
Last Updated: November 9, 2025
Introduction to I-Cell disease
I-cell disease (also called mucolipidosis IIA, or mucolipidosis II alpha/beta: ML-IIα/β) is an autosomal recessive disorder that belongs to the family of disorders identified as lysosomal storage disorders. I-cell disease results as a consequence of defective targeting of lysosomal hydrolases to the lysosomes. The disorder is so called because fibroblasts from afflicted patients contain numerous phase-dense inclusion bodies in the cytosol. The lysosomal targeting defect results from mutations in the gene (GNPTAB) that encodes two of the subunits of UDP-N-acetylglucosamine:lysosomal-enzyme N-acetylglucosaminyl-1-phosphotransferase (GlcNAc-phosphotransferase).
I-cell disease was first described in 1967 and was originally thought to be a variant of Hurler syndrome. However, I-cell disease patients present with symptoms earlier than do Hurler syndrome patients and in addition, I-cell patients do not exhibit mucopolysacchariduria. The inclusion bodies seen in I-cell disease are also observed in another condition called pseudo-Hurler polydystrophy (also called mucolipidosis III alpha/beta; ML-IIIα/β) that presents later and with milder symptoms compared to I-cell disease. The use of the term mucolipidosis for these related disorders is to denote diseases whose combined clinical manifestations are common to both the mucopolysaccharidoses and the sphingolipidoses.
Molecular Biology of I-Cell Disease
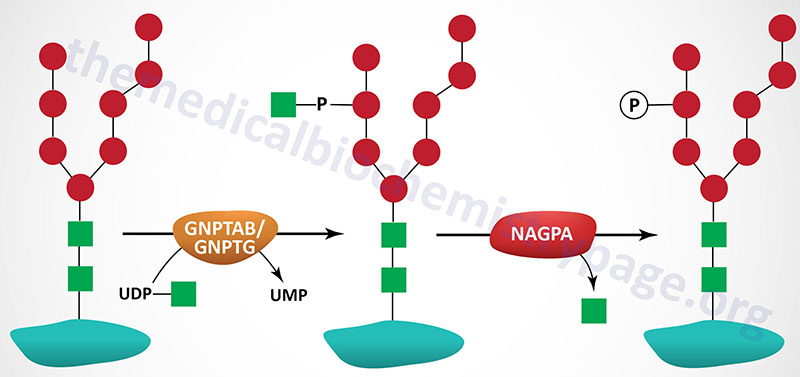
The targeting of lysosomal enzymes to lysosomes is mediated by receptors that bind mannose 6-phosphate recognition markers on the enzymes (see Glycoproteins page). The recognition marker is synthesized in a two-step reaction in the Golgi complex. The enzyme that catalyzes the first step in this process is UDP-N-acetylglucosamine:lysosomal-enzyme N-acetylglucosaminyl-1-phosphotransferase (GlcNAc-phosphotransferase). GlcNAc-phosphotransferase is an α2β2γ2 hexameric complex whose protein subunits are encoded by two genes.
The phosphotransferase is a low abundance membrane-associated enzyme possessing two activities. One activity is responsible for the recognition of enzymes that need to be targeted to the lysosomal compartment and the other activity is the catalytic component. The α- and β-subunits of the phosphotransferase are encoded by the GNPTAB gene and the γ-subunits are encoded by the GNPTG gene.
The GNPTAB gene is located on chromosome 12q23.3 and is composed of 23 exons that encode a 1256 amino acid precursor protein. The protein is proteolytically processed generating a 928 amino acid fragment from the N-terminus (the α-subunit) and a 328 amino acid fragment from the C-terminus (the β-subunit).
The GNPTG gene is located on chromosome 16p13.3 and is composed of 12 exons that encode a 305 amino acid precursor protein.
Both I-cell disease and pseudo-Hurler polydystrophy result from defects in the GNPTAB gene. However, whereas the mutations in the GNPTAB gene that cause Pseudo-Hurler polydystrophy result in reduced enzyme activity, the mutations that cause I-cell disease are null mutations, i.e. no enzyme activity is present. As of 2025 a total of 240 pathogenic mutations have been identified in the GNPTAB gene that cause I-cell disease or Pseudo-Hurler polydystrophy.
A variant disorder called mucolipidosis III gamma (ML-IIIγ) results from defects in the GNPTG gene. As of 2025 a total of 85 pathogenic mutations have been identified in the GNPTG gene resulting in mucolipidosis III gamma.
Clinical Features of I-Cell Disease
I-cell disease is characterized by severe psychomotor retardation that rapidly progresses leading to death between 5 and 8 years of age. Although there are similar signs and symptoms, the earlier onset of symptoms and the lack of mucopolysacchariduria distinguish I-cell disease from Hurler syndrome. I-cell patients exhibit coarse facial features, craniofacial abnormalities, and severe skeletal abnormalities. These skeletal abnormalities include kyphoscoliosis (an abnormal curvature of the spine in both the coronal and sagittal planes), widening of the ribs, lumbar gibbus deformity (refers to a hump or swelling or enlargement on one side of a body surface), anterior beaking and wedging of the vertebral bodies, and proximal pointing of the metacarpals.
A clinically unique feature seen in I-cell disease patients that allows easy distinction from Hurler syndrome is a striking gingival hyperplasia. In addition, due to the loss of proper targeting of lysosomal enzymes, I-cell disease patients have measurable levels of numerous lysosomal enzymes in the blood and urine, a feature not shared with any other lysosomal storage disorders. Additional clinical symptoms of I-cell disease include hepatomegaly, cardiomegaly, umbilical hernias, and recurrent upper respiratory infections. The disease progresses rapidly with developmental delay and failure to thrive. Psychomotor retardation is evident in almost all patients by 6 months of age.