
Last Updated: July 7, 2026
Introduction to Heme Biosynthesis
The processes of heme biosynthesis and degradation to bilirubin constitute the pathways of heme and bilirubin metabolism.
Heme molecules are derived from the porphyrins. Porphyrins are complex chemical compounds that are large heterocyclic organic ring structures. The complex ring structures of porphyrins are composed of four modified pyrrole (5-membered organic ring) subunits connected by methine (carbon with one double bond and two single bonds: =CH-) bridges. The naturally occurring porphyrins of biological significance are the hemes.
Hemes in biological systems consist of ferrous iron (Fe2+) complexed with four nitrogens of the specific porphyrin molecule identified as protoporphyrin IX. The major function of heme in humans is its role in the coordination of O2 molecules in hemoglobin. There are three structurally distinct hemes in humans identified as heme a, heme b, and heme c (discussed below). In addition to the heme b of hemoglobin and its role in oxygen transport, hemes are critical for the biological functions of several enzymes such as the cytochromes of oxidative phosphorylation and the xenobiotic metabolizing enzymes of the cytochrome P450 family (CYP).
Aside from its importance as the prosthetic group of hemoglobin and the cytochromes, heme is clinically significant because a number of genetic disease states are associated with deficiencies of the enzymes used in its biosynthesis and catabolism. Some of these disorders are readily diagnosed because they cause δ-aminolevulinic acid (ALA) and other abnormally colored heme intermediates to appear in the circulation, the urine, and in other tissues such as teeth and bones. Some disorders of heme biosynthesis are more insidious such as the various porphyrias, a list of which can be found below. In addition to the clinical consequences of defects in heme biosynthesis, defects in heme catabolism can lead to potentially lethal elevations in the primary catabolic byproduct, bilirubin (see below)
An important feature of the intermediates in heme biosynthesis, as well as in heme degradation, is their chromophoric character, some are colored while others are not. An easy way to distinguish which will have a color and which will not is to look at the suffix of the compound name. All heme intermediates and degradation products that end in -ogen (e.g. porphobilinogen) will be colorless while those that end in -in (e.g. bilirubin) will be colored.
Reactions of Heme Biosynthesis
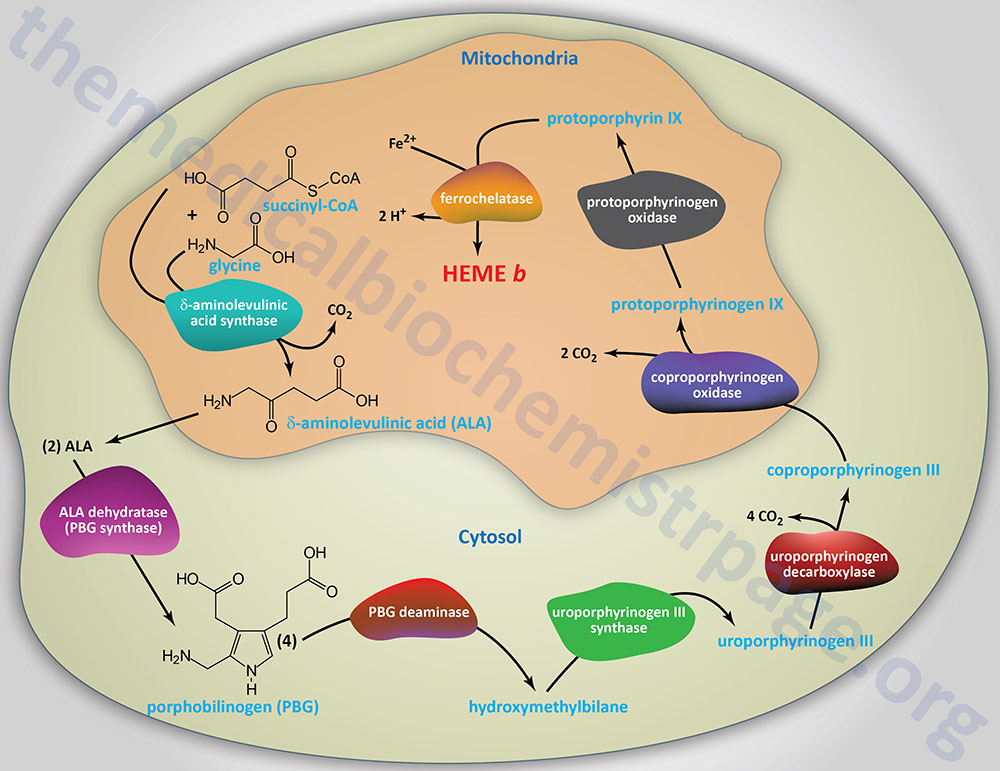
Origin of Succinyl-CoA Incorporated into Heme
The succinyl-CoA used by the initial and rate-limiting enzyme of heme biosynthesis, δ-aminolevulinic acid synthase (ALAS), can be derived from the TCA cycle, but can also be derived from the series of reactions that convert propionyl-CoA to succinyl-CoA. Propionyl-CoA is a product of the catabolism of the amino acids, methionine, threonine, valine, and isoleucine, as well as from the oxidation of fatty acids with an odd number of carbon atoms and from the synthesis of the bile acids in the liver. With respect to heme biosynthesis, particularly in adipose tissue, the major source of propionyl-CoA used in the synthesis of heme is derived from the catabolism of valine and isoleucine.
Delta-Aminolevulinic Acid Synthase
The first reaction in heme biosynthesis takes place in the matrix of the mitochondria and involves the condensation of one glycine and one succinyl-CoA by the pyridoxal phosphate-requiring (vitamin B6) enzyme, δ-aminolevulinic acid synthase (ALAS) forming the compound δ-aminolevulinic acid. Delta-aminolevulinic acid (ALA) is also called 5-aminolevulinic acid. This reaction is both the rate-limiting reaction of heme biosynthesis, and the most highly regulated reaction (see Regulation of Heme Biosynthesis below).
The glycine used in heme synthesis is transported across the inner mitochondrial membrane into the matrix by the SLC family transporter encoded by the SLC25A38 gene. Mutations in the SLC25A38 gene are associated with an autosomal recessive form of congenital sideroblastic anemia (CSA).
Due to the loss of ALAS activity in vitamin B6 deficiency there is an associated microcytic hypochromic anemia where the erythroblasts are histopathologically associated with siderosomes (punctate staining due to iron deposition on mitochondrial membranes of erythroblasts).
There are two forms of ALAS encoded by two different genes. ALAS1 is considered a house-keeping gene and is expressed in all cells. ALAS2 is an erythroid-specific form of the enzyme and is expressed only in fetal liver and adult bone marrow.
The ALAS1 gene is located on chromosome 3p21.2 and is composed of 12 exons that generate four alternatively spliced mRNAs that collectively encode two distinct precursor proteins. ALAS1 isoform 1 is composed of 640 amino acids and ALAS1 isoform 2 is composed of 657 amino acids.
The ALAS2 gene is located on the X chromosome (Xp11.21) and is composed of 12 exons that generate three alternatively spliced mRNAs, each of which encode a distinct protein isoform. ALAS2 isoform a is composed of 587 amino acids, ALAS2 isoform b is composed of 550 amino acids, and ALAS2 isoform c is composed of 574 amino acids.
Inherited defects in the ALAS2 gene result in the disorder called X-linked sideroblastic anemia, XLSA. Sideroblasts are erythroblasts with non-heme iron-containing conjugates (predominantly associated with the mitochondria), called siderosomes. XLSA has also been called congenital sideroblastic anemia, hereditary sideroblastic anemia, hereditary iron-loading anemia, X-linked hypochromic anemia, hereditary hypochromic anemia, and hereditary anemia.
Delta-Aminolevulinic Acid Dehydratase
Following synthesis, the mitochondrial ALA is transported to the cytosol by an as yet undetermined transporter. Although it may be that either SLC25A38 or ABCB10 function to transport ALA to the cytosol, no definitive verification has yet been made. Within the cytosol ALA dehydratase (also called porphobilinogen synthase) dimerizes two molecules of ALA to produce the pyrrole ring compound porphobilinogen. Functional ALA dehydratase is a multi-subunit complex composed of eight identical subunits (homooctamer) and is also a clinically significant zinc– (Zn2+) requiring enzyme.
ALA dehydratase is encoded by the ALAD gene. The ALAD gene is located on chromosome 9q32 and is composed of 15 exons that generate three alternatively spliced mRNAs, each of which encode a distinct protein isoform. Mutations in the ALAD gene result in the autosomal recessive hepatic porphyria called ALAD deficient porphyria, ADP.
Porphobilinogen Deaminase
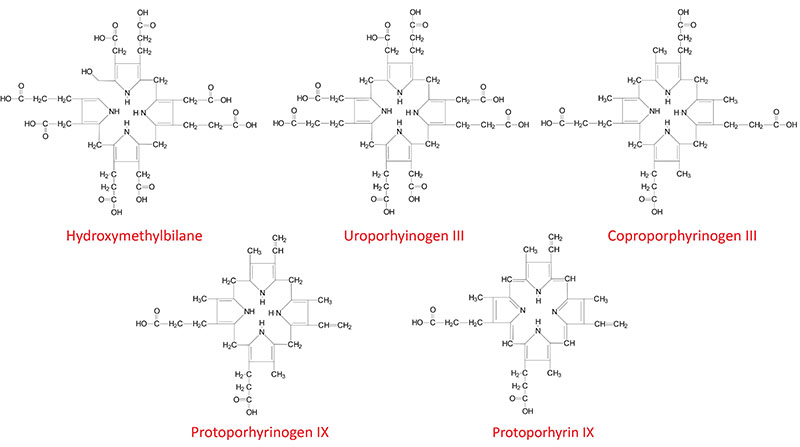
The next step in the pathway involves the head-to-tail condensation of four molecules of porphobilinogen to produce the linear tetrapyrrole intermediate, hydroxymethylbilane. The enzyme for this condensation is porphobilinogen deaminase (PBG deaminase). This enzyme is also called hydroxymethylbilane synthase or uroporphyrinogen I synthase.
PBG deaminase is encoded by the HMBS (hydroxymethylbilane synthase) gene. The HMBS gene is located on chromosome 11q23.3 and is composed of 15 exons that generate four alternatively spliced mRNAs, each encoding different isoforms of the enzyme. PBG deaminase isoform 1 is composed of 361 amino acids, PBG deaminase isoform 2 is composed of 344 amino acids, PBG deaminase isoform 3 is composed of 321 amino acids, and PBG deaminase isoform 4 is composed of 304 amino acids. PBG deaminase isoform 2 is an erythroid cell-specific form of the enzyme.
Once produced, hydroxymethylbilane has two main fates, one is due to enzymatic action, the other is non-enzymatic. Non-enzymatic alteration of hydroxymethylbilane is a cyclization to the compound called uroporphyrinogen I (urogen I). The non-enzymatic fate of hydroxymethylbilane is of significance only in patients with defects in enzymes downstream of PBG deaminase. Of significance to patients harboring a defective heme biosynthetic enzyme is the fact that defects prior to hydroxymethylbilane synthesis ARE NOT associated with photosensitivity, whereas, defects from this point on ARE associated with photosensitivity. Defects in the PBG deaminase gene result in the autosomal dominant hepatic porphyria called acute intermittent porphyria, AIP.
Uroporphyrinogen-III Synthase
The most important fate of hydroxymethylbilane is the regulated, enzymatic conversion to uroporphyrinogen III (urogen III), the next intermediate on the pathway to heme synthesis. This step is catalyzed by the enzyme, uroporphyrinogen-III synthase.
Uroporphyrinogen-III synthase is encoded by the UROS gene. The UROS gene is located on chromosome 10q26.2 and is composed of 17 exons that generate five alternatively spliced mRNAs, each of which encode a distinct protein isoform. Defects in the UROS gene are associated with the autosomal recessive erythroid porphyria called congenital erythropoietic porphyria, CEP.
Uroporphyrinogen Decarboxylase
In the cytosol, the acetate substituents of uroporphyrinogen (normal uroporphyrinogen III or abnormal uroporphyrinogen I) are all decarboxylated in a step-wise process by the enzyme uroporphyrinogen decarboxylase which is encoded by the UROD gene. The resultant products have methyl groups in place of acetate and are known as coproporphyrinogens (coprogens), with coproporphyrinogen III (coprogen III) being the important normal intermediate in heme synthesis.
The step-wise action of uroporphyrinogen decarboxylase on uroporphyrinogen (an 8-carboxylic acid porphyrinogen) is the formation of a 7-carboxylic acid porphyrinogen (heptacarboxylate porphyrinogen) with the release of a molecule of CO2. The enzyme then removes a second CO2, converting heptacarboxylate porphyrinogen to hexacarboxylic porphyrinogen (a 6-carboxylic acid porphyrinogen). The enzyme then remove the third CO2, converting heptacarboxylate porphyrinogen into pentacarboxylate porphyrinogen (a 5-carboxylic acid porphyrinogen). In the final decarboxylation the enzyme removes a CO2 from pentacarboxylate porphyrinogen forming coproporphyrinogen III (a 4-carboxylic acid porphyrinogen).
The UROD gene is located on chromosome 1p34.1 and is composed of 10 exons that encode a protein is 367 amino acids. Mutations in the UROD gene are associated with various forms of the autosomal dominant porphyria called porphyria cutanea tarda, PCT. Mutations in the UROD gene that are directly associated with the pathogenesis of PCT are only associated with the type II version of the disorder. The type I and type III forms of PCT are most likely the result of multifactorial effects that secondarily negatively affect the function of the UROD enzyme.
Coproporphyrinogen III Oxidase
Following its synthesis, coproporphyrinogen III is transported across the outer mitochondrial membrane into the intermembrane space most likely by the ABC family transporter ABCB6. Coproporphyrinogen III then interacts with the inner mitochondrial membrane localized enzyme, coproporphyrinogen III oxidase (encoded by the CPOX gene).
Coproporphyrinogen-III oxidase decarboxylates two propionate residues yielding vinyl substituents on the two pyrrole rings. The colorless product is protoporphyrinogen IX. Similar to the step-wise decarboxylation of uroporphyrinogen by uroporphyrinogen decarboxylase, coproporphyrinogen III oxidase catalyzed the decarboxylation of propionate residues in coproporphyrinogen III in a two-step process. In the first step the enzyme decarboxylates the propionate group on ring III (positions 6 and 7 of the ring) generating the tricarboxylic intermediate known as harderoporphyrinogen. In the next step the enzyme decarboxylates the propionate group on ring IV (position 2) generating protoporphyrinogen IX.
The CPOX gene is located on chromosome 3q11.2 and is composed of 11 exons that encode a precursor protein of 454 amino acids. Mutations in the CPOX gene result in the autosomal dominant acute hepatic porphyria called hereditary coproporphyria, HCP.
Protoporphyrinogen IX Oxidase
Protoporphyrinogen IX is then transported across the inner mitochondrial membrane into the matrix. This transport process is carried out by the transporter identified as transmembrane protein 14C which is encoded by the TMEM14C gene. Protoporphyrinogen IX is then converted to protoporphyrin IX (structure shown below) by protoporphyrinogen IX oxidase. The oxidase reaction requires molecular oxygen and results in the loss of six protons and six electrons, yielding a completely conjugated ring system, which is responsible for the characteristic red color of the hemes.
Protoporphyrinogen IX oxidase is encoded by the PPOX gene. The PPOX gene is located on chromosome 1q23.3 and is composed of 18 exons that generate ten alternatively spliced mRNAs. These ten mRNAs encode five distinct protein isoforms. Mutations in the PPOX gene result in the autosomal dominant acute hepatic porphyria called variegate porphyria, VP.
Ferrochelatase
The final reaction in heme synthesis also takes place in the mitochondrion and involves the insertion of the ferrous iron (Fe2+) atom into the ring system generating heme b. The enzyme catalyzing this reaction is known as ferrochelatase. The Fe2+ iron used by ferrochelatase is transported across the inner mitochondrial membrane by the SLC family transporters encoded by the SLC25A28 and/or SLC25A37 genes. The SLC25A37 encoded transporter is also known as mitoferrin 1 (MFRN1). The SLC25A28 encoded transporter is also known as mitoferrin 2 (MFRN2).
Ferrochelatase is encoded by the FECH gene. The FECH gene is located on chromosome 18q21.31 and is composed of 12 exons that generate five alternatively spliced mRNAs, each of which encoded a distinct protein isoform. Mutations in the FECH gene are associated with the autosomal dominant erythroid porphyria called erythropoietic protoporphyria, EPP.
Once produced, heme is transported out of the mitochondria to the cytosol by the transporter encoded by the FLVCR1 (feline leukemia virus subgroup C receptor 1) gene. The FLVCR1 encoded protein is a member of the SLC family of transporters and as such is also identified as SLC49A1. Two isoforms of FLVCR1 have been characterized, FLVCR1a and FLVCR1b. The FLVCR1b isoform is required for the regulation of erythropoiesis by controlling mitochondrial heme efflux.
Structures of Heme Biosynthesis Intermediates
Heavy Metal Inhibition of Heme Metabolism
The enzymes ferrochelatase, ALA synthase, and ALA dehydratase (a sulfhydryl containing enzyme) are sensitive to inhibition by heavy metal poisoning, with inhibition of ferrochelatase being the most sensitive and most significant to the clinical manifestations of heavy metal poisoning. A characteristic of lead poisoning is an increase in ALA in the circulation in the absence of an increase in heme. Indeed, due to the inhibition of ferrochelatase and the associated loss of heme synthesis, the feed-back inhibition of ALA synthase is no longer active leading to increased ALA synthesis even in the presence of a heavy metal such as lead.
The consequences of heavy metal inhibition of ferrochelatase are an acquired porphyria (as opposed to an inherited disease) referred to as plumbism (so-called because the symbol for lead is Pb). Due to the lack of enzymatic incorporation of iron into protoporphyrin IX, by heavy metal inhibited ferrochelatase, erythroblasts will acquire siderosomes. Siderosomes are histologically observable structures that result from iron deposition on mitochondria in the erythroblasts.
In addition, zinc atoms will non-enzymatically incorporate into protoporphyrin IX forming Zn-protoporphyrin (ZPP). Zn-protoporphyrin imparts a fluorescence capability that can be visualized by observing blood under appropriate wavelength light which causes the erythroid cells to “glow”. This same phenomenon is observable in patients with iron deficient anemia. Indeed, the measurement of ZPP is used as a screening tool for both heavy metal (e.g. lead) poisoning and iron deficiency.
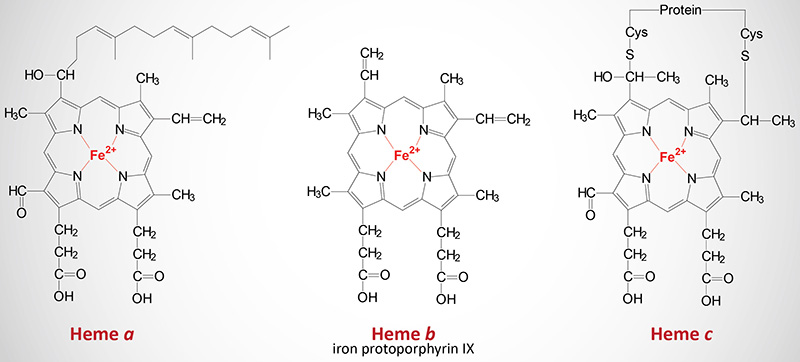
The Various Heme Molecules
In addition to the heme b found in hemoglobin, there are two additional forms of heme found in cytochromes such as those involved in the process of oxidative phosphorylation. Cytochromes of the c type contain a modified iron protoporphyrin IX known as heme c. In heme c the 2 vinyl (C=C) side chains are covalently bonded to cysteine sulfhydryl residues of the apoprotein. Only cytochromes of the c type contain covalently bound heme. Heme a is also a modified iron protoporphyrin IX. Heme a is found in cytochromes of the a type such as those of complex IV of the oxidative phosphorylation pathway.
Regulation of Heme Biosynthesis
Although heme is synthesized in virtually all tissues, the principal sites of synthesis are erythroid cells (≈85%) and hepatocytes (accounting for nearly all the rest of heme synthesis). The differences in these two tissues and their needs for heme result in quite different mechanisms for regulation of heme biosynthesis.
In hepatocytes, heme is required for incorporation into the cytochromes, in particular, the P450 class of cytochromes (CYP) that are important for xenobiotic detoxification. In addition, numerous cytochromes of the oxidative-phosphorylation pathway contain heme. The rate-limiting step in hepatic heme biosynthesis occurs at the ALA synthase catalyzed step, which is the committed step in heme synthesis. Heme itself functions as a co-repressor in the inhibition of ALA synthase gene expression. The Fe3+ oxidation product of heme is termed hemin. Heme itself, and hemin acts as a feed-back inhibitors on ALA synthase. Hemin also inhibits transport of ALA synthase from the cytosol (its site of synthesis) into the mitochondria (its site of action).
Because certain pharmaceutical drugs are metabolized by the hepatic CYP system, which requires heme, increased utilization of heme occurs upon administration of these drugs. Of particular significance are the barbiturates. Use of barbiturates should NEVER be prescribed for the pain associated with certain types of porphyrias. This is because the administration of barbiturates leads to their degradation by CYP enzymes in the liver, resulting in a reduction in overall heme levels as the heme needs to be incorporated into the CYP enzymes for their function. This results in de-repression of ALA synthase with the result being an exacerbation of the symptoms of the porphyria due to increased ALA synthesis and subsequent heme biosynthesis products upstream of the defective enzyme.
In erythroid cells all of the heme is synthesized for incorporation into hemoglobin and occurs only upon differentiation when synthesis of hemoglobin proceeds. When red cells mature both heme and hemoglobin synthesis ceases. The heme (and hemoglobin) must, therefore, survive for the life of the erythrocyte (normally this is 120 days). In reticulocytes (immature erythrocytes) heme regulates protein synthesis. The mechanism of this mode of heme-mediate regulation of protein synthesis is described in the Protein Synthesis (Translation) Processes and Regulation page.
Additionally, control of heme biosynthesis in erythrocytes occurs at numerous sites other than at the level of ALA synthase. Control has been shown to be exerted on ferrochelatase, the enzyme responsible for iron insertion into protoporphyrin IX, and on porphobilinogen deaminase.
Heme Metabolism and Disposition of Bilirubin
The largest repository of heme in the human body is in red blood cells, which have a life span of about 120 days. There is thus a turnover of about 6 g/day of hemoglobin, which presents 2 problems. First, the porphyrin ring is hydrophobic and must be solubilized to be excreted. Second, iron must be conserved for new heme synthesis.
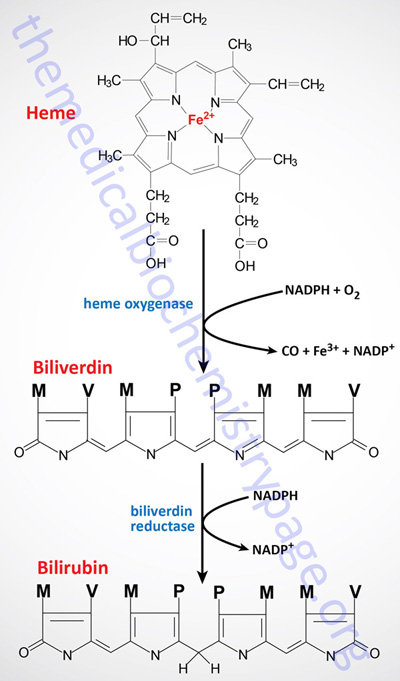
Normally, senescent red blood cells and heme from other sources are engulfed by cells of the reticuloendothelial system (phagocytic macrophages primarily of the spleen but also of the liver, lymph, and bone marrow). The globin is recycled or converted into amino acids, which in turn are recycled or catabolized as required. Heme is oxidized, with the heme ring being opened by the endoplasmic reticulum enzyme, heme oxygenase. The oxidation step requires heme as a substrate, and any hemin (Fe3+) is reduced to heme (Fe2+) prior to oxidation by heme oxygenase.
The heme oxygenase-mediated oxidation occurs on a specific carbon producing the linear tetrapyrrole biliverdin, ferric iron (Fe3+), and carbon monoxide (CO). This is the only reaction in the body that is known to produce CO. Most of the CO is excreted through the lungs, with the result that the CO content of expired air is a direct measure of the activity of heme oxygenase in an individual.
Humans express two distinct heme oxygenase genes identified as HMOX1 and HMOX2. The heme oxygenase enzyme encoded by the HMOX1 gene is the rate-limiting enzyme of heme catabolism. Both HMOX1 and HMOX2 genes are constitutively expressed, however, the activity of the HMOX1 encoded enzyme is inducible by heme, heavy metals, and conditions of stress such as hypoxia.
The HMOX1 gene is located on chromosome 22q12.3 and is composed of 5 exons encoding a protein of 288 amino acids. Mutations in the HMOX1 gene are the causes of a very rare autosomal recessive disorder that is associated with progressive multi-organ system pathology. Patients with HMOX1 deficiency experience persistent, direct antibody-negative intravascular hemolytic anemia, systemic inflammation as a result of persistent macrophage activation, and disseminated intravascular coagulation (DIC). Hallmark organ system pathology includes nephritis and hepatitis, in part due to excessive iron deposition in these organs.
The HMOX2 gene is located on chromosome 16p13.3 and is composed of 13 exons that generate nine alternatively spliced mRNAs that collectively encode three isoforms identified as isoform a (370 amino acids), isoform b (316 amino acids), and isoform c (287 amino acids).
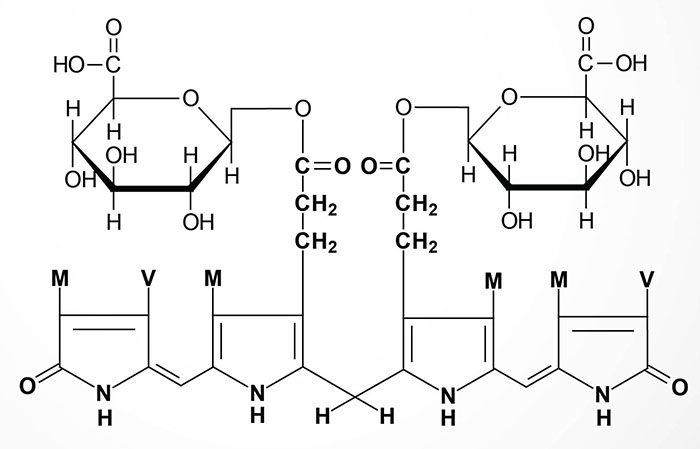
In the next reaction a second bridging methylene (between rings III and IV) is reduced by biliverdin reductase, producing bilirubin. Bilirubin is not just an intermediate in the catabolism of heme as it functions as a potent endogenous antioxidant.
There are two biliverdin reductase genes in humans identified as BLVRA and BLVRB. The enzyme encoded by the BLVRA gene is principally responsible for the catabolism of biliverdin. The enzyme encoded by the BLVRB gene catalyzes the reduction of not only biliverdin but also a variety of flavins, such as riboflavin, FAD or FMN, and methemoglobin. The enzyme encoded by the BLVRB gene is also called NADPH-dependent flavin reductase.
There are two biliverdin reductase genes in humans identified as BLVRA and BLVRB. The enzyme encoded by the BLVRA gene, although responsible for the catabolism of biliverdin, exerts numerous functions related to metabolic regulation, particularly with respect to insulin receptor signaling.
The enzyme encoded by the BLVRB gene catalyzes the reduction of not only biliverdin but also a variety of flavins, such as riboflavin, FAD or FMN, and methemoglobin. The enzyme encoded by the BLVRB gene is also called NADPH-dependent flavin reductase.
The BLVRA gene is located on chromosome 7p13 and is composed of 12 exons generate two alternatively spliced mRNAs, both of which encode the same 296 amino acid precursor protein.
The BLVRB gene is located on chromosome 19q13.2 and is composed of 5 exons that encode a protein of 206 amino acids. The BLVRB gene belongs to the large family of short chain dehydrogenase/reductase (SDR) encoding genes that includes 77 family members.
Bilirubin is significantly less extensively conjugated than biliverdin causing a change in the color of the molecule from blue-green (biliverdin) to yellow-red (bilirubin). The latter catabolic changes in the structure of tetrapyrroles are responsible for the progressive changes in color of a hematoma, or bruise, in which the damaged tissue changes its color from an initial dark blue to a red-yellow and finally to a yellow color before all the pigment is transported out of the affected tissue. Peripherally arising bilirubin is transported to the liver in association with albumin, where the remaining catabolic reactions take place.
Role of Biliverdin Reductase A in Signaling and Metabolism
Although the protein encoded by the BLVRA gene was initially characterized as being involved in the catabolism of heme, it was subsequently found that the enzyme also exhibits dual specificity kinase activity, being able to phosphorylate numerous substrates on either Ser/Thr or Tyr residues. The primary substrates for the kinase activity of biliverdin reductase A (BVRA) are involved in the signaling cascades of the insulin receptor pathway as well as the MAPK signaling pathways. The ability of BVRA to modulate these signaling cascades places the enzyme in the context of the regulation of glucose uptake, the modulation of stress responses, and the regulation of expression of numerous genes.
The ability of BVRA to exert these numerous metabolic regulatory effects is imparted by the consensus regulatory motifs and protein-protein interaction motifs present in the protein in addition to the catalytic reductase domain and the kinase domain. BVRA also possesses basic leucine zipper (bZip) domain located in the middle of the protein. The binding of ATP, as well as NADH and NADPH, is effected via an adenine-binding domain present in the N-terminus of the protein, a domain characteristic of all protein kinases. BVRA also possesses two domains in the C-terminal half of the protein that are SH2 domain-binding sites, allowing for protein-protein interaction.
BVRA can move between the cytosol and the nucleus due to the presence of a nuclear localization signal sequence (NLS) and a nuclear export signal sequence (NES). BVRA can homodimerize which allows the bZip domain to interact with DNA following import into the nucleus vis the NLS. Importantly, the biliverdin reductase B (BVRB) protein, encoded by the BLVRB gene, does not possess the bZip domain nor the NLS nor NES domains indicating that the BVRB protein does not participate in the metabolic regulatory processes in which BVRA is involved.
Activation of the insulin receptor, via binding of insulin, results in tyrosine (Y) phosphorylation of BVRA. When BVRA is phosphorylated on Y198 it’s Ser/Thr kinase activity is activated. One important substrate for BVRA phosphorylation is IRS-1 which results in decreased affinity of IRS-1 for the insulin receptor. One important metabolic consequence of increased BVRA phosphorylation of IRS-1 is decreased insulin-mediated glucose uptake.
Bilirubin Glucuronidation
In hepatocytes, bilirubin UDP-glucuronosyltransferase (bilirubin-UGT: a member of the large UDP glucuronosyltransferase family of enzymes) adds two equivalents of glucuronic acid to bilirubin to produce the more water soluble, bilirubin diglucuronide derivative. The increased water solubility of the tetrapyrrole facilitates its excretion with the remainder of the bile as the bile pigments.
Bilirubin-UGT is encoded by the UGT1A1 gene of the UGT1A gene complex. The UGT1A gene complex is located on chromosome 2q37.1. Several UGT1A enzymes, including bilirubin-UGT, are encoded by the UGT1A gene complex. The 5′ region of the UGT1A complex contains 13 tandemly arrayed first exons, including four pseudo exons. These tandemly arrayed exons are identified as 1A1, 1A2, 1A3, etc. Exons 2, 3, 4, and 5 are located in the UGT1A 3′ region. All UGT isoforms contain the same C-terminal domain encoded by exons 2 through 5. Each first exon has its own promoter element. The 9 viable first exons are independently spliced to the common exons 2 through 5 to generate nine UGT1A transcripts with unique 5′ ends and identical 3′ ends. The N-terminal region encoded by each unique first exon determines acceptor substrate specificity, while the 246-amino acid C-terminal region encoded by the four common exons specifies interactions with the common donor substrate, UDP-glucuronic acid. The bilirubin-UGT isoform (UGT1A1) consists of 533 amino acids.
In newborn infants, or in individuals with abnormally high red cell lysis, or liver damage with obstruction of the bile duct, the bilirubin and its precursors accumulate in the circulation; the result is hyperbilirubinemia, the cause of the abnormal yellowish pigmentation of the eyes and tissues known as jaundice. All newborn infants undergo turnover of the red blood cells that contain fetal hemoglobin (HbF) so that new red blood cells containing adult hemoglobin (HbA) can be produced. In some cases the activation of the UGT1A gene at birth is insufficient to handle all the red cell turnover resulting in neonatal jaundice, apparent around day 2 or 3.
If the blood levels of bilirubin do not decline in a short period of time these infants will need to be treated by phototherapy. The blue-green wavelength light (460-490 nm) used in biliblankets and bililamps is sufficient to induce breakdown of bilirubin in the skin so that it can be cleared from the blood. In rare cases the phototherapy does not work fast enough and in these cases it is appropriate to treat the infants with phenobarbital which enhances the induction of the UGT1A gene.
In normal individuals, intestinal bilirubin is acted on by bacteria to produce the final porphyrin products, urobilinogens and stercobilins, that are found in the feces. The stercobilins oxidize to brownish pigments which impart the brown to brown-black color to normal feces. Indeed, the color of the stool can be quite diagnostic since chalky clay colored feces are indicative of a defect in the hepato-biliary circulation, such as in bile obstruction. Some of the urobilinogen produced by intestinal bacteria is reabsorbed from the gut and enters the circulation. These urobilinogens are converted to the urobilins which are then excreted in the urine. Oxidation of the urobilins imparts the yellowish coloration to urine.
Clinical Aspect of Heme Metabolism and Bilirubin
Clinical problems associated with heme metabolism are of two types. Disorders that arise from defects in the enzymes of heme biosynthesis are termed the porphyrias and cause elevations in the serum and urine content of intermediates in heme synthesis. Inherited disorders in bilirubin metabolism lead to hyperbilirubinemia.
Hyperbilirubinemias
Bilirubin levels are measured in the serum by an assay utilizing Ehrlich diazo reagent and results in the formation of an azobilirubin product. Conjugated bilirubin does not require addition of alcohol to promote the azotization reaction and thus, this is referred to as measurement of direct bilirubin. The reaction with unconjugated bilirubin requires the addition of alcohol and thus is referred to as the measurement of indirect bilirubin. Normal bilirubin measurements in a healthy adult are 0.3–1.2md/dL for total (indirect + direct). Direct type bilirubin does not exist in the plasma, however, a small portion of indirect type bilirubin may present as direct reacting type and thus the serum measurement may show a direct bilirubin but this is never above 0.3mg/dL in a normal individual.
Excess circulation and accumulation of bilirubin (hyperbilirubinemia) results in a yellow-orange discoloration of the tissues and is most easily visible as icteric (yellowish) discoloration in the sclera of the eyes. Bilirubin toxicity (bilirubin encephalopathy) can be life threatening in neonates. Bilirubin encephalopathy is characterized by yellow discoloration of the basal ganglia in babies with intense jaundice and was first described over a century ago and the term “kernicterus” was coined to describe these physical changes. Any increase in plasma bilirubin above 20mg/dL is considered dangerous in neonates. However, individual differences in bilirubin sensitivity can result in kernicterus at lower bilirubin levels. Kernicterus occurs in infants with severe unconjugated hyperbilirubinemia and in young adults with high serum levels of unconjugated bilirubin. The latter is the result of inherited deficiencies in the enzyme responsible for bilirubin conjugation to glucuronic acid, bilirubin UDP glucuronosyltransferase (bilirubin-UGT).
Bilirubin has been shown to inhibit DNA synthesis, uncouple oxidative phosphorylation, and inhibit ATPase activity in brain mitochondria. Bilirubin also inhibits a variety of different enzyme classes including dehydrogenases, electron transport proteins, hydrolyases, and enzymes of RNA synthesis, protein synthesis and carbohydrate metabolism. All of these toxic effects of bilirubin are reversed by binding to albumin. In fact, albumin plays a vital role in the disposition of bilirubin in the body by keeping the compound in solution and transporting it from its sites of production (primarily bone marrow and spleen) to its site of excretion which is the liver.
Several inherited disorders in bilirubin metabolism have been identified. Gilbert syndrome and the Crigler-Najjar syndromes result from predominantly unconjugated hyperbilirubinemia. Dubin-Johnson syndrome and Rotor syndrome result from conjugated hyperbilirubinemia. Once conjugated to glucuronate, bilirubin is water soluble, therefore, conjugated hyperbilirubinemias are less severe in their symptomology than are the unconjugated hyperbilirubinemias.
Porphyrias
The porphyrias are both inherited and acquired disorders in heme synthesis. These disorders are classified as either erythroid or hepatic, depending upon the principal site of expression of the enzyme defect. Eight different porphyrias have been classified encompassing defects in each of the enzymes of heme synthesis. Defects in the function of hepatic uroporhyrinogen decarboxylase (UROD) result in type I porphyria cutanea tarda (PCT I), whereas mutations in the UROD gene result in type II PCT (PCT II). PCT is the most commonly occurring type of porphyria. It should be noted that no porphyria has been identified resulting from defects in the house-keeping form of ALAS (ALAS1). The most commonly occurring hepatic porphyria is acute intermittent porphyria, AIP, which is caused by a defect in porphobilinogen deaminase, (PBG deaminase). This enzyme is also called hydroxymethylbilane synthase (official gene symbol: HMBS) or also but rarely, uroporphyrinogen I synthase.
All of the porphyrias lead to excretion of heme biosynthetic byproducts that turn the urine red and when deposited in the teeth turn them reddish brown. Accumulation of these byproducts in the skin renders it extremely sensitive to sunlight causing ulceration and disfiguring scars. Increased hair growth (hypertrichosis) is also a symptom of the porphyrias leading to appearance of fine hairs over the entire face and on the extremities. This latter symptom lends to the description of “werewolf syndrome” in many porphyria patients.
In many cases the treatment protocols for the intermittent attacks of the various porphyrias, in particular in the case of acute intermittent porphyria, include the use of hemin or hematin and glucose supplementation. Hemin is a form of iron protophorphyrin IX in which the associated iron has an additional chloride ligand. Hematin is similar except that instead of a chloride ion there is an hydroxide ion liganded to the iron. The rationale for the use of these agents is that they act as analogs of heme and strongly inhibit the activity of ALAS resulting in reductions in the heme biosynthetic intermediates that precipitate the porphyria attack.
The use of glucose to treat porphyrias was a serendipitous observation but for a long time the mechanism by which glucose infusion alleviated the symptoms of a porphyria attack were not understood. Quite often there is an association between fasting, low serum glucose, and the precipitation of an acute porphyria attack which suggested the utility of glucose infusion. The molecular mechanism was ascertained when it was found that the transcription factor, PGC-1α, is activated in the liver in response to hypoglycemia. Indeed, activation of PGC-1α is required to initiate hepatic gluconeogenesis via the activation of several genes in this metabolic pathway. In addition to gluconeogenic genes, the ALAS1 gene is activated in the liver via PGC-1α. Thus, hypoglycemia leads to increased ALAS1 activity and results in accumulation of heme intermediates that result in the precipitation of the attack.
Table of the Porphyrias
| Porphyria | Enzyme Defect – Gene | Primary Symptoms – Comments |
| Erythroid Class | ||
| X-linked sideroblastic anemia, XLSA | δ-aminolevulinic acid synthase 2: ALAS2 | microcytic hypochromic anemia; erythroblast present with sidersomes; wide variability in age of presentation; progressive iron accumulation, fatal if not treated |
| Congenital erythropoietic porphyria, CEP (Günther disease) | uroporphyrinogen III synthase: UROS | photosensitivity evidenced by blistering on the back of the hands and other sun-exposed areas of skin, skin friability after minor trauma, facial hypertrichosis (excessive hair growth), skin hyperpigmentation, reddish discoloration of the teeth (erythrodontia); mild to severe hemolytic anemia; wide variability in phenotypic presentation from fetal lethality to late onset |
| Erythropoietic protoporphyria, EPP | ferrochelatase: FECH | hypersensitivity to sunlight and fluorescent lighting resulting in burning and itching sensations in skin, severe blistering and scarring |
| Hepatic Class | ||
| ALA dehydratase deficient porphyria, ADP | ALA dehydratase (also called porphobilinogen synthase): ALAD | neurovisceral symptoms that are very similar to those experienced by acute intermittent porphyria patients |
| Acute intermittent porphyria, AIP | PBG deaminase (also called hydroxymethylbilane synthase or rarely uroporphyrinogen I synthase): HMBS | neurovisceral symptoms including severe abdominal pain, nausea, vomiting, tachycardia, hypertension, anxiety, depression, convulsions, peripheral neuropathy; chronic complications include hepatocellular carcinoma (HCC) and renal failure |
| Hereditary coproporphyria, HCP | coproporphyrinogen III oxidase: CPOX | neurovisceral symptoms similar to those experienced by acute intermittent porphyria patients; seizures; peripheral neuropathy with ascending paralysis; some photosensitivity |
| Variegate porphyria, VP | protoporphyrinogen IX oxidase: PPOX | neurovisceral symptoms and photosensitivity; most commonly adult-onset; cutaneous blistering skin on photoexposed surfaces; crusty slowly healing skin lesions; occasional facial hypertrichosis (excessive hair growth) and hyperpigmentation; abdominal pain; constipation; back, chest, and extremity pain; anxiety; seizures; peripheral neuropathy associated with progressive muscle weakness that may progress to respiratory paralysis |
| Porphyria cutanea tarda type I, PCT type I, also called the sporadic type PCT | hepatic uroporphyrinogen decarboxylase activity | photosensitivity; referred to as the sporadic type of PCT; associated with reduced UROD activity in liver; not associated with direct mutations in the UROD gene; most likely due to multifactorial causes |
| Porphyria cutanea tarda type II, PCT type II, also called the familial type PCT, may also be referred to as hepatoerythropoietic porphyria, HEP | uroporphyrinogen decarboxylase in non-hepatic tissues: UROD | photosensitivity evidenced by blistering on the back of the hands and other sun-exposed areas of skin, skin friability after minor trauma, facial hypertrichosis (excessive hair growth), skin hyperpigmentation, severe thickening of affected skin areas (pseudoscleroderma) |
Differential Diagnosis: Microcytic Anemias
| Deficiency/Defect | Characteristics |
| B6 Deficiency | Pyridoxal phosphate (PLP) required for the rate-limiting enzyme in heme biosynthesis: δ-aminolevulinic acid synthase (ALAS); deficiency results in loss of protoporphyrin IX synthesis, therefore, there will be a significant reduction in measurable ALAS product (δ-aminolevulinic acid, δ-ALA) and protoporphyrin in these patients; loss of heme production leads to hypochromic microcytic anemia; lack of protoporphyrin results in iron deposits on mitochondria in bone marrow erythroblasts resulting in the formation of ringed sideroblasts; loss of iron incorporation into protoporphyrin IX leads to increased serum and intracellular iron concentration; increase in intracellular iron results in increased translation of ferritin as a means to prevent iron toxicity |
| Iron Deficiency | iron deficiency is the leading cause of microcytic anemia; loss of iron results in reduced production of heme, thus, the result is a hypochromic microcytic anemia; lack of heme production results in loss of feed-back inhibition of ALAS, therefore these patients will have an associated increase in measurable protoporphyrin; loss of iron intake means reduced iron in the serum and reduced intracellular iron, the latter resulting in reduced ferritin translation; loss of iron for incorporation into protoporphyrin IX results in spontaneous, non-enzymatic incorporation of Zn2+ forming Zn-protoporphyrin (ZPP), ZPP causes erythrocytes to fluoresce under ultraviolet illumination and is the basis of the ZPP test for iron deficiency or lead poisoning |
| Heavy Metal Poisoning | heavy metals, such as lead, inhibit several enzymes of heme biosynthesis and metabolism with the most significant toxic effects resulting from inhibition of ferrochelatase, the enzyme that incorporates iron into protoporphyrin IX generating heme; similar to B6 deficiency, lead poisoning leads to increased intracellular iron in bone marrow erythroblasts causing the formation of ringed sideroblasts; because there is no heme, the ALAS reaction is not inhibited, as in the case of iron deficiency, this results in increased production of δ-ALA and protoporphyrin; lack of iron incorporation into protoporphyrin results in increased serum and intracellular iron concentrations, with the latter leading to increased ferritin synthesis as in the case of iron-deficient anemia; loss of iron for incorporation into protoporphyrin IX results in spontaneous, non-enzymatic incorporation of Zn2+ forming ZPP as in the case of iron-deficient anemia the ZPP test is diagnostic for lead poisoning |