions rises in erythrocytes, an osmotic imbalance occurs. The imbalance is relieved as bicarbonate ion leaves the erythrocytes in exchange for chloride ions from the plasma. This phenomenon is known as the chloride shift which is also shown in the diagram above. Therefore, the majority of the bicarbonate ion formed from CO2 leaves the peripheral tissues and is transported by the plasma to the lungs.
Around 15% of CO2 transport from the tissues to the lungs occurs through a reversible combination with non-ionized amino groups (–NH2) of hemoglobin forming hemoglobin carbamate.
Hemoglobin-NH2 + CO2 ↔ Hemoglobin-NH-COO– + H+
The formation of hemoglobin carbamate results in a reduced affinity of hemoglobin for O2 thus favoring dissociation of bound oxygen in the tissues where the concentration of CO2 is high. The process is reversed when the erythrocytes enter the lungs and the partial pressure of O2 is elevated.
The partial pressure of O2 (PO2) in the pulmonary alveoli is higher than the PO2 of the entering erythrocytes that contain predominantly deoxygenated hemoglobin. This increased PO2 leads to oxygenation of hemoglobin and release of H+ ions from the hemoglobin. The released H+ ions combine with the bicarbonate ions to form H2CO3. Cellular carbonic anhydrase then catalyzes the reverse reaction, leading to release of CO2 from erythrocytes. Owing to the PCO2 gradient (described above), the CO2 diffuses from the blood to the alveoli where it is expelled.
The great utility of bicarbonate as a physiological buffer stems from the fact that if excess acid is added to the blood the concentration of bicarbonate ion declines and the level of CO2 increases. The CO2 then passes from capillaries in the pulmonary alveoli and is expelled. As a consequence, the H+ ion concentration drives the reaction to the left and bicarbonate ion acts as a buffer until all of the hydrogen ion is consumed. Conversely, when excess base is added to the blood, CO2 is consumed by conversion to carbonic acid and replaced by metabolic reactions within the body.
If blood is not adequately buffered, the result may be metabolic acidosis or metabolic alkalosis. These physiological states can be reached if a metabolic defect results in the inappropriate accumulation or loss of acidic or basic compounds. These compounds may be ingested, or they may accumulate as metabolic by-products such as acetoacetic acid and lactic acid. Both of these will ionize, thereby increasing the level of H+ ions that will in turn remove bicarbonate ions from the blood and alter blood pH. The predominant defect in acid or base elimination arises when the excretory system of the kidneys is impaired. Alternatively, if the lungs fail to expel accumulated CO2 adequately and CO2 accumulates in the body, the result will be respiratory acidosis. If a decrease in PCO2 within the lungs occurs, as during hyperventilation, the result will be respiratory alkalosis.
Anion Gap
The anion gap is measured to determine acid-base balance of the body fluids. The anion gap defines the difference between the measured anions and cations and can be calculated by subtracting the concentrations of bicarbonate (HCO3–) and chloride (Cl–) from sodium (Na+) and potassium (K+). However, because normal K+ levels are very low relative to the other ions in the calculation, the value for K+ can be omitted. The anion gap equation is normally written as:
anion gap = [Na+ – (Cl– + HCO3–)]
Using this formula a normal anion gap is less than 15 mEq/L (normal is 6-15 mEq/L). The anion gap can be, normal, high, or low. However, a low anion gap is extremely rare and so the anion gap determination is used for analysis of metabolic acidosis. If the anion gap is elevated, then there are anions in the bodily fluid that have not been accounted for in the equation. Common anions that result in an elevated anion gap include lactate and the ketone bodies (β-hydroxybutyrate and acetoacetate). Organic acids such as propionic acid and methylmalonic acid that accumulate in inborn errors of metabolism can also lead to an increased anion gap.
The most common causes of a high anion gap metabolic acidosis are lactic acidemia and the ketoacidosis associated with excessive ketone body synthesis as is typical in diabetic ketoacidosis. Numerous drugs can also lead to an increased anion gap such as aspirin and isoniazid (tuberculosis treatment). Toxins such as methanol, ethylene glycol, and cyanide will cause an increased anion gap. Renal failure will lead to an increased anion gap metabolic acidosis due to reduced acid (H+) excretion and reduced bicarbonate reabsorption.
When considering a diagnosis in a patient with a high anion gap metabolic acidosis there is a useful mnemonic to remember the most common causes. There are two commonly applied mnemonics MUDPILES (methanol, uremia, diabetic ketoacidosis, paraldehyde, infection, lactic acidosis, ethylene glycol, and salicylates) and GOLDMARK (glycols, oxoproline, L-lactate, D-lactate, methanol, aspirin, renal failure, ketoacidosis). Glycols are commonly ethylene glycol and propylene glycol. Oxoproline is a metabolite of acetaminophen (also called paracetamol). GOLDMARK is most useful for the recognition of the more common causes of high anion gap metabolic acidosis.
Ampholytes, Polyampholytes, pI and Zwitterion
Many substances in nature contain both acidic and basic groups as well as many different types of these groups in the same molecule. (e.g. proteins). These are called ampholytes (one acidic and one basic group) or polyampholytes (many acidic and basic groups). Proteins contains many different amino acids some of which contain ionizable side groups, both acidic and basic. Therefore, a useful term for dealing with the titration of ampholytes and polyampholytes (e.g. proteins) is the isoelectric point, pI. This is described as the pH at which the effective net charge on a molecule is zero.
For the case of a simple ampholyte like the amino acid glycine the pI, when calculated from the Henderson-Hasselbalch equation, is shown to be the average of the pK for the α-COOH group and the pK for the α-NH2 group:
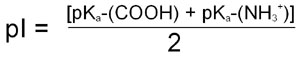
For more complex molecules such as polyampholytes the pI is the average of the pKa values that represent the boundaries of the zwitterionic form of the molecule. The pI value, like that of pK, is very informative as to the nature of different molecules. A molecule with a low pI would contain a predominance of acidic groups, whereas a high pI indicates predominance of basic groups.
Solvation and Hydration Shells
Depending on the pH of a solution, macromolecules such as proteins which contain many charged groups, will carry substantial net charge, either positive or negative. Cells of the body and blood contain many polyelectrolytes (molecules that contain multiple same charges, e.g. DNA and RNA) and polyampholytes that are in close proximity. The close association allows these molecules to interact through opposing charged groups. The presence, in cells and blood, of numerous small charged ions (e.g. Na+, Cl–, Mg2+, Mn2+, K+) leads to the interaction of many small ions with the larger macro-ions. This interaction can result in a shielding of the electrostatic charges of like-charged molecules. This electrostatic shielding allows macro-ions to become more closely associated than predicted based upon their expected charge repulsion from one another. The net effect of the presence of small ions is to maintain the solubility of macromolecules at pH ranges near their pI. This interaction between solute (e.g. proteins, DNA, RNA, etc.) and solvent (e.g. blood) is termed solvation or hydration. The opposite effect to solvation occurs when the salt (small ion) concentration increases to such a level as to interfere with the solvation of proteins by H2O. This results from the H2O forming hydration shells around the small ions.
Role of the Kidneys in Acid-Base Balance
The kidneys function to filter the plasma that passes through the nephrons. Filtration of the plasmas occurs in the glomerular capillaries of the nephron. These capillaries allow the passage of water and low molecular weight solutes (less than 70 kDa) into the capsular space. The filtrate then passes through the proximal and distal convoluted tubules where reabsorption of water and many solutes takes place. In the course of glomerular filtration and tubule reabsorption the composition of the plasma changes generating the typical composition of urine. From a biochemical standpoint the kidneys serve important roles in the regulation of plasma acid-base balance (explained in greater detail in the Renal Transporters: Biochemistry, Physiology, Pharmacology, Pathology page) and the elimination of nitrogenous wastes.
Sodium Bicarbonate Reabsorption
Regulation of plasma acid-base balance is primarily effected within the kidney through control over HCO3– reabsorption and secretion of H+. Secretion of H+, in excess of its capacity to react with HCO3– in the tubular luminal fluid, requires the presence of other buffers (see below). The generation of HCO3– and H+ occurs by dissociation of carbonic acid (H2CO3), formed in the epithelial cells of the proximal convoluted tubule (PCT) from H2O and CO2, through the action of carbonic anhydrase (CA). As indicated above, there are 12 isoforms of CA and at least six of these are found in the kidneys including cytosolic, membrane-bound, and mitochondrial versions. Cytosolic (CAII) and membrane-bound (CAIV) carbonic anhydrases in the PCT function to prevent the rapid development of limiting pH gradients in this segment and contribute to high rates of bicarbonate reabsorption.
Secretion of H+ into the lumen of the tubule is accompanied by an exchange for Na+. The reabsorption of Na+, within the PCT, occurs by an antiport mechanism coupled to the efflux of H+ primarily through the action of the Na+/H+ exchangers identified as NHE3 and NHE8. Both of these transporters belong to the solute carrier (SCL) family of transporters, and they are encoded by the SLC9A3 and SLC9A8 genes, respectively. Reduction in the intracellular concentration of Na+ occurs by an active transport process involving a Na+/K+-ATPase pump which pumps the excess Na+ out of the tubule epithelial cells into the interstitial spaces for uptake by the blood. The reabsorbed HCO3– is then transported from the PCT epithelial cell into the interstitial fluid.
Humans express several bicarbonate transporter genes, not all of which are expressed in the kidney. Many of these bicarbonate transporter genes also belong to the solute carrier (SLC) family of transporters, specifically the SLC4A and SLC9A families. The SLC4A4 gene encodes an electrogenic Na+/HCO3––cotransporter (also called NBCe1) found in the basolateral membranes of PCT cells. This transporter is responsible for about 80% of the renal bicarbonate reabsorption. Within the PCT the SCL9A3 encoded transporter is involved in Na+ reabsroption in exchange for HCO3– efflux to the tubular lumen.
Another important PCT bicarbonate transporter is encoded by the SLC26A6 gene. This transporter is expressed in the apical membranes of PCT epithelial cells and effluxes HCO3– in exchange for reabsorption of Cl–. The SLC26A6 transporter is also expressed in apical membranes of epithelial cells of the thick ascending limb (TAL) in the loop of Henle and the distal convoluted tubule (DCT). of the nephron.
Regulation of bicarbonate excretion and reabsorption also occurs within the connecting tubule (CNT) and collecting ducts of the distal nephron. Within this segment of the nephron the basolateral membranes of epithelial cells express the SLC4A1, SLC4A2, SLC4A3, SLC4A9, and SLC26A7 transporters that efflux bicarbonate to the tubular lumen. Of these CNT located basolateral transporters, the SCL4A1 encoded transporter may be the most significant in the context of renal transport of reabsorbed bicarbonate back to the blood. The SLC4A1 encoded protein is more commonly called anion exchanger 1, AE1. Within the apical membranes of the CNT and collecting ducts the SLC4A8, SLC26A4, and SLC26A6 encoded transporters are involved in bicarbonate reabsorption from (SLC4A8) or efflux to (SLC26A4 and SLC26A6) the tubular lumen.
The capacity of the kidney to secrete H+ is regulated by the maximal H+ gradient that can form between the tubule and lumen and still allow transport mechanisms to operate. This gradient is determined by the pH of the urine which in humans is near 4.5. The capacity to secrete H+ would be rapidly reached if it were not for the presence of buffers within the interstitial fluid. The H+ secreted into the tubular lumen can undergo three different fates depending upon the concentration of the three primary buffers of the interstitial fluid. These buffers are HCO3–, HPO42– and NH3. Reaction of H+ with HCO3– forms H2O and CO2 which diffuse back into the tubule cell. The net result of this process is the regeneration of HCO3– within the tubule cell. This is a mechanism of sodium bicarbonate reabsorption. As indicated, most of the bicarbonate resorption occurs in the PCT with the remainder of bicarbonate reabsorption occurring in the TAL of the loop of Henle and in the connecting tubule (CNT).
Excretion of Acid
As the concentration of HCO3– in the tubular lumen drops, the pH of the fluid drops due to an increasing concentration of H+. The pH of the tubular fluid gradually approaches the pKa for the dibasic/monobasic phosphate buffering system (pKa = 6.8). The excess H+ reacts with dibasic phosphate (HPO42–) forming monobasic phosphate (H2PO4–). The H2PO4– so formed is not reabsorbed and its excretion results in the net excretion of H+. The greatest extent of H2PO4– formation occurs within the distal convoluted tubule (DCT) and the connecting tubule (CNT).
Ammonia Secretion
Buffering of H+ is also accomplished within the blood and the kidney by reaction with ammonia, NH3, to form ammonium ion, NH4+. Indeed, renal ammoniagenesis is a critical process required by the kidneys in response to metabolic acidosis. Elimination of NH4+ is the major contributory factor in the ability of the body to excrete acid. Because the pKa of NH4+ is 9.3, excretion of acid in this form can be accomplished without lowering the pH of the urine. Additionally important is the fact that excretion of acid in the form of NH4+ occurs without depleting Na+ nor K+.
Two principal reactions, occurring within epithelial cells of the proximal tubule, result in the generation of NH3: conversion of glutamine to glutamate and conversion of glutamate to 2-oxoglutarate (α-ketoglutarate). These reactions are catalyzed by glutaminase and glutamate dehydrogenase, respectively, as shown in the Figures below.
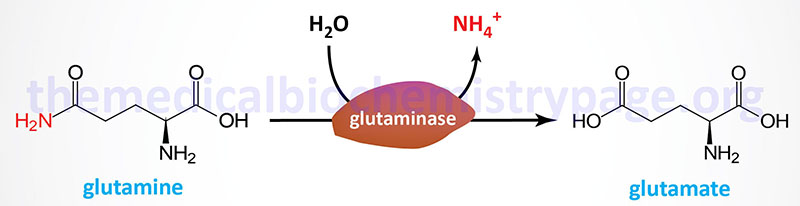
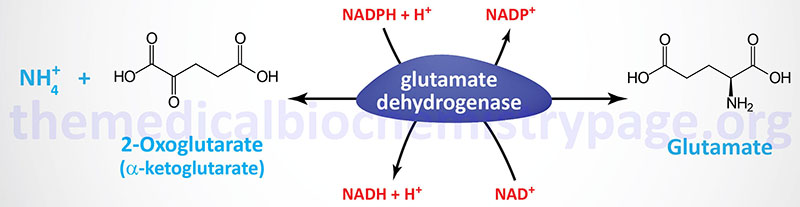
Both of these enzymes are abundant in proximal tubule cells. The ammonia released through the actions of these enzymes ionizes to ammonium ion, 50% of which is effluxed to the tubular lumen for excretion in the urine through the actions of the SLC9A3 encoded transporter commonly called Na+/H+ exchanger 3 (NHE3). The remainder of the ammonia/ammonium ion is effluxed from the PCT epithelial cells to the blood where it is delivered to the liver for incorporation into urea.
Acidosis and Alkalosis
As discussed above (and in greater detail in the Renal Transporters: Biochemistry, Physiology, Pharmacology, Pathology page) the kidneys play an important role in the control of acidosis by responding with an increase in the excretion of H+. When H+ is excreted as a titratable acid such as H2PO4– or when the anions of strong acids such as acetoacetate are excreted there is a requirement for simultaneous excretion of cations to maintain electrical neutrality. The principal cation excreted is Na+. As the level of excretable Na+ is depleted excretion of K+ increases. In conditions of acidosis the kidney will increase the production of NH3 from tubular amino acids or amino acids absorbed from the plasma. As indicated above the NH3 ionizes by picking up H+ and a is excreted in the urine. This demonstrates that an inability of the kidney to generate NH3 would rapidly lead to fatal acidosis.
When the kidneys fail to modulate HCO3– excretion, metabolic alkalosis will develop. Alkalosis is normally countered quite effectively by the kidney allowing HCO3– to freely escape. Alkalosis generally only becomes problematic if the kidneys are restricted in their ability to secrete HCO3–. This situation can occur in patients taking diuretics since several of this class of drug cause a reduction in the ability of the kidney to reabsorb an anion (e.g. Cl–) concomitant with the reabsorption of Na+.
Neurotoxicity of Ammonia
Excess ammonia is severely neurotoxic. Marked brain damage is seen in cases of failure to make urea via the urea cycle or to eliminate urea through the kidneys. The result of either of these events is a buildup of circulating levels of ammonium ion. Aside from its effect on blood pH, ammonia readily traverses the brain blood barrier and in the brain is converted to glutamate via glutamate dehydrogenase, depleting the brain of 2-oxoglutarate (α-ketoglutarate). As the 2-oxoglutarate is depleted, oxaloacetate falls correspondingly, and ultimately TCA cycle activity comes to a halt. In the absence of aerobic oxidative phosphorylation and TCA cycle activity, irreparable cell damage and neural cell death ensue.
In addition, the increased glutamate leads to glutamine formation. This depletes glutamate stores which are needed in neural tissue since glutamate is both a neurotransmitter and a precursor for the synthesis of γ-aminobutyrate: GABA, another neurotransmitter. Therefore, reductions in brain glutamate affect energy production as well as neurotransmission. For detailed information on the role of the glutamate-glutamine cycle in the brain and its role in regulating neural cell ammonia levels visit the Nitrogen Metabolism and the Urea Cycle page.
Additional untoward consequences of hyperammonemia have been attributed to an increase in neural glutamine concentration. Astrocyte cell volume is controlled by intracellular organic osmolyte metabolism. One important organic osmolyte in these cells is glutamine. As glutamine levels rise in the brain concomitant with increased ammonia uptake it was hypothesized that the volume of fluid within glial cells would increase resulting in the cerebral edema seen in infants with hyperammonemia caused by urea cycle defects.
However, there are some problems with this particular model. First, glutamine accounts for no more than 1.5% of the sum of all the osmolytes within the in the brain. Second, a significant proportion of the newly synthesized glutamine can rapidly exit astrocytes due to both diffusion and through the action of specific glutamine transporters. The primary astrocyte glutamine transporter is the sodium-coupled neutral amino acid (system N/A) transporter 3 (SNAT3: a member of the solute carrier family of transporters also identified as SLC38A3). The result of diffusion and transport is a reduction in the glutamine overload within astrocytes. Additionally, an increase in brain glutamine during other causes of hyperammonemia, such as hepatic encephalopathy, has been shown to be accompanied by loss of different low MW osmolytes, including myo-inositol, taurine, and betaine.
A current concept of the role of brain glutamine concentration in the neurotoxicity associated with hyperammonemia relates to its adverse effects on mitochondrial function. In laboratory animals it has been shown that inhibition of glutamine synthesis by methionine sulfoximine (MSO) results in suppression of ammonia-induced formation of reactive oxygen species (ROS). In addition, when glutamine is added directly to astrocyte cell cultures there is a resultant induction of mitochondrial permeability transition (mPT) and swelling of the mitochondria. This effect of glutamine can be blocked by addition of cyclosporine A (CsA), which is a known inhibitor of mPT, as well as by inhibition of mitochondrial glutamine uptake by histidine. Of note is that fact that histidine administration can prevent brain edema during drug-induced liver failure in rodents.
Additionally, studies have demonstrated that when glutamine is administered in the absence of excess ammonia there is a dose-dependent increase in ROS production by astrocytes. This latter effect can be blocked by administration of CsA. Conversely, ammonia itself does not induce mPT or mitochondrial swelling indicating that its direct entry into the mitochondria may not be efficient enough to exert a deleterious effect on mitochondrial function. However, elevation in intracellular ammonia does lead to increased mitochondrial uptake of glutamine.
Within the mitochondria the glutamine is degraded to glutamate and ammonia by a glutaminase encoded by the GLS2 gene. The GLS2 gene encoded glutaminase was originally thought to be liver specific but is in fact expressed in numerous tissues and is important in the glutamate-glutamine cycle in the brain. The GLS2 encoded glutaminase is dependent on inorganic phosphate (Pi) for activity and is, therefore, also referred to as phosphate-activated glutaminase, PAG. The action of mitochondrial glutaminase in the astrocytes leads to further increases in intramitochondrial ammonia levels. That the PAG action is significant to glutamine-induced astrocyte swelling is demonstrated by the fact that inhibition of PAG prevents swelling of mitochondria under hyperammonemic conditions. These results suggest a model whereby, under conditions of hyperammonemia, the accumulation of ammonia in brain cells (in particular astrocytes) potentiates glutamine toxicity by facilitating its uptake into mitochondria. The increased mitochondrial uptake of glutamine leads to increased mitochondrial ammonia production triggering mitochondrial impairment, and an ensuing chain of deleterious events leading to astrocytic dysfunction, swelling and brain edema.