
Last Updated: November 9, 2025
Introduction to the Hurler and Scheie Syndromes
Hurler and Scheie syndromes are autosomal recessive disorders that belong to the family of disorders identified as lysosomal storage disorders, and historically as the mucopolysaccharidoses (MPS). Due to the association with the term MPS the Hurler and Scheie syndromes are collectively identified as MPS I (or MPS1). The MPS I disorders are characterized by the lysosomal accumulation of dermatan and heparan sulfates as a consequence of defects in the lysosomal hydrolase, α-L-iduronidase. This enzyme hydrolyzes terminal α-L-iduronic acid residues from the two classes (dermatan and heparan sulfates) of glycosaminoglycans.
Molecular Biology of Hurler and Scheie Syndromes
The α-L-iduronidase gene (IDUA) is located on chromosome 4p16.3 spanning 19 kb comprising 18 exons that generate two alternatively spliced mRNAs that encode two distinct protein isoforms; isoform a is a 653 amino acid precursor protein and isoform b is a 521 amino acid protein. Removal of the 26 amino acid signal peptide from the isoform a preprotein generates the enzymatically functional glycoprotein that is active as a monomer.
As of 2025 a total of 217 pathogenic mutations have been identified in the IDUA gene resulting in MPS I diseases. The most commonly occurring mutations seen in the Caucasian population are two that cause truncation of the functional enzyme. One mutation truncates the protein at amino acid 402 following a tryptophan residue (the mutation is designated as: W402X or W402Ter) and the other at amino acid 70 following a glutamine residue (the mutation is designated as: Q70X or Q70Ter).
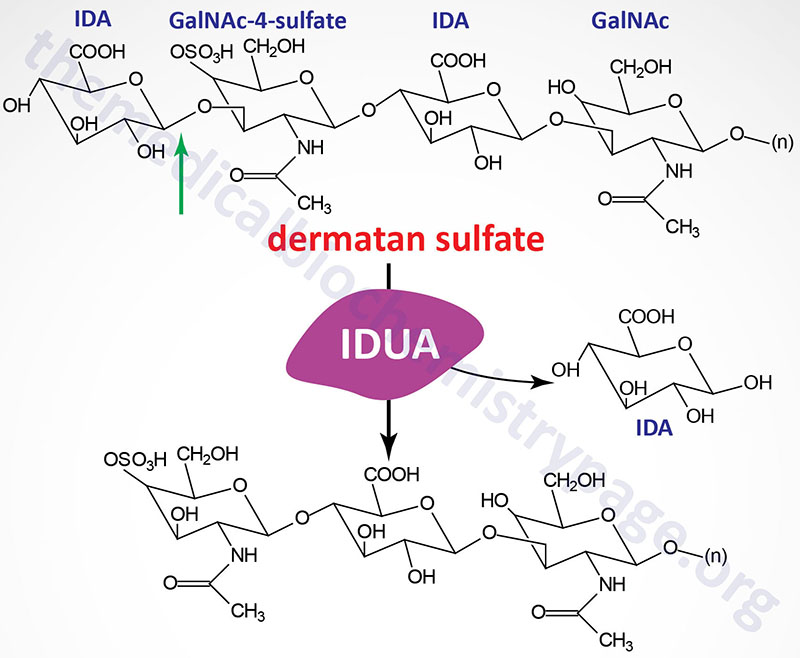
Deficiencies in α-L-iduronidase results in a wide spectrum of clinical manifestations that are classified into three clinical diseases. Hurler disease represents the most severe end of the spectrum and Scheie the least severe with the Hurler-Scheie syndrome representing the phenotype that is intermediate between the other two.
Clinical Features of Hurler Syndrome, MPS IH
Hurler syndrome is a progressive disorder manifesting with multiple organ and tissue involvement leading to death in early childhood. Most Hurler infants succumb to cardiomyopathy. Diagnosis of Hurler syndrome is usually made between 4 and 18 months of age in infants that appeared normal at birth.
Early symptoms that prompt medical attention are hepatosplenomegaly, coarse facial features and an enlarged tongue. Developmental delay is apparent by 12 months with maximum attainment of a functional age of 2 to 4 years. Progressive deterioration is seen as the disease progresses. Hearing loss is also typical in Hurler syndrome. As the disease progresses there is an increase in the degree of corneal clouding. Most patients have recurrent ear and upper respiratory tract infections, persistent nasal discharge and noisy breathing.
Typical of other mucopolysaccharidoses, Hurler patients exhibit the constellation of skeletal abnormalities referred to dystosis multiplex. Dystosis multiplex is characterized by an enlarged skull, thickened calvarium, premature closure of lamboid and sagittal sutures, shallow orbits, enlarged J-shaped sella turcica (a saddle-shaped skull structure into which sits the bottom of the pituitary gland), and abnormal spacing of the teeth with dentigerous cysts. There is anterior hypoplasia of the lumbar vertebrae, the long bone diaphyses are enlarged and an irregular appearance of the metaphyses. The epiphyseal centers are not well developed, the pelvis is poorly formed with small femoral heads and coxa valga. The clavicles are short, thick and irregular and the ribs are oar shaped. Phalanges are shortened and trapezoidal in shape. Hurler patients have a short neck and odontoid hypoplasia (decreased ossification of the odontoid bone which is the anterior process of the second vertebra). This latter symptom can result in atlantoaxial subluxation which is a misalignment between the 1st and 2nd cervical vertebrae.
Clinical Features of Hurler-Scheie Syndrome, MPS IHS
The clinical phenotype associated with Hurler-Scheie syndrome is intermediate between that of Hurler and Scheie syndromes. This form of the disease is characterized by progressive dystosis multiplex with no deficit in intellect. The onset of symptoms is usually observed between the ages of 3 and 8 years. Patients with Hurler-Scheie syndrome often survive into adulthood. In addition to the skeletal abnormalities, patients exhibit progressive corneal clouding, deafness, aortic valve abnormalities and joint stiffness.
Clinical Features of Scheie Syndrome, MPS IS
Scheie syndrome is the mild form of mucopolysaccharidosis type I. This form of the disease is characterized by corneal clouding, aortic valve abnormalities, and joint stiffness with few of the other features seen in the more severe forms of the disease. The onset of symptoms in Scheie syndrome does not usually occur until after the age of 5. Diagnosis is often made only when a patient is between 10 and 20 years of age.
Treatment of Hurler and Scheie Syndromes
Enzyme replacement therapy (ERT), bone marrow transplantation, hematopoietic stem cell transplantion, and gene transfer to patient bone marrow have all had a level of success in treating several patients suffering from MPS I. A current clinically approved ERT for Hurler syndrome is the Sanofi drug Aldurazyme® (iaronidase).