
Last Updated: June 15, 2026
Introduction to Glycoproteins
Membrane associated carbohydrate is exclusively in the form of oligosaccharides covalently attached to proteins forming glycoproteins, and to a lesser extent covalently attached to lipid forming the glycolipids. The co- and post-translational attachment of carbohydrate to proteins plays a critical role in overall biochemical complexity in humans (as well as all eukaryotes). The importance of these protein modifications can be emphasized by the fact that approximately 50% of all proteins are known to be glycosylated and at least 1% of the human genome is represented by glycan biosynthesis genes.
The glycan biosynthetic gene superfamily represents the enzymes referred to as glycosyltransferases. The glycosyltransferase superfamily is composed of 26 subfamilies in humans, including for example the UDP glucuronosyltransferase family, members of which are responsible for hepatic bilirubin metabolism, and the glycogen phosphorylases. Many glycosyltransferases are specific for glycan attachment and modification on proteins while others are specific to the synthesis of glycosaminoglycans (GAG) and GAG modifications in proteoglycans.
The predominant sugars found in glycoproteins are glucose (Glc), galactose (Gal), mannose (Man), fucose (Fuc), N-acetylgalactosamine (GalNAc), N-acetylglucosamine (GlcNAc) and N-acetylneuraminic acid (NANA). NANA is also called sialic acid (Sia). The distinction between proteoglycans and glycoproteins resides in the level and types of carbohydrate modification. Proteoglycans (as well as glycosaminoglycans) also contain the sugar acids glucuronic acid (GlcA) and iduronic acid. The carbohydrate modifications found in glycoproteins are rarely as extensive as that of proteoglycans but nonetheless they can be quite complex in their composition.
The large diverse family of glycoproteins found in humans is generated via one of several pathways that includes N-glycosylation, O-glycosylation, O-mannosylation, C-mannosylation (of Trp residues), or glycosylphosphatidylinositol (GPI) linkage. In the GPI-linked family of glycoproteins the sugar residues are not directly attached to the proteins but are linked via a phosphoethanolamine attachment.
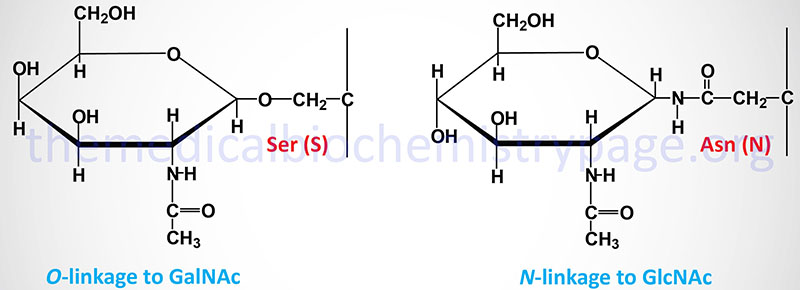
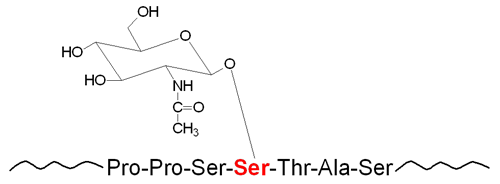
The carbohydrates of glycoproteins are linked to the protein component through either O-glycosidic or N-glycosidic bonds. The N-glycosidic linkage is through the amide group of asparagine (Asn, N). The O-glycosidic linkage is to the hydroxyl of serine (Ser, S), threonine (Thr, T), or hydroxylysine (hLys).
In N-linked glycoproteins, the sugar directly attached to the amide nitrogen of the R-group of Asn residue is always GlcNAc. When attached to Ser or Thr, the sugar directly attached to the protein of O-linked glycoproteins is most often GalNAc. This most common O-glycoprotein type is also commonly referred to as a mucin-type glycan.
The linkage of carbohydrate to hLys is generally found only in the collagens. The linkage of carbohydrate to hLys is either the single sugar galactose or the disaccharide glucosylgalactose.
The mannosylation of tryptophan (Trp, W) residues (C-mannosylation) occurs in proteins of the thrombospondin type-I repeat family (e.g. thrombospondin and complement protein 6) and proteins of the type I cytokine receptor family (e.g. IL-12β). Incorporation of mannose into Trp residues occurs in the first Trp of the consensus Trp-X-X-Trp (WXXW).
N-Linked Glycoproteins
The predominant carbohydrate attachment in glycoproteins of mammalian cells is via N-glycosidic linkage. The site of carbohydrate attachment to N-linked glycoproteins is the amide nitrogen of the R-group of Asn (N) residues found within a consensus sequence of amino acids, Asn-X-Ser/Thr (N-X-S/T), where X is any amino acid except proline. Analysis of human protein sequences demonstrates that approximately 65% of all the proteins contain at least one occurrence of the N-X-S/T consensus. All sugar attachments in N-linked glycoproteins are carried out by the numerous enzymes of the glycosyltransferase superfamily.
O-Linked Glycoproteins
The synthesis of O-linked glycoproteins occurs via the stepwise addition of nucleotide-activated sugars directly onto the polypeptide. The nucleotide-activated sugars are coupled to either UDP, GDP (as with mannose) or CMP (for instance, NANA). As is the case for N-linked glycoproteins, all sugar attachments in O-linked glycoproteins are carried out by the numerous enzymes of the glycosyltransferase superfamily.
Most proteins that are secreted, or bound to the plasma membrane, are modified by carbohydrate attachment. The part that is modified, in plasma membrane-bound proteins, is the extracellular portion of the protein. Intracellular proteins are less frequently modified by carbohydrate attachment. However, the attachment of carbohydrate to intracellular proteins confers unique functional activities on these proteins.
Linkage of carbohydrate to cytosolic and/or nuclear proteins occurs via O-linkage and involves attachment of GlcNAc to serine or threonine residues. The linkage is catalyzed by the enzyme O-GlcNAc transferase, OGT. Numerous signal transduction proteins, nearly all transcription factors (including members of the nuclear receptor family), as well as RNA polymerase II have been shown to be modified by O-GlcNAc linkage. As a means for modification of protein activity, the addition and removal of GlcNAc is considered to be as wide-spread and significant as protein phosphorylation/dephosphorylation. The process of, and the significances to, O-GlcNAc addition (called O-GlcNAcylation) and removal is discussed in detail below.
The protein component of all glycoproteins is synthesized from polyribosomes that are bound to the endoplasmic reticulum (ER). The processing of the sugar groups occurs co- and post-translationally in the lumen of the ER and continues in the Golgi apparatus for N-linked glycoproteins. Attachment of sugars in O-linked glycoproteins occurs post-translationally in the Golgi apparatus. Sugars used for glycoprotein synthesis (both N-linked and O-linked) are activated by coupling to nucleotides. Glucose and GlcNAc are coupled to UDP and mannose is coupled to GDP.
Nucleotide Sugar Biosynthesis
The monosaccharides that are attached to proteins (as well as lipids in the formation of glycolipids) are first activated by coupling to a nucleotide. These nucleotide-activated sugars are derived from dietary sources and salvage pathways. Glucose and fructose are the two major forms of sugar in humans from which all other monosaccharides can be synthesized. Through the actions of different enzymes these two sugars are phosphorylated, epimerized, and acetylated converting them into the various high-energy nucleotide sugar donors used in the synthesis of complex glycans. The synthesis of nearly all nucleotide-activated sugars occurs in the cytosol. The exception to this is the synthesis of CMP-activated N-acetylneuraminic acid (NANA, also NeuAc or Sia) which takes place in the nucleus.
Although nucleotide-activated sugars are synthesized in the cytosol they are utilized within the lumen of the ER and/or the Golgi network. Therefore, they must be translocated into these organelles before they can be used in the process of glycan synthesis. Since nucleotide-activated sugars cannot freely pass through the ER or Golgi membranes, specific transport systems are responsible for their translocation. In general, the transport of a nucleotide-activated sugar occurs only into the organelle in which the corresponding glycosyltransferase is localized. Some nucleotide sugars enter only the lumen of ER, others only enter the lumen of the Golgi apparatus, and a few nucleotide-activated sugars are transported into both organelles.
There are two mechanisms for the importation of nucleotide-activated sugars into the ER and/or Golgi apparatus. The first mechanism involves dolichol phosphate and it is used to transport mannose and glucose. This dolichol-mediated transport system is only functional in the ER. On the cytosolic side of the ER membrane, the mannose from GDP-Man and the glucose from UDP-Glc are linked to dolichol with concomitant cleavage of the nucleotide moiety. The enzymes that catalyze these two reactions are referred to as Dol-P-Man and Dol-P-Glc synthases, respectively. The sugar is then released into the lumen of the ER by a “flippase” activity allowing the monosaccharide to be used by ER-localized glycosyltransferases.
The second mechanism of nucleotide-sugar transport into the ER and/or Golgi involves specific nucleotide sugar transporters (NSTs). NSTs belong to the solute carrier 35 (SLC35) family of membrane transporters. These SLC35 transporters reside in the ER and/or Golgi membranes and function as typical antiporters. Some NSTs can transport more than one substrate. For example, the UDP-Gal (SLC35A2) transporter transports both UDP-Gal and UDP-GalNAc. In contrast, for example, the GDP-Fuc (SLC35C1, also known as FUCT1) and CMP-NANA (SLC35A1) transporters are monospecific.
When the nucleotide-activated sugar is transported into the lumen of the ER or Golgi there is a concomitant equimolar exit of a corresponding nucleoside monophosphate from the ER/Golgi lumen to the cytosol. It is the nucleotide portion of the nucleotide-activated sugar that serves as the recognition feature required for initial binding to the NST. Following entry into the ER or Golgi, a glycosyltransferase will transfer the monosaccharide to a target glycan concomitant with removal of the nucleoside diphosphate. The nucleoside diphosphates are then converted to nucleoside monophosphates and inorganic phosphate by a nucleoside diphosphatase. The resultant nucleoside monophosphate can then be transported out into the cytosol via the action of the SLC35 transporters. The inorganic phosphate is transported into the cytosol by a specific transporter. Both nucleoside diphosphates and monophosphates can inhibit the nucleotide-activated sugar transport process as well as the activity of luminal glycosyltransferases.
The synthesis of nucleotide-activated sugars is under tight regulatory control. Loss of this control, due to enzyme deficiency, can result in serious clinical manifestation. However, because of the interconnected pathways of nucleotide-activated sugar metabolism, the results of an individual enzyme deficiency can be difficult to predict. Due to the tight regulation of nucleotide-activated sugar synthesis, the alteration in production of only a single nucleotide sugar can significantly impair glycosylation with potentially profound effects. That disruption in the processes of nucleotide-activated sugar synthesis can result in severe clinical symptomology is evident from a number of disorders classified as congenital disorders of glycosylation, CDG (see below and the CDG page). For example, patients with CDG-IIc, caused by a deficient GDP-Fuc transporter (FUCT1, SLC35C1), manifest with unique facial features, recurrent infections, persistent leukocytosis, defective neutrophil chemotaxis and severe growth and intellectual impairment.
Mechanism of N-Glycosylation
In contrast to the step-wise addition of sugar groups to the O-linked class of glycoproteins, N-linked glycoprotein synthesis requires a lipid intermediate: dolichol phosphate. Dolichol phosphate is a polyisoprenoid compound synthesized from the isoprenoid intermediates of the de novo cholesterol biosynthesis pathway. The function of dolichol phosphate is to serve as the foundation for the synthesis of the precursor carbohydrate structure, termed the lipid-linked oligosaccharide, LLO (also referred to as the en bloc oligosaccharide), required for the attachment of carbohydrate to asparagine residues in N-linked glycoproteins.
Dolichol Phosphate Synthesis
Dolichol is derived from intermediates in the biosynthetic pathway commonly referred to as the mevalonate pathway. The mevalonate pathway is involved in the pathway of cholesterol biosynthesis as well. The intermediates in this pathway, geranylpyrophosphate (GPP) and isopentenylpyrophosphate (IPP) are condensed into farnesylpyrophosphate (FPP) through the action of the farnesyl diphosphate synthase enzyme which is encoded by the FDPS gene.
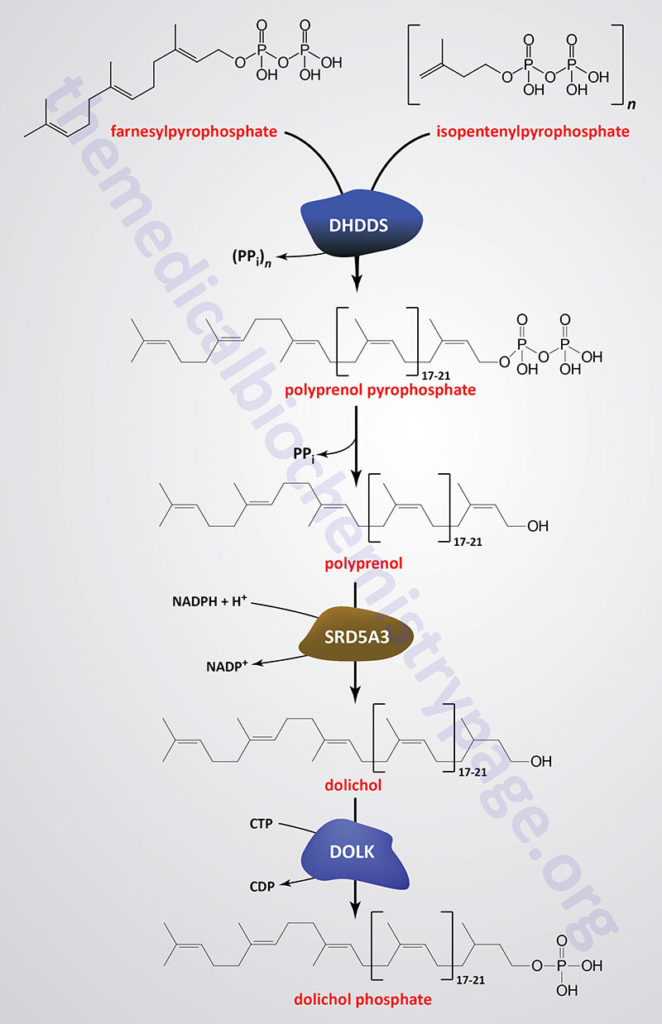
Through the action of the ER-localized enzyme, dehydrodolichyl diphosphate synthase, farnesylpyrophosphate is elongated via the sequential head-to-tail addition of multiple isopentenylpyrophosphate groups in a reaction referred to as cis-prenylation. The number of IPP substrates added ultimately determines the overall number of isoprene units in dolichol which in humans ranges from 17 to 21.
Dehydrodolichyl diphosphate synthase is encoded by the DHDDS gene. The DHDDS gene is located on chromosome 1p36.11 and is composed of 9 exons that generate five alternatively spliced mRNAs, each of which encode a distinct protein isoform.
The product(s) of the DHDDS reaction is referred to as a polyprenolpyrophosphate. The pyrophosphate is removed by an as yet uncharacterized enzyme activity that may be either a polyprenol pyrophosphate phosphatase or a polyprenol phosphatase resulting in the formation of a polyprenol.
The resultant polyprenol(s) is a substrate for steroid 5-α reductase 3 (also called polyprenol reductase) which is encoded by the SRD5A3 gene. Steroid 5-α reductase 3 belongs to the polyprenol reductase subfamily of the steroid 5-α reductase family. The SRD5A3 encoded enzyme reduces the carbon-carbon double bond closest to the hydroxyl end of the polyprenol generating dolichol. In addition to participating in the synthesis of dolichol, the SRD5A3 encoded enzyme synthesizes 5-α-dihydrotestosterone from testosterone.]
The SRD5A3 gene is located on chromosome 4q12 and is composed of 6 exons that encode a 318 amino acid protein. Mutations in the SRD5A3 gene are associated with the congenital disorder of glycosylation (CDG) identified as CDG-1q (SRD5A3-CDG).
Dolichol phosphate is then synthesized from dolichol through the action of the ER-localized enzyme dolichol kinase. The phosphate donor for dolichol kinase is CTP and not ATP as is the case for most kinases. Dolichol kinase is encoded by the DOLK gene.
The DOLK gene is located on chromosome 9q34.11 which is an intronless gene that encodes a 538 amino acid protein. Mutations of the DOLK gene are associated with the CDG identified as CDG-1m (DOLK-CDG).
Synthesis of the LLO
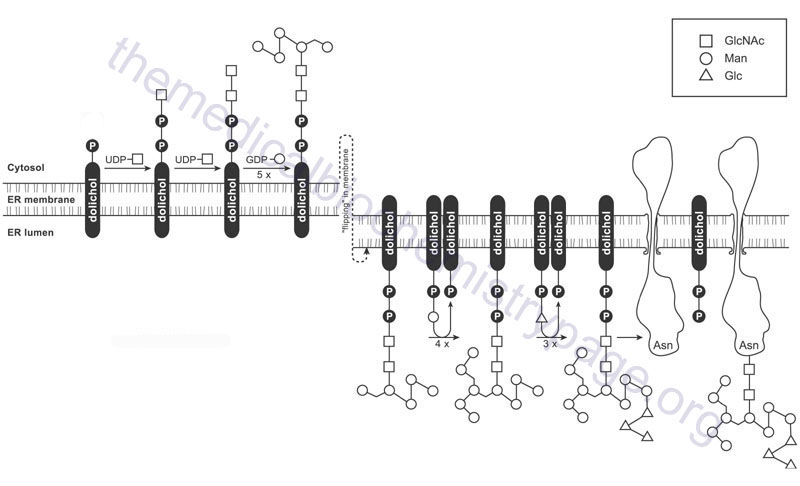
As indicated, the formation of the GlcNAc-β-Asn linkage in proteins occurs in the endoplasmic reticulum (ER) through co-translational addition of a pre-assembled carbohydrate core structure that is delivered via the carbohydrate-dolichol lipid intermediate. The pre-assembled carbohydrate core structure comprises three terminal residues of glucose attached to a branched cluster of nine mannose residues that are in turn attached to two GlcNAc residues attached to dolicholpyrophosphate. The structure is abbreviated Glc3Man9GlcNAc2–PP–Dol. This structure is commonly referred to as the lipid-linked oligosaccharide (LLO) whereas the oligosaccharide structure itself is termed the en bloc oligosaccharide.
In mammalian cells the importance of the terminal glucose residues is evident from the fact that transfer of Man9GlcNAc2–PP–dolichol is some 25-times less efficient than the complete structure. In addition, structures that contain three terminal glucose residues, but not the complete Man9GlcNAc2 structure, are efficiently transferred to protein by the oligosaccharyltransferase (OST) complex (details discussed below).
Synthesis of the en bloc dolichol–PP–oligosaccharide unit begins on the cytoplasmic face of the ER membrane. The addition of GlcNAc-phosphate, from UDP-GlcNAc, to dolichol phosphate (Dol-P) generates the initiating GlcNAc-PP-Dol structure. This reaction is catalyzed by the enzyme dolichyl-phosphate N-acetylglucosaminephosphotransferase 1 encoded by the DPAGT1 gene. The DPAGT1 gene is also known as ALG7 (ALG refers to Asparagine-Linked Glycosylation). The DPAGT1 gene is located on chromosome 11q23.3 and is composed of 9 exons that encode a 408 amino acid protein.
The additional GlcNAc and the five initial mannose (Man) residues are added sequentially to GlcNAc-PP-Dol utilizing the nucleotide activated sugars, UDP-GlcNAc and GDP-Man, respectively. The formation of the cytoplasmic oriented Man5GlcNAc2-PP-Dol structure is catalyzed by two multienzyme complexes.
The first complex uses UDP-GlcNAc as its substrate and is composed of proteins that includes the DPAGT1 encoded enzyme as well as two enzymes encoded by genes that are members of the asparagine-linked glycosylation (ALG) family of glycosyltransferase encoding genes. These two additional enzymes are encoded by the ALG13 and ALG14 genes.
The second complex uses GDP-Man as its substrate and is composed of the glycosyltransferases encoded by the ALG1, ALG2, and ALG11 genes. The clinical significance of these initial steps in the synthesis of the LLO is that mutations in five of the six genes encoding the proteins in these two complexes (DPAGT1, ALG1, ALG2, ALG11, and ALG13) are associated with the development of diseases that are members of the congenital disorders of glycosylation (CDG) family of diseases. Mutations in DPAGT1 are associated with CDG-Ij, ALG1 with CDG-Ik, ALG2 with CDG-Ii, ALG11 with CDG-Ip, and ALG13 with CDG-Is.
When the structure linked to dolichol phosphate contains these five Man and two GlcNAc residues (Man5GlcNAc2-PP-Dol) the structure is “flipped” to the luminal side of the ER membrane. The membrane flipping of Man5GlcNAc2-PP-Dol is catalyzed by the human homolog of the yeast enzyme identified as RFT1 (Requiring Fifty-Three protein 1).
The RFT1 gene is located on chromosome 3p21.1 and is composed of 19 exons that encode a 541 amino acid protein. The significance of RFT1 encoded enzyme is evidenced by the fact that mutations in the gene result in the congenital disorder of glycosylation identified as CDG-In (RFT1-CDG).
Within the lumen of the ER the remaining four Man and the three Glc residues are added to the structure via the action of various glycosyltransferases generating the complete LLO structure. The mannose and glucose residues are added utilizing the dolicholphosphate activated sugars, Dol-P-Man and Dol-P-Glc, respectively. Both Dol-P-Man and Dol-P-GLc are synthesized on the cytoplasmic side of the ER membrane. Dol-P-Man is synthesized by the action of the Dol-P-Man synthase complex (DPM synthase) and Dol-P-Glc is synthesized by the Dol-P-Glc synthase complex (DPG synthase).
The DPM synthase complex is composed of three subunits encoded by three distinct genes identified as DPM1, DPM2, and DPM3. The DPM1 encoded protein is the catalytic activity of the complex. The DPM1 protein is stabilized via interaction with the DMP3 protein and the interaction of these two proteins is stabilized by the function of the DPM2 protein. Mutations in the DPM1 gene are associated with the congenital disorder of glycosylation identified as CDG-Ie (DPM1-CDG). The Dol-P-Glc synthase is encoded by the ALG5 gene which is a member of the asparagine-linked glycosylation (ALG) family of enzymes. Following their synthesis, the Dol-P-Man and Dol-P-Glc structures are flipped so that the Man and Glc residues are present in the lumen of the ER accessible to the glycosyltransferases that incorporate these sugars into complex glycans.
The enzymes that add the Man and Glc residues to the growing LLO, within the lumen of of the ER, are members of the ALG family of enzymes. The four mannose residues are sequentially added through the actions of ALG3, ALG9, and ALG12, respectively, where ALG9 sequentially adds two Man residues. The sequential addition of the three Glc residues is catalyzed by ALG6, ALG8, and ALG10, respectively. Clinical significance to the process of the synthesis of the en bloc oligosaccharide complex of the LLO can be seen from the fact that mutations in many of the genes involved in this synthesis pathway have been associated with severe diseases of the congenital disorders of glycosylation (CDG) family of disorders. For example ALG3 mutations result in CDG-Id, ALG6 mutations result in CDG-Ic, ALG8 mutations result in CDG-Ih, ALG9 mutations result in CDG-IL, and mutations in ALG12 result in CDG-Ig.
Upon completion of the synthesis of the LLO the carbohydrate complex is transferred to an asparagine (Asn, N) residue in a substrate protein via the action of the oligosaccharyltransferase (OST) complex. The mammalian OST complex contains at least two catalytic subunits and at least six accessory subunits. The catalytic subunits are encoded by the STT3A and STT3B genes. The accessory subunits of the OST complex include dolichyl-diphosphooligosaccharide-protein glycosyltransferase non-catalytic subunit (encoded by the DDOST gene; also known as OST48), oligosaccharyltransferase complex subunit 4, non-catalytic (encoded by the OST4 gene), ribophorin I (encoded by the RPN1 gene), ribophorin II (encoded by the RPN2 gene), defender against cell death 1 (encoded by the DAD1 gene; also known as OST2), and tumor suppressor candidate 3 (encoded by the TUSC3 gene; also known as N33). The clinical significance of the OST can be evidenced by the fact that mutations in several of the genes encoding subunits of the complex are associated with congenital disorders of glycosylation, CDG. For example mutations in the DDOST gene result in CDG-Ir (DDOST-CDG).
Immediately following transfer of the en bloc oligosaccharide unit to the protein, processing and alteration of the composition of the oligosaccharide ensues and continues as the protein passes through the ER then into and through the Golgi apparatus. Initially, the terminal glucose is removed through the action of processing α-glucosidase I (GluI or GCS1), a membrane bound enzyme recognizing α-1,2-linked glucose. GluI is a single pass type II transmembrane protein with the catalytic domain contained within the lumen of the ER. GluI will cleave the terminal glucose residue from the substrate, Glc3Man9GlcNAc2, found linked to dolichol or protein or as a free oligosaccharide. The official name for GluI is mannosyl-oligosaccharide glucosidase which is encoded by the MOGS gene. Mutations in the MOGS gene result in the congenital disorder of glycosylation identified as CDG-IIb (GCS1-CDG).
The remaining two glucose residues are then removed by α-glucosidase II (GluII), a soluble heterodimeric enzyme recognizing α-1,3-linked glucose. Functional GluII is composed of an α-subunit and a β-subunit. The GluII α-subunit gene is GANAB (glucosidase II alpha subunit) and the β-subunit gene is PRKCSH (protein kinase C substrate 80K-H).
After removal of the glucose residues, the action of α-mannosidases removes several mannose residues as the protein progresses to the Golgi. Humans express several genes encoding mannosidase enzymes and these genes are divided into two families called mannosidases type alpha and mannosidases type beta.
The mannosidase type alpha family is further divided into three subfamilies identified as alpha class 1, alpha class 2, and endo-alpha. The alpha class 1 subfamily contains 7 genes. The alpha class 2 subfamily contains 5 genes, and the endo-alpha subfamily contains 2 genes.
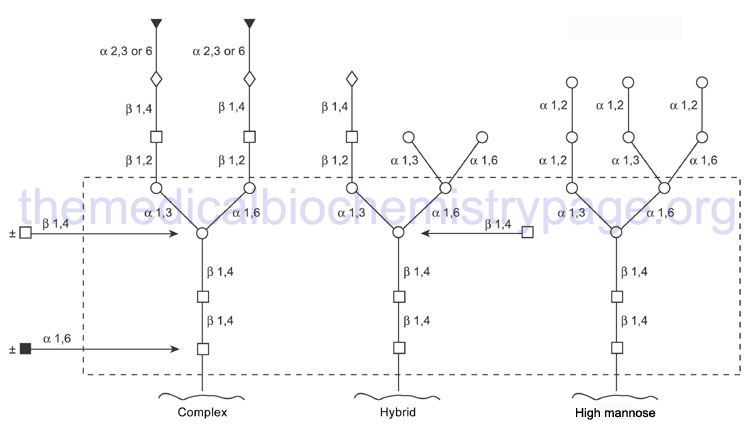
The mannosidase type beta family is composed of 2 gene. The action of the various glucosidases and mannosidases leaves N-linked glycoproteins containing a common core of carbohydrate consisting of three mannose residues and two GlcNAc.
Through the action of a wide range of glycosyltransferases and glycosidases a variety of other sugars are attached to this core as the protein progresses through the Golgi. Upon completion of glycan processing, all N-linked glycoproteins will contain a common core of carbohydrate attached to the polypeptide. This core consists of three mannose residues and two GlcNAc. A variety of other sugars are attached to this core resulting in the generation of the three major N-linked glycoprotein families:
- High-mannose type contains all mannose outside the core in varying amounts
- Hybrid type contains various sugars and amino sugars
- Complex type is similar to the hybrid type, but in addition, contains sialic acids to varying degrees
O-Glycosylation & Mucin-Type O-Glycans
The attachment of carbohydrate to the hydroxyl group of serine (Ser, S) or threonine (Thr, T) residues in proteins, as well as hydroxylysine (hLys) in collagens, constitutes the bulk of O-linked glycans (O-glycans) found in humans. Carbohydrate addition in O-glycans occurs via the stepwise addition of nucleotide activated sugars as the modified proteins traverse the ER and the Golgi network. There are seven major types of O-glycans in humans that are defined by the first sugar residue that is attached to either Ser, Thr or hLys.
The Table below lists these seven O-glycan types. The (R)- symbol represents the fact that a broad array of additional carbohydrates can be found attached to these basic glycan structures. The ± symbol indicates that the additional carbohydrate structure is found in some but not all species of that particular O-glycan type. Xyl is the sugar xylulose.
Table of Major O-Glycan Types in Humans
| O-Glycan Type | Structure of Linkage | Glycoprotein Type |
| O-linked GlcNAc | GlcNAc-β1–Ser/Thr | nuclear and cytoplasmic |
| mucin-type | (R)-GalNAc-α1–Ser/Thr | plasma membrane and secreted |
| O-linked mannose | NANA-α2–3Gal-β1–4GlcNAc-β1–2Man-α1–Ser/Thr | α-dystroglycan |
| O-linked fucose | NANA-α2–6Gal-β1–4GlcNAc-β1–3±Fuc-α1-Ser/Thr | EGF domains; this particular O-fucosylation is critical in the function of the receptor protein Notch |
| O-linked fucose | Glc-β1–3Fuc-α1–Ser/Thr | thrombospondin 1 repeats (TSR) |
| O-linked glucose | Xyl-α1–3Xyl-α1–3±Glc-β1–Ser | EGF domains |
| O-linked galactose | Glc-α1–2±Gal-β1-O–Lys | collagens |
| glycosaminoglycan (GAG) | (R)-GlcA-β1–3Gal-β1–4Xyl-β1–Ser | proteoglycans |
Of the seven different types of O-glycans, by far the most common in humans is the mucin-type. Mucin glycoproteins are so-called because of their abundance in the mucous secretions on cell surfaces and in body fluids. Mucin-type O-glycans all have the amino sugar GalNAc attached to the Ser or Thr residue of the modified protein. The process of O-GalNAc glycosylation occurs via two distinct steps that consist of initiation and processing. The initiation step controls the pattern and the density of the carbohydrate structures attached to the protein. The processing step determines the ultimate O-glycan structure that is present in the fully modified protein.
The attachment of the initial GalNAc residue occurs in the Golgi network after the target protein has obtained its’ native folded state. Attachment of GalNAc to Ser or Thr residues in a target protein is catalyzed by a family of enzymes known as UDP-N-acetylgalactosamine:polypeptide N-acetylgalactosaminyltransferases (ppGalNAcTs or ppGaNTases). This family of enzymes is most commonly referred to as the polypeptide N-acetylgalactosaminyltransferases (GALNT) family. Humans express 20 genes in the GALNT family.
The primary GALNT family enzyme responsible for the majority of GalNAc attachment to Ser or Thr residues in human proteins is encoded by the GALNT2 gene. The addition of the GalNAc residue forms what is referred to as the Tn antigen (Tn Ag). Subsequent to the formation of the Tn Ag in mucin-type O-glycans, numerous glycosyltransferases begin the process of modification of the carbohydrate structure that then results in the fully modified protein.
The GALNT2 gene is located on chromosome 1q42.13 and is composed of 19 exons that generate two alternatively spliced mRNAs encoding precursor proteins of 571 amino acids (isoform 1) and 533 amino acids (isoform 2).
One common modification of the Tn Ag is the addition of sialic acid forming the structure referred to as the sialyl-Tn antigen. In addition to the Tn and the sialyl-Tn structures, the further modifications result in the formation of eight additional mucin-type core structures. These eight mucin-type core structures are defined based upon the second sugar attached to the GalNAc and the linkage of that attachment (e.g. α or β and 1–3 or 1–6). The core 1 through core 4 structures detailed in the Table below represent the majority of mucin-type O-glycan structures produced in human tissues.
Table of Major Mucin-Type O-Glycan Structures
| Core Type | Carbohydrate Structure of Core | Comments |
| 1 | Galβ1–3GalNAc-α1–Ser/Thr | most cells, secreted proteins; most abundant mucin-type O-glycan structure, referred to as the Tn antigen; there is a single core 1 β1–3 galactosyltransferase (C1Gal-T1 or T synthase) in mammals; highest expression in liver, kidney, heart, and placenta |
| 2 | Galβ1–3(GlcNAcβ1–6)GalNAc-α1–Ser/Thr | all blood cells; represents a branching glycan of the core 1 structure; catalyzed by one of three β1–6 N-acetylglucosaminyltransferases (C2GnT-1 to -3 or β6GlcNAc-T); each with distinct expression patterns; two synthesize the core 2 structure, one (C2GnT-2)synthesizes both core 2 and core 4 structures |
| 3 | GlcNAcβ1–3GalNAc-α1–Ser/Thr | colon and saliva; synthesized by β1–3 N-acetylglucosaminyltransferase (C3GnT-1 or β3Gn-T6), single enzyme in humans expressed primarily in small intestine and stomach |
| 4 | GlcNAcβ1–3(GlcNAcβ1–6)GalNAc-α1–Ser/Thr | mucin-secreting cell types; represents a branching glycan of the core 3 structure; catalyzed by β1–6 N-acetylglucosaminyltransferases (β6GlcNAc-T); three β6GlcNAc-Ts in humans, each with distinct expression patterns; two synthesize the core 2 structure, one synthesizes both core 2 and core 4 structures |
| 5 | GalNAcα1–3GalNAc-α1–Ser/Thr | adenocarcinomas, embryonic gut-derived meconium |
| 6 | GlcNAcβ1–6GalNAc-α1–Ser/Thr | embryonic gut, mucins from ovarian cysts; core 6 glycans may represent β-galactosidase degradation products of core 2 glycans |
| 7 | GlcNAcα1–6GalNAc-α1–Ser/Thr | not described in humans |
| 8 | Galα1–3GalNAc-α1–Ser/Thr | bronchial tissue mucins |
Recent evidence has demonstrated that human core 1 β1–3-galactosyltransferase (C1Gal-T1) requires a molecular chaperone present in the ER for its enzymatic function. This molecular chaperone has been named core 1 β1–3-Gal-T-specific molecular chaperone, Cosmc, which is encoded by the C1GALT1C1 (C1GALT1 specific chaperone 1) gene. Cosmc is a member of the type II transmembrane protein family. The role of Cosmc in C1Gal-T1 activity is in ensuring that the enzyme is properly folded within the ER. Loss of Cosmc activity results in C1Gal-T1 being degraded in the proteasome. Given these results it is possible, but as yet not demonstrated, that additional chaperones specific for other glycosyltransferases may exist.
The attachment of carbohydrates to proteins forming O-glycans serves multiple critical functions with respect to the structure and activity of the modified proteins. O-linked glycans are important for protein stability, modulation of enzyme activity, receptor-mediated signaling, immune function and immunity, protein-protein interactions, as well as many other functions. Mucin-type O-glycans are also important for binding water and are often found on the outer surfaces of tissues such as the gastrointestinal, urogenital and respiratory systems. The interaction of water with the mucin-type O-glycans results in the formation of a viscous solution or gel that forms a protective barrier harboring antibacterial properties.
O-Mannosylation
Incorporation of mannose into target proteins by attachment to Ser and/or Thr, referred to as O-mannosylation, was originally thought to only occur in fungi. However, it is now known that this particular form of protein glycosylation serves a critical function in mammals. In mammals, O-mannosylation has been identified on α-dystroglycan (α-DG) from nerve and muscle, chondroitin sulfate proteoglycans (CSPG), and several members of the cadherin family of calcium-dependent cell-cell adhesion proteins.
Incorporation of mannose into O-glycans involves the transfer of GDP-activated mannose (GDP-Man) to dolichol-phosphate forming Dol-P-Man. Synthesis of Dol-P-Man takes place on the cytosolic face of the ER membrane via the action of the Dol-P-Man synthase complex (DPM synthase) as described above for the synthesis of the lipid-linked oligosaccharide (LLO) complex involved in N-linked glycoprotein synthesis. As indicated, the DPM synthase complex is composed of three subunits encoded by three distinct genes identified as DPM1, DPM2, and DPM3. The DPM1 encoded protein is the catalytic activity of the complex. The DPM1 protein is stabilized via interaction with the DMP3 protein and the interaction of these two proteins is stabilized by the function of the DPM2 protein.
Following synthesis of Dol-P-Man the orientation of the structure is flipped so that the Man residue is present in the lumen of the ER accessible to the enzymes catalyzing transfer to protein or for the glycosyltransferases that incorporate the Man into complex glycans. Transfer of the mannose from Dol-P-Man to the substrate protein is catalyzed by enzymes of the dolichyl-phosphate-D-mannose:protein O-mannosyltransferase (PMT) family. In mammals, POMT1 (O-mannosyltransferase-1) and POMT2 (O-mannosyltransferase-2) are the known PMTs involved in O-mannosylation reactions. Evidence shows that the POMT1 and POMT2 proteins form a complex and these complexes are crucial for mannosyltransferase activity. Since it occurs in the ER, the actual process of O-mannosylation is distinct from most other O-glycosylation reactions that take place exclusively in the Golgi apparatus.
After mannosylation of the target protein further extension of the O-linked mannose residue takes place in the Golgi apparatus. The addition of GlcNAc residues to O-mannosylated glycans is catalyzed by the enzyme encoded by the POMGNT1 (O-mannose β-1,2-N-acetylglucosaminyltransferase) gene. The vast majority of mammalian O-mannosyl glycans represent variations of the tetrasaccharide NANAα2–3Galβ1–4GlcNAcβ1–2Manα1–Ser/Thr with different lengths (e.g., asialo) and variable fucose (α1,3- linked to GlcNAc) contents. In addition to the conserved tetrasaccharide, O-mannosyl glycans containing the human natural killer-1 (HNK-1) epitope [HSO4-3GlcAβ1–3Galβ1–4GlcNAcβ1–2Man-Ser/Thr] have been detected in significant amounts on PTPRZ1 (RPTPβ) from neuroblastoma cells.
To date, the best-studied O-mannosylated protein in humans is α-DG. It comprises two globular domains separated by a Ser/Thr-rich mucin-like region that is substantially O-mannosylated. α-DG is an essential component of the dystrophin-glycoprotein complex (DGC) in skeletal muscle. Highlighting the significance of O-mannosylated α-DG is the fact that most of the defects associated with impaired O-mannosylation can be explained by reduced function of α-DG.
In humans, defective O-mannosylation is associated with a group of autosomal recessive muscular dystrophies termed congenital muscular dystrophies (CMD) and also referred to as muscular dystrophy-dystroglycanopathies (MDDG). There are at least six related syndromes identified as MDDG (MDDGA1, MDDGA2, MDDGA3, MDDGA4 (and MDDGB4), MDDGA5 (and MDDGB5), and MDDGA6 (and MDDGB6). The letter “A” and “B” in the designations relate to disease severity with “A” indicating severe, and “B” indicating intermediate.
In addition to muscle dysfunction, many of these related disorders are associated with variable brain and ocular abnormalities. The CMD are also referred to as secondary α-dystroglycanopathies since their common pathological feature is the hypoglycosylation of α-DG. CMD patients exhibit both clinical and genetic variability. Several of these related disorders resulting from defects in O-mannosylation were originally classified as Walker-Warburg syndrome (WWS; most severe forms) and the muscle-eye-brain diseases (MEB; less severe forms). The WWS disorders are associated with multiple malformations and these patients often die within the first year of life. The less severe MEB disorders (which also includes Fukuyama congenital muscular dystrophy, FCMD) are characterized by being associated with severe brain malformations and abnormalities of the eye. The mildest forms of CMD may not present until adulthood such as in limb-girdle muscular dystrophy (LGMD) which manifests with neither brain or eye abnormalities. Since all of these disorders are the result of mutations in genes whose encoded enzymes function in processes of protein glycosylation the diseases also belong to the large family of disorders called the congenital disorders of glycosylation, CDG.
The proposed nomenclature for CDG that cause congenital muscular dystrophy spectrum due to defects in POMT1 (POMT1-CDG), POMT2 (POMT2-CDG), and POMGNT1 (POMGNT1-CDG) is MDDGA1, MDDGA2, and MDDGA3, respectively. These three MDDG disorders are associated with characteristic brain and eye malformations, profound intellectual impairment, congenital muscular dystrophy, and early death.
MDDGA4 is also commonly referred to as Fukuyama-type congenital muscular dystrophy and is caused by mutations in the fukutin gene (symbol: FKTN) whose encoded protein is thought to be a glycosyltransferase involved in brain development.
MDDGA5 (also referred to as congenital muscular dystrophy type 1C) results from mutations in the fukutin-related protein gene (FKRP).
MDDGA6 (also referred to as congenital muscular dystrophy type 1D) results from mutations in the LARGE1 (like-acetylglucosaminyltransferase 1) gene which encodes the bifunctional enzyme identified as LARGE xylulose- and glucuronyltransferase 1. The LARGE1 encoded enzyme belongs to the N-acetylglucosaminyltransferase family of enzymes. The LARGE1 encoded enzyme is important in the glycosylation of α-dystroglycan.
C-Mannosylation
The process of C-mannosylation of proteins occurs post-translationally within the lumen of the endoplasmic reticulum, ER. This type of carbohydrate modification of proteins involves a single mannose residue being attached to the first Trp residue that is found to reside in the motif, W-X-X-W/C, where X is any amino acid. The C-C bond that is generated in the process of C-mannosylation of Trp residues is a unique form of sugar–amino acid linkage when compared to the more commonly occurring C–O bond and C–N bond in O-glycans and N-glycans, respectively.
The enzymes that catalyze the C-mannosylation are members of the DPY19 family of C-mannosyltransferases. The DPY19 nomenclature is derived from the C. elegans gene, dpy-19, that refers to “dumpy” body type identified in various mutants in this organism. Humans express two genes encoding C-mannosyltransferases, DPY19L1 and DPY19L3.
Following the initial identification of C-mannosylation in the RNase 2 enzyme, numerous (at least 40) human proteins have been found to be C-mannosylated. These proteins include members of the thrombospondin type 1 repeat (TSR) family of proteins and type 1 cytokine receptor family. Proteins of the TSR family that are C-mannosylated include thrombospondin-1, several complement system component proteins (C6, C7, C8, and C9), and several proteins of the A Disintegrin and Metalloproteinase with Thrombospondin type 1 motif (ADAMTS) family. Proteins of the type 1 cytokine receptor family that are C-mannosylated include the erythropoietin receptor, the interleukine-21 (IL-21) receptor, and the granulocyte colony-stimulating factor receptor (G-CSFR).
The process of C-mannosylation first involves the attachment of mannose, from GDP-mannose, to dolichol-phosphate present in the ER membrane. The mannose attachment to dolichol-phosphate involves the same DPM synthase complex, specifically DPM1, DPM2, and DPM3, that are described in the Synthesis of the LLO and the O-Mannosylation sections above. Following mannose attachment to dolichol at the cytoplasmic side of the ER membrane the complex is flipped to the lumen and then the ER membrane-associated C-mannosyltgransferases transfer the mannose to an appropriate Trp residue in the ER lumen.
The major function for C-mannosylation of proteins is to facilitate proper folding of the modified protein as well as proper intracellular transport and also secretion, if the protein is a secreted protein such as the protein encoded by the ADAMTS1 gene.
Mutation in a C-mannosylation site of the ADAMTSL1 (which was originally termed punctin) protein has been shown to be associated with congenital glaucoma, craniofacial abnormalities, and other systemic features demonstrating the clinical significance of this type of post-translational modification.
Hexosamine (O-GlcNAc) Signaling in Metabolism & Development
Hexosamine Biosynthesis
Multiple nuclear and cytoplasmic proteins, including transcription factors, cytoskeletal proteins, oncogenes, and kinases, are post-translationally modified on serine and threonine residues with β-N-acetylglucosamine (O-GlcNAc).
Until the discovery of O-GlcNAc modification (referred to as O-GlcNAcylation: pronounced “Oh glook knack ill ation”) on nuclear and cytoplasmic proteins, more than 25 years ago, it was believed that protein glycosylation was restricted to the luminal compartments of the secretory machinery and to the cell surface and extracellular matrix. Early studies on the subcellular localization of O-GlcNAc showed that it was highly concentrated at the nuclear envelope, particularly at the nuclear pore complex, as well as throughout the chromosomes. Additionally, several cytosolic and cytoskeletal proteins were also found to be O-GlcNAcylated.
The gamut of O-GlcNAcylated proteins includes enzymes involved in the metabolism of amino acids, nucleotides (e.g. thymidine kinase), and carbohydrates (e.g. glucose-6-phosphatase), regulating overall metabolic processes (e.g. AMPK), cell growth and maintenance (e.g. MYC, Sp1, β-catenin), DNA damage responses, intracellular transport, transcription (e.g. RNA polymerase II), and translation (e.g. eIF-5). The carboxy-terminal domain of a subpopulation of RNA polymerase II is extensively O-GlcNAcylated, and almost all RNA polymerase II transcription factors are O-GlcNAcylated. Thus far, more than 600 proteins have been shown to be O-GlcNAcylated.
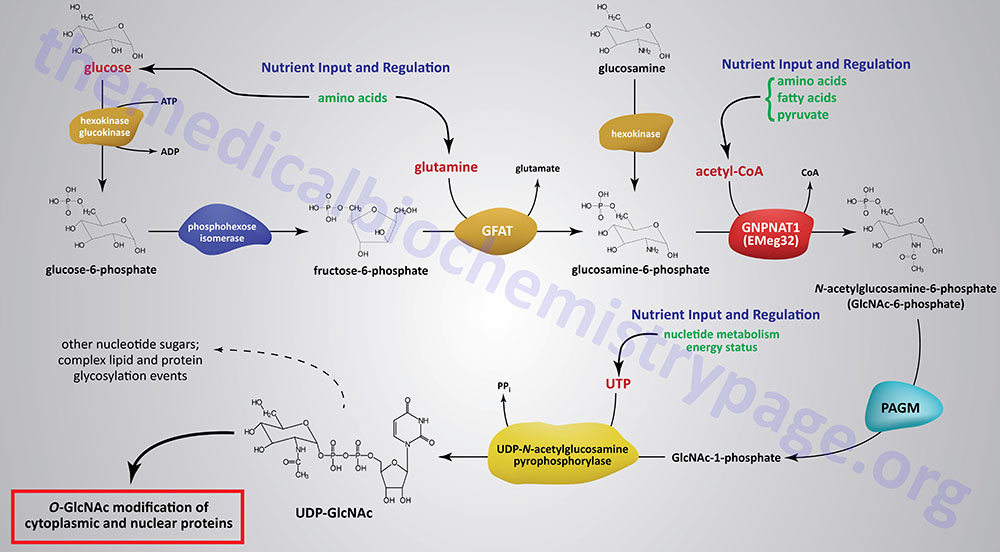
The synthesis of O-GlcNAc occurs via the metabolic pathway called the hexosamine biosynthesis pathway, HBP. Synthesis of O-GlcNAc initiates from glucose. Upon entering cells, glucose is converted to glucose-6-phosphate (G6P) via the action of glucokinase/hexokinase. Most of the G6P formed is oxidized in the process of glycolysis. However, a portion of the G6P can be shuttled into either glycogen synthesis or the pentose phosphate pathway, depending upon the metabolic needs of the cell. During the process of glycolysis, phosphohexose isomerase converts G6P into fructose-6-phosphate (F6P). Whereas, the majority of F6P enters glycolysis, 2–5% enters the hexosamine biosynthesis pathway (HBP) to generate uridine diphospho-N-acetylglucosamine (UDP-GlcNAc). The end product of the HBP, UDP-GlcNAc, is referred to as an activated nucleotide sugar. The nucleotide sugar is an essential intermediate in the synthesis of a wide array of complex glycans.
The first and rate-limiting enzyme in the HBP is glutamine:fructose-6-phosphate aminotransferase 1, GFAT (also abbreviated GFPT1). GFAT utilizes glutamine and F6P for the formation of glucosamine-6-phosphate (GlcN6P) and glutamate. It is possible for glucosamine to enter the HBP directly by conversion to GlcN6P via the action of hexokinase, thus bypassing the rate-limiting step of the pathway. This has been shown to be true when glucosamine is infused into rats or mice.
GlcN6P is acetylated forming N-acetylglucosamine-6-phosphate (GlcNAc6P) from acetyl-CoA via the action of glucosamine-6-phosphate N-acetyltransferase (gene symbol GNPNAT1). This enzyme is sometimes referred to as EMeg32 due to the fact that it was clone 32 originally isolated from a screen for genes specific for precursors of Erythroid and Megakaryocytic lineages.
GlcNAc6P is converted to GlcNAc-1-phosphate (GlcNAc1P) via the action of phosphoacetylglucosamine mutase (PAGM). The PAGM protein is encoded by the phosphoglucomutase 3 gene (PGM3). GlcNAc1P is finally nucleotide activated via the action of UDP-N-acetylglucosamine pyrophosphorylase (UAP1) generating UDP-GlcNAc.
Like many metabolic pathways, the product, in this case UDP-GlcNAc, serves to regulate the rate of flux through the pathway by feedback inhibition of the rate-limiting enzyme, GFAT. Once synthesized the UDP-GlcNAc can be converted into other types of nucleotide sugars or directly incorporated into a variety of carbohydrate modified macromolecules such as lipids and proteins.
Role of Mannose in Hexosamine Signaling
Dietary mannose, in the free state, is obtained primarily from fruits, berries, and legumes. Small amounts of mannose are obtained from heteropolymers such as galactomannans following digestion by intestinal bacteria as well as from ingested glycoproteins.
Mannose is taken up into intestinal enterocytes and delivered to the circulation via GLUT-mediated transport and also transported into cells via GLUT transporters. Whether a particular GLUT is specific for mannose has yet to be determined. Once inside cells mannose is phosphorylated to mannose-6-phosphate via hexokinases.
Mannose-6-phosphate is utilized in the N-glycosylation and O-mannosylation of proteins following its conversion to mannose-1-phosphate by phosphomannomutase (PMM2) followed by activation to GDP-mannose. Mannose-6-phosphate is also a substrate for mannose phosphate isomerase (MPI) which interconverts mannose-6-phosphate and fructose-6-phosphate. Indeed, approximately 90% of ingested mannose is converted to fructose-6-phosphate via the actions of MPI.
Conversion of mannose-6-phosphate to fructose-6-phosphate allows mannose to directly contribute to energy production via the glycolytic pathway. In addition to oxidation via glycolysis, mannose converted to fructose-6-phosphate can also contribute to carbon storage as glycogen.
The conversion of mannose-6-phosphate to fructose-6-phosphate also allows mannose to contribute to the synthesis of UDP-GlcNAc via the hexosamine biosynthesis pathway. Physiological significance of the incorporation of mannose into the pathway of protein O-GlcNAcylation has been demonstrated in T-cells where this pathway enhances T-cell anti-tumor activity as a result of increased O-GlcNAcylation of β-catenin.
Hexosamine Cycling
Similar to serine and threonine phosphorylation, which can cycle between phosphorylation and dephosphorylation, thereby effecting fluctuating regulation of enzyme activity, O-GlcNAc modification at serine and threonine residues also cycles. The cycling of O-GlcNAc is a nutrient-responsive, post-translational modification that, like phosphorylation, impacts target protein activity. The enzymes responsible for the addition and removal O-GlcNAc have been identified and cloned from several model organisms. Additionally, upstream modulators that control the level of the donor sugar, UDP-GlcNAc, have also been identified.
Studies of hexosamine cycling have been greatly aided by a variety of inhibitors and sensors of the process. Research on O-GlcNAc cycling has identified this molecule as a key regulator of nutrient sensing. Additionally, disruption of HBP has a profound impact on diseases of nutrient sensing, such as type 2 diabetes, and neurodegeneration. That O-GlcNAcylation is a critical protein modification is demonstrated by the fact that blocking its synthesis or attachment to target proteins causes embryonic stem cell lethality in mice.
O-GlcNAc cycling at serine and threonine residues is maintained by the action of two enzymes. Addition of GlcNAc at serine and threonine residues is referred to as O-GlcNAcylation and is catalyzed by the enzyme UDP-N-acetylglucosamine:polypeptide β-N-acetylglucosaminyltransferase or more commonly just O-GlcNAc transferase, OGT. Removal of the O-GlcNAc modification is catalyzed by β-N-acetylglucosamindase or more commonly O-GlcNAcase, OGA. The OGA gene is also referred to as the MGEA5 gene due to the fact that it was originally isolated as the meningioma-expressed antigen 5 gene.
Whereas there are hundreds of different kinases and phosphatases that add and remove phosphate, respectively, from serine and threonine residues in various target proteins, there is but a single OGT and single OGA gene in the human genome.
The OGT gene is located on the X-chromosome (Xq13.1) and is composed of 22 exons that generate two alternatively spliced mRNAs encoding proteins of 1046 amino acids (isoform 1) and 1036 amino acids (isoform 2). The physical location of the OGT gene places it in close proximity to the Xist locus and the X-inactivation center (XIC). The close proximity of the OGT gene to known heterochromatin boundaries may have significance for the regulation of its expression.
The OGA gene is located on chromosome 10q24.32 and is composed of 18 exons that generate two alternatively spliced mRNAs encoding proteins of 916 amino acids (isoform a) and 863 amino acids (isoform b).
The HBP integrates the nutrient status of the cell by utilizing glucose, acetyl-CoA, glutamine, and UTP to produce UDP-GlcNAc. In turn, OGT will transmit this nutrient information throughout the cell changing the level of GlcNAcylated target proteins. While UDP-GlcNAc is used throughout the secretory pathway as a building block for the synthesis of N-linked and O-linked glycans, as well as the assembly of GPI-anchors, nuclear and cytosolic O-GlcNAc-modified proteins appear to be particularly sensitive to physiological flux of the UDP-GlcNAc pools.
How then does a complex array of protein substrates in a variety of different tissues come under the concerted control of a single enzyme? It turns out that OGT is composed of a modular structure generated by tetratricopeptide repeat (TPR) domains. These domains produce a super helical structure containing an asparagine ladder that is critical for protein recognition. Alternative splicing produces multiple isoforms, each with varying number of TPRs. This difference allows each isoform to modify a select subset of substrates. Alternative splicing also targets OGT to different subcellular compartments. Additionally, there have been a wide array of OGT-interacting proteins (OIP) identified that provide another layer of regulation by bridging the interaction of the enzyme with its with target substrates.
Hexosamine Signaling in Development
As indicated above O-GlcNAc cycling is essential for viability since its deletion results in extreme developmental defects, most often leading to death of the developing embryo. By tissue selective knockout of the OGT gene, profound changes in all cell types examined have been found. For example, within the brain neuronal-specific knockout of OGT leads to mice that are smaller at birth and exhibit aberrant locomotor activity. EMeg3, which is essential for the efficient production of UDP-GlcNAc is also a critical enzyme controlling early development. Knocking out EMeg32 in mice results in neonatal lethality and the animals exhibit pronounced developmental delay.
Hexosamine cycling is also known to be critical in the regulation of transcriptional repression. Using Drosophila melanogaster as a model organism it has been shown that the polycomb group proteins (PcG) repress the transcription of HOX genes as a multimeric complex referred to as the PcG repressive complex (PRC). This PRC-mediated transcriptional repression effects control over anterior-posterior segmentation of the developing larva. The PcG repression of HOX genes, thereby, prevents homeotic transformations. The PcG family proteins are conserved in mammals and carry out similar transcriptional repressive functions. The unifying theme of PcG function is to act as an epigenetic regulator of cell fate and maintain cellular identity through many rounds of cell division.
It turns out that in Drosophila OGT is the same as the super sex combs (sxc) gene which is an essential component of PcG complexes in this organism. The lethal phenotype associated with sxc mutations can be rescued by expression of the human OGT gene. Therefore, OGT plays an integral role in the ability of the PcG proteins to repress genes appropriately. In addition, since the C-terminal domain (CTD) of RNA polymerase II is extensively O-GlcNAcylated, the cycling could alter the activity of RNA Pol II at the promoter of many of these same PRC regulated genes.
With respect to epigenetic changes in gene expression it is interesting that the OGT gene is located very close to the XIST locus which encodes the long non-coding RNA (lncRNA) involved in X-chromosome inactivation, XCI. In addition, the OGT gene is one of a small number of genes that are highly regulated in embryonic stem cells during the process of XCI, owing to its close proximity to XIST. Experiments in Drosophila demonstrate that PcG regulates expression of the NK cluster on chromosome 10 in which the OGA gene resides. Since OGT regulates the activity of the PRC, OGT could, in turn, control the expression of OGA. Therefore, OGT acting through PcG would repress OGA expression and bias the cell towards sustained levels of O-GlcNAc.
Hexosamine Signaling and Insulin Resistance
Numerous proteins involved in insulin signaling and the downstream targets of these signaling cascades have been shown to be O-GlcNAcylated. With respect to insulin receptor signaling proteins, insulin receptor substrate 1 (IRS-1), phosphatidylinositol-3-kinase (PI3K), PKB/AKT, PIP3-dependent kinase 1 (PDPK1), and glycogen synthase kinase 3β (GSK3β) are all known to be O-GlcNAcylated. These modifications have all been observed in adipocytes which are a major target for the actions of insulin.
PKB (protein kinase B)/AKT (AK strain transforming) was originally identified as the tumor inducing gene in the AKT8 retrovirus found in the AKR strain of mice. Humans express three genes in the AKT family identified as AKT1 (PKBα), AKT2 (PKBβ), and AKT3 (PKBγ).
Insulin-stimulated glucose uptake into adipocytes occurs via insulin-mediated mobilization of GLUT4 to the plasma membrane. Increased glucose uptake, in response to insulin can, therefore, significantly modify the flux through the HBP. Evidence linking the correlation between the HBP and insulin resistance in adipocytes was demonstrated at least 20 years ago.
Using cultured rat adipocytes experiments demonstrated that chronic exposure to both insulin and glucose was required for the adipocytes to become insulin-resistant. For more information of insulin resistance go to the Insulin Function, Insulin Resistance, and Food Intake Control of Secretion page. This is now a common theme underlying insulin resistance in other insulin-sensitive tissues such as skeletal muscle. In these early experiments it was shown that the impairment in insulin-stimulated glucose uptake, under hyperglycemic and hyperinsulinemic conditions, was exclusively dependent on the presence of the amino acid glutamine. Remember that glutamine is required as a substrate for GFAT, the rate-limiting enzyme in the HBP.
Inhibition of GFAT activity was observed in the hyperglycemic and hyperinsulinemic conditions likely due to feedback inhibition by UDP-GlcNAc as the HBP product was shown to accumulate in the treated cells. However, if GFAT was inhibited with the use of various amidotransferase inhibitors the hyperglycemia-induced insulin resistance was prevented. Additionally, if cells are treated with glucosamine, which enters the HBP after the GFAT catalyzed reaction, there was a greater reduction in insulin-mediated glucose uptake compared to the hyperglycemic condition. As expected, since GFAT is bypassed, the glucosamine-induced insulin resistance does not require glutamine. Although glucose and glutamine metabolism are key inducers of flux through the HBP, free fatty acids (FFA) and uridine are also potent modulators of the HBP.
Utilizing experiments in whole animals, as opposed to cell culture, has provided additional direct evidence that excess flux through the HBP leads to modulation of insulin sensitivity in adipocytes. When GFAT is over-expressed in mice under the control of a GLUT4 promoter the animals develop classical insulin-resistant phenotype with hyperinsulinemia and reduction in whole-body glucose disposal rate. Because GLUT4 is highly expressed in adipose tissue and skeletal muscle, two major insulin-responsive tissues, it is not surprising that defective whole-body glucose disposal was observed. Elevation in serum leptin level was also observed in these GFAT over-expressing mice. Interestingly, muscle explants from GLUT4-GFAT mice showed normal insulin-stimulated glucose uptake. This latter observation is strong evidence that adipocytes play a major regulatory role in the HBP-mediated whole-body insulin resistance.
Another strain of mice has been utilized for studies on the role of HBP in insulin sensitivity that express GFAT specifically in adipose tissue by the use of an aP2 (adipocyte lipid binding protein) promoter driving its expression. Adipose tissue-restricted elevations in O-GlcNAc levels are detected in these mice and this is associated the development of whole-body insulin resistance. The results in these animals is characterized by a reduction in both glucose disposal rate and skeletal muscle glucose uptake. An increase in serum leptin and a decrease in serum adiponectin levels were also found in these mice.
As pointed out above, numerous proteins downstream of the insulin receptor that are critical to insulin-mediated signal transduction are known to be O-GlcNAcylated. Therefore, it is not difficult to assume that HBP-mediated glucose desensitization will occur at multiple stages, in particular through insulin-mediated signal transduction. Under high glucose-induced insulin resistance, there is a reduction in insulin-stimulated phosphorylation of PKB/AKT. There has been some discrepancy in determining precisely how HBP flux affects PKB/AKT phosphorylation in response to insulin binding its receptor. Recent research has shown that when cells are exposed to chronically high glucose and insulin there is a concomitant reduction in PIP3 which is a product of activated PI3K, a target of the activated insulin receptor. This reduction in PIP3 levels is correlated with an increase in PTEN (phosphatase and tensin homolog deleted on chromosome 10) levels. PTEN is a known inhibitor of PI3K.
In addition, it was shown that there is an increase in IRS1 phosphorylation on Ser636 and Ser639. Since rapamycin treatment inhibits the alteration of PIP3 and PTEN levels under insulin-resistant conditions, it is believed that mechanistic target of rapamycin (mTOR) complex 1 (mTORC1) is involved in negatively regulating the IRS1–PI3K–AKT signaling cascade downstream of the insulin receptor. The sites on IRS1 seen to be phosphorylated by chronic hyperglycemic and hyperinsulinemic conditions (S636/S639) are known to be substrates of mTORC1.
The regulation of insulin-stimulated GLUT4 translocation is also affected by changes in the flux rate through the HBP. Several cytoskeletal proteins involved in mobilization of GLUT4 to the plasma membrane are known to be O-GlcNAcylated. In addition, several of the proteins involved in the translocation process are targets of signaling proteins downstream of the insulin receptor. In cell culture models of both glucose- and glucosamine-induced insulin-resistance a reduction in the acute insulin-stimulated GLUT4 translocation is associated with a significant alteration in membrane redistribution of vesicle proteins such as t-(target membrane) SNARE, v-(vesicle membrane) SNARE and Munc18c (mammalian uncoordinated). SNARE stands for soluble-N-ethylmaleimide-sensitive factor attachment protein receptor. Munc18c is a negative regulator of both t- and v-SNAREs. Munc18c is known to be a target for O-GlcNAcylation. These results suggest a direct involvement of excess HBP flux in desensitizing the fusion between GLUT4-containing intracellular vesicles and the plasma membrane.
In addition to GLUT4 translocation, insulin-mediated PI3K and PKB/AKT activation also stimulates glycogen synthesis. The net effect is to balance the level of glucose metabolism in response to excess glucose influx. Insulin-dependent glycogen synthesis is mediated via the activation of of glycogen synthase (GS). Like other downstream targets of the insulin receptor, GS regulation involves a PKB/AKT-mediated inhibition of GSK3β which normally phosphorylates and inhibits GS. The insulin-stimulated increase in glycogen synthesis decreases the pool of G6P and subsequently F6P, thereby restricting flux through the HBP. PKB/AKT activation also leads to reduced dephosphorylation of GS via protein phosphatase 1 (PP1). Exposing cells to either high glucose or glucosamine results in a reduction in insulin-stimulated GS activity. Additionally, GS is a known O-GlcNAcylated protein and as might be expected it has been shown that GS becomes more resistant to dephosphorylation by PP1 under conditions of excess HBP flux.
While increased global O-GlcNAc levels are implicated in the development of insulin resistance, OGT is also regulated by insulin in adipocyte cell cultures. OGT is tyrosine phosphorylated by the insulin receptor upon acute insulin stimulation and this phosphorylation increases the activity of the enzyme. In addition there is an observed shift in OGT localization from the nucleus to the cytosol in response to insulin stimulation. This OGT translocation to the plasma membrane is PI3K-dependant in response to acute insulin stimulation.
In summary, given that genetic and pharmacologic elevation in O-GlcNAc levels in cultured adipocytes and mouse models is associated with insulin-resistant phenotypes, it is likely that reducing O-GlcNAc levels in adipocytes should reverse the HBP-induced insulin resistance. A proof-of-concept experiment in transgenic mice (the insulin-resistant db/db mouse model which harbors a mutated leptin receptor) showed that over-expression of OGA, which reduces the level of O-GlcNAcyaltion, significantly improves whole-body glucose tolerance and insulin sensitivity. This result suggests that lowering O-GlcNAc levels in vivo should be of significant clinical beneficial.
O-GlcNAcylation and Glucose Homeostasis
Numerous lines of evidence, as discussed above, demonstrate that over-expression of the rate-limiting enzyme of the HBP, GFAT, leads to peripheral insulin resistance. In addition, over-expression of O-GlcNAc transferase (OGT) in skeletal muscle and adipose tissue results in elevated circulating insulin levels and insulin resistance. Insulin resistance is a major contributing factor to the hyperglycemia typical of type 2 diabetes. However, additional factors exacerbate the hyperglycemic consequences of insulin resistance such as aberrantly regulated hepatic gluconeogenesis.
Hyperglycemia is also associated with O-GlcNAcylation of transcription factors and cofactors such as FOXO1, FOXO3, CREB-regulated transcription coactivator 2 (CRTC2), and PGC-1α that are involved in the modulation of the expression of gluconeogenic genes. PGC-1α is a key transcriptional coactivator that regulates mitochondrial biogenesis as well as hepatic gluconeogenesis.
Increased levels of glucose can stimulate an increased level of glycolysis via O-GlcNAcylation. The initiation of glycolysis occurs when glucose is phosphorylated thereby activating it and trapping it within the cell. Humans express four isoforms of the enzymes that phosphorylate glucose identified as hexokinases, HK1, HK2, HK3, and HK4 (HK4 is most commonly termed glucokinase). With respect to HK1, the O-GlcNAcylation of the regulatory domain promotes the assembly of HK1 to the outer mitochondrial membrane. The synthesis of UDP-GlcNAc, the substrate of O-GlcNAc transferase (OGT), is enhanced by increasing levels of glucose which leads directly to an increase in the state of HK1 O-GlcNAcylation. Since O-GlcNAcylation of HK1 enhances association with the mitochondrial outer membrane, there is a direct correlation between glucose levels, increased glycolysis, and increased ATP production. The increased ATP production, coupled with HK1 association with the mitochondria, allows increased HK1 access to the ATP required for it to activate glucose for oxidation.
As indicated above, humans express a single OGT gene as well as a single OGA gene involved in the attachment and removal of O-GlcNAc from target proteins, respectively. Currently, even though hundreds of proteins and enzymes are known to be modified by O-GlcNAcylation, the details of how OGT and OGA achieve substrate specificity is largely unknown. It has been proposed that OGT recognizes numerous different substrates primarily though the tandem tetratricopeptide repeats (TPRs) present in the protein. Indeed, different OGT isoforms with various lengths in TPRs show different substrate specificities.
Recent evidence has shown that OGT regulates gluconeogenesis through O-GlcNAcylation of PGC-1α via interaction with host cell factor C1 (HCF1, also HCFC1). HCF1 is an essential transcriptional coactivator shown to be required for herpes virus gene expression, cell-cycle regulation, and stem cell growth. In addition to interacting with OGT, HCF1 is also highly O-GlcNAcylated. O-GlcNAcylation of PGC-1α stabilizes the transcriptional coactivator by inhibiting its ubiquitylation by recruiting the deubiquitylase BRCA1-associated protein 1 (BAP1). BAP1 encodes a nuclear ubiquitin carboxy-terminal hydrolase (UCH). Stabilization of PGC-1α thereby, results in enhanced gluconeogenesis.
The term, glucose effectiveness, describes the ability of high glucose, by itself, to suppress endogenous glucose production. This phenomenon has an important role in overall glucose homeostasis. One important pathway through which glucose determines its own production involves the regulation of OGT/HCF1 complex formation and subsequent O-GlcNAcylation and stabilization of PGC-1α resulting in enhanced gluconeogenesis. Compared to no glucose, low levels of glucose stimulate gluconeogenesis, whereas, hyperglycemia inhibits gluconeogenesis.
Under euglycemic conditions, OGT interaction with HCF1 leads to O-GlcNAcylation of PGC-1α and enhanced expression of gluconeogenic genes. Thus, the OGT/HCF-1/PGC-1α pathway is critical in maintaining normal glucose levels and mediating glucose effectiveness at the level of enhanced gluconeogenesis. Conversely, the hyperglycemia, typical of that seen in type 2 diabetes, leads to hyperactivation of the OGT/HCF-1/PGC-1α pathway which results in diminished glucose effectiveness. The potential for pharmacological inhibition of this pathway represents a potential novel strategy for treating the hyperglycemia associated with type 2 diabetes.
O-GlcNAcylation in the Epigenetic Regulation of Gene Expression
Due to the intermediates in the HBP originating from important nutritional components (e.g. glucose, amino acid, fatty acids), the process of O-GlcNAcylation is highly responsive to nutrient availability as well as to nutrient excess. Recent evidence has conclusively demonstrated that the synthesis of O-GlcNAc and the activities of the two enzymes responsible for the addition (O-GlcNAc transferase: OGT) and removal (OGA) of O-GlcNAc contribute to the maintenance of epigenetic states within the chromatin and to the etiology of epigenetic related disease states. The primary control of epigenesis is cytidine methylation (within the context of CpG dinucleotides) in DNA and histone modifications.
With respect to histone modification, all four histones present in the nucleosome have been shown to be O-GlcNAcylated with histone H2B being the most highly modified. At least six Ser residues and one Thr residue in H2B have been shown to be O-GlcNAcylated under various conditions. The O-GlcNAcylation of S112 in H2B is increased in response to DNA double-strand breaks. The significance of this modification to the normal cellular response to DNA damage has been demonstrated with either H2B mutants that contain an Ala residue at position 112 (S112A) or where the OGT gene has been downregulated. In both instances non-homologous end joining (NHEJ) and homologous repair processes are impaired. When H2A is O-GlcNAcylated on Thr 101 (T101) there is reduced dimerization with H2B which promotes an open chromatin structure leading to increased transcriptional activity.
A link between the energy/nutritional state and regulation of epigenetic modifications of histone proteins has been defined by the observations that the master metabolic regulatory kinase, AMPK, phosphorylates OGT on Thr 444 (T444) which alters the ability of OGT to O-GlcNAcylate histone H2B. When AMPK phosphorylates OGT there is a reduced level of O-GlcNAcylation of S112 in H2B leading to reduced levels of expression of genes that are normally activated by the presence of histone H2B S112 O-GlcNAcylation. Concomitant with the AMPK-mediated phosphorylation of OGT is an increase in the level of histone H3 acetylation on Lys 9 (K9).
During nutrient deprivation or energy limitation, AMPK phosphorylates histone H2B on S36 which is one of the sites O-GlcNAcylated by OGT. The phosphorylation of H2B on S36 is essential for the transcriptional response to changes in energy and nutrient content. The interplay between changing OGT activity in nutrient excess and AMPK activity during nutrient deprivation can be shown by the fact that OGT O-GlcNAcylates AMPK on the α-subunit and all three γ-subunits. The consequences of O-GlcNAcylation of AMPK is an increase in its activity indicating a regulatory feedback loop exist between these two important metabolic regulators.
Changes in epigenetic marks on DNA can also be affected by the level of OGT activity. As discussed in detail in the DNA: Chromatin Structure, Replication, DNA Damage Repair page, the level of methyl cytidine (5mC) in DNA results from the activities of DNA methyltransferases that add the methyl group to cytidine and the demethylases that remove the methylation marks. The cytidine demethylases of DNA are the TET (ten eleven translocation) enzymes. All three of the TET enzymes are extensively O-GlcNAcylated with TET2 and TET3 having been shown to have at least 20 different O-GlcNAcylation sites consisting of both Ser and Thr residues. The O-GlcNAcylation of TET proteins allows for these DNA demethylases to target OGT to chromatin, thereby, increasing the level of O-GlcNAcylation of chromatin-associated proteins.
Energy excess, as evidenced by hyperglycemia, has been correlated to increased DNA demethylation, in large part due to increased O-GlcNAcylation of TET1 and TET3. When TET3 is O-GlcNAcylated it is transported out of the nucleus resulting in reduced DNA demethylation. This effect of O-GlcNAcylation of TET3 is suspected to play a role in the hyperglycemia-induced changes in expression of the insulin gene in the β-cells of the pancreas. One of the products of TET activity on methylated CpG dinucleotides is 5-hydroxymethylcytidine, 5hmC. The presence of 5hmC at transcription start sites and on promoters of genes is correlated with increased transcriptional activity of these genes. Therefore, increased OGT activity can lead to changes in the distribution of 5mC epigenetic marks and consequently changes in the level of gene expression.
Lysosomal Targeting of Enzymes
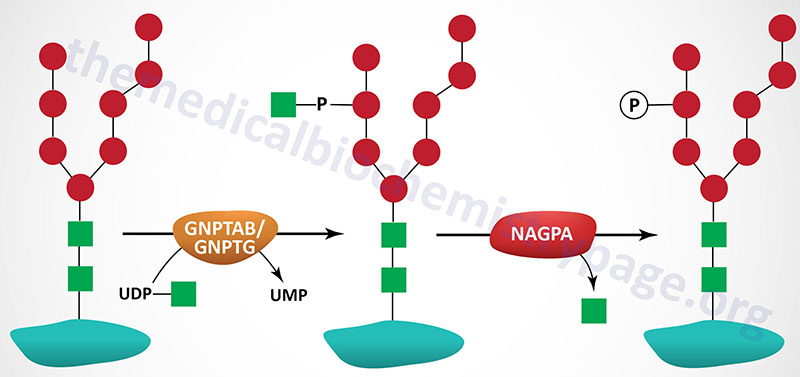
The majority of enzymes that are destined for the lysosomes (lysosomal enzymes) are directed there by a specific carbohydrate modification. During transit through the Golgi apparatus a residue of GlcNAc-1-phosphate (GlcNAc-1-P) is added to the carbon-6 hydroxyl group of one or more specific mannose residues that have been added to these enzymes.
The GlcNAc is activated by coupling to UDP and is transferred by UDP-GlcNAc:lysosomal enzyme GlcNAc-1-phosphotransferase complex (commonly called GlcNAc-phosphotransferase), yielding a phosphodiester intermediate: GlcNAc-1-P-6-Man-protein. The GlcNAc-phosphotransferase complex is a heterohexameric complex composed of two α-, two β-, and two γ-subunits.
The α- and β-subunits are both derived from the same gene following proteolysis of the encoded protein. This gene is identified as N-acetylglucosamine-1-phosphate transferase alpha and beta subunits (GNPTAB). The GNPTAB gene is located on chromosome 12q23.2 and is composed of 23 exons that encode a 1256 amino acid precursor protein. Processing of the precursor protein results in the generation of the α- and β-subunits.
Mutations in the GNPTAB gene result in the lethal lysosomal storage disease identified as I-cell disease (mucolipidosis II; also known as mucolipidosis II alpha/beta) and the less severe related disorder, pseudo-Hurler polydystrophy (mucolipidosis IIIA; also known as mucolipidosis III alpha/beta). More than 70 mutations in the GNPTAB gene have been found in I-cell disease patients with the most common (40%) being a nonsense mutation: R1189X.
The γ-subunits are encoded by the N-acetylglucosamine-1-phosphate transferase gamma subunit (GNPTG) gene. The GNPTG gene is located on chromosome 16p13.3 and is composed of 11 exons that encode a 305 amino acid precursor protein. Mutations in the GNPTG gene are associated with a disease referred to as mucolipidosis IIIC (also called mucolipidosis III gamma).
A second reaction, catalyzed by N-acetylglucosamine-1-phosphodiester α-N-acetylglucosaminidase (encoded by the NAGPA gene), removes the GlcNAc leaving mannose residues phosphorylated in the 6 position, identified as Man-6-P-protein. The NAGPA gene is also located on chromosome 16p13.3 and is composed of 11 exons that encode a 515 amino acid precursor protein.
The protein encoded by the NAGPA gene is often referred to as uncovering enzyme (UCE). The preproprotein encoded by the NAGPA gene is processed into a functional homotetrameric complex composed of two disulfide bonded homodimers. A specific mannose-6-phosphate receptor (MPR) is present in the membranes of the Golgi apparatus and binding of Man-6-P to this receptor targets proteins to the lysosomes.
Two distinct MPRs have been identified and both are members of the P-type lectin family. Both are type I integral membrane glycoproteins that contain an N-terminal extracellular domain, a single transmembrane domain and a C-terminal cytoplasmic domain. One receptor is large with a molecular weight of approximately 300kDa, the other receptor is smaller with a molecular weight of approximately 46kDa. Structural similarities between these two receptors indicates they are derived from a single ancestral gene with the larger receptor arising through multiple gene duplications. The extracellular portion of the larger receptor contains 15 repeating elements, each of which is highly similar to the extracellular domain of the smaller receptor. Both receptors exist as dimers embedded in the membrane.
The large receptor binds two moles of Man-6-P and the smaller binds one mole of Man-6-P per subunit, thus 4 and 2 moles of Man-6-P per dimer, respectively. The bovine and murine versions of the smaller receptors require divalent cations for ligand binding and thus the receptor has been termed the cation-dependent Man-6-P receptor (CD-MPR). However, the human counterpart may not require cations for its activity. The CD-MPR protein is encoded by the M6PR (mannose-6-phosphate receptor, cation dependent) gene.
The larger receptor does not require divalent cations for ligand binding and is therefore, commonly referred to as the cation-independent Man-6-P receptor (CI-MPR). However, the CI-MPR has been shown to bind the non-glycosylated polypeptide hormone, insulin-like growth factor 2 (IGF-2) and as such the larger MPR is more frequently identified as the IGF-2 receptor (encoded by the IGF2R gene). The IGF-2 receptor is available at the cell surface and its role in binding IGF-2 is to target this hormone for degradation in the lysosomes. In addition to IGF-2, the IGF-2 receptor has been shown to bind a diverse array of Man-6-P-containing proteins as well as several non-glycosylated proteins. Although the IGF-2 receptor and CD-MPR exhibit distinct activities, both receptors function to target newly synthesized lysosomal enzymes to the lysosomes.
Glycosylphosphatidylinositol Anchored Proteins (GPI-Linkage)
Many membrane-associated glycoproteins belong to neither the peripheral nor the transmembrane class. These glycoproteins are tethered to the outer leaflet of the plasma membrane via a glycosylphosphatidylinositol linkage to their C-termini. This type of membrane attachment is referred to as a GPI linkage and the glycoproteins are termed glypiated proteins. At least 150 human proteins are known to be anchored to the plasma membrane via the GPI linkage. The basic structure of the GPI linkage is shown in the Figure below. This class of membrane anchored protein was initially discovered due to their release from cells following treatment with crude bacterial phospholipase C (PLC).
The first mammalian glypiated proteins characterized were alkaline phosphatase and 5′-nucleotidase. Two clinically important glypiated proteins are the erythrocyte surface glycoproteins, decay-accelerating factor, (DAF; also known as CD55: cluster of differentiation protein 55) and CD59. Both DAF and CD59 prevent erythrocyte lysis by the complement system of immune surveillance involved in responses to pathogen invasion. Other important GPI linked proteins include the ferroxidase, ceruloplasmin, the acetylcholinesterases, the cell adhesion molecule N-CAM-120 (neural cell adhesion molecule-120), and the T-cell markers Thy-1 and LFA-3 (lymphocyte function associated antigen-3).
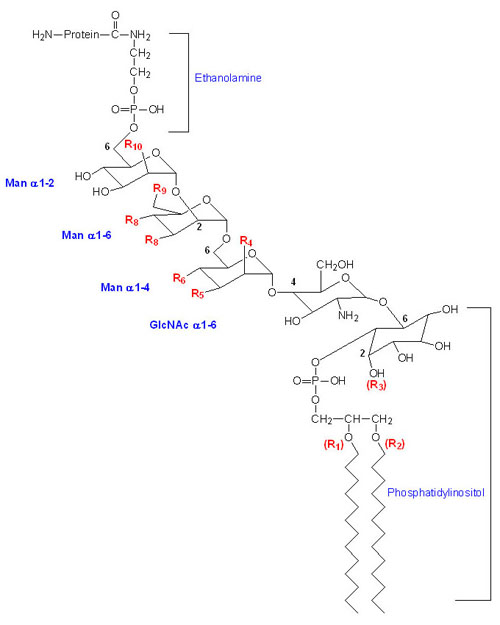
Virtually all protein-linked GPI anchors share the common core structure that is depicted in the Figure above. The final structures of GPI anchors are quite diverse, depending on both the protein to which they are attached and the organism in which they are synthesized. The use of the GPI-anchor has been found in a functionally diverse array of mammalian proteins that includes hydrolytic enzymes, adhesion molecules, complement system regulatory proteins, and receptors. In several mammalian mRNAs that encode GPI-anchored proteins, the process of alternative splicing results in the expression of transmembrane and/or soluble forms of the same gene product. In addition, some of these variants are developmentally regulated. For example, neural cell adhesion molecule (NCAM) exists in GPI-anchored and soluble forms when expressed in muscle and in GPI-anchored and transmembrane forms when expressed in brain. Another important example of tissue-specific differences is the ferroxidase, ceruloplasmin. The liver synthesizes ceruloplasmin as a secreted soluble plasma protein whereas in the brain and numerous other tissue ceruloplasmin is attached to the cell via a GPI anchor.
The biosynthesis of GPI anchors occurs via a complex series of reactions involving numerous enzymes. The majority of the enzymes involved in the synthesis of the GPI anchor are referred to as phosphatidylinositol glycan (PIG) anchor biosynthesis class enzymes. A total of 21 PIG genes encode the enzymes of GPI anchor synthesis. Initially there is the pre-assembly of a GPI anchor precursor in the ER membrane with synthesis beginning on the cytosolic face of the membrane followed by transfer to the luminal side for completion prior to protein attachment. When completed, the attachment of the GPI anchor to the newly synthesized protein takes place within the lumen of the ER and is catalyzed by the enzyme complex called GPI transamidase.
The GPI transamidase complex is composed of at least five subunits. These proteins are encoded by the GPAA1 (glycosylphosphatidylinositol anchor attachment 1), PIGK, PIGS, PIGT, and PIGU genes. The protein encoded by the PIGK gene is the enzymatic component of the complex that attaches the GPI anchor to a target protein and cleaves the GPI attachment signal peptide. Finally the GPI-anchor undergoes lipid remodeling and/or carbohydrate side-chain modifications in the ER and the Golgi. Some GPI-anchored proteins possess two fatty acid chains (e.g. diacylglycerol, alkylacylglycerol, alkenylacylglycerol, or ceramide) which allows for a stable association of the protein with the lipid bilayer. The process of GPI anchor remodeling following attachment to a target protein is carried out by at least four enzymes encoded by post GPI attachment protein (PGAP) genes.
GPI-anchored proteins play significant roles in the function of the immune system. For example, transmembrane signaling is known to occur as a result of the cross-linking of GPI-anchored proteins with antibody followed by clustering with a second antibody on various cells such as leukocytes. The types of intracellular responses initiated by GPI protein-induced signaling include tyrosine phosphorylation, cytokine production, oxidative burst, increased intracellular Ca2+ release and cell proliferation.
Glycosylphosphatidylinositol specific phospholipase D1: GPLD1
The protein components of GPI-anchored proteins can be released to the circulation where they can exert numerous effects that includes cell signaling, cell-cell and host-pathogen interactions, immunorecognition, and complement regulation. Removal of the protein component is catalyzed by the GPLD1 encoded enzyme, glycosylphosphatidylinositol specific phospholipase D1. Expression of the GPLD1 gene is highest in the liver and the release of this enzyme from the liver classifies the protein in the family of hepatokines, secreted substances that act locally or at a distance but that are not defined by the classic definition of a hormone. In addition, GPLD1 secretion increases in response to exercise indicating that it also functions as an exerkine.
The GPLD1 gene is located on chromosome 6p22.3 and is composed of 27 exons that encode a precursor protein of 840 amino acids. Although expression is highest in the liver, the GPLD1 gene is also expressed in the adipose tissue, brain, kidney, skeletal muscle, testis, heart, lung, and pancreas.
The function of the GPLD1 encoded enzyme is to catalyze the removal of GPI anchored proteins from their anchor by hydrolyzing the inositol phosphate bonds in the GPI anchor. Removal of the protein allows for it to circulate in the blood. In addition to functioning at the plasma membrane, GPLD1 can cleave GPI-anchored intermediates within cells in the endoplasmic reticulum, the Golgi apparatus, and likely the lysosomes as well.
Several proteins have been identified as targets for the action of GPLD1 including tissue-nonspecific alkaline phosphatase (TNAP), DAF (decay accelerating factor; encoded by the CD55 gene), the Thy-1 glycoprotein, and the glycoproteins Glypican 4 (encoded by the GPC4 gene), and bFGF [within the heparan sulfate proteoglycan (HSPG) complex].
TNAP was identified as a GPI-anchored protein on endothelial cells of capillaries at the blood brain barrier (BBB) whose levels increased with aging. Exercise-mediated increases in liver release of GPLD1 lead to increased release of TNAP from brain capillary endothelial cells without the enzyme having to enter the brain. The consequences of this action of GPLD1 are enhanced remodeling of the brain vasculature, which in turn is associated with fortification of the integrity of the BBB. Several studies have demonstrated that exercise contributes to reduced levels of aging in the brain and enhanced cognitive capacity. These effects are most likely to be the result of GPLD1 action at the BBB.
Clinical Significances of Glycoproteins
Glycoproteins on cell surfaces are important for communication between cells, for maintaining cell structure and for self-recognition by the immune system. The alteration of cell-surface glycoproteins can, therefore, produce profound physiological effects, of which several are listed below.
1. The ABO blood group antigens are the carbohydrate moieties of glycolipids on the surface of cells as well as the carbohydrate portion of serum glycoproteins. When present on the surface of cells the ABO carbohydrates are linked to sphingolipid and are therefore of the glycosphingolipid class. When the ABO carbohydrates are associated with protein in the form of glycoproteins they are found in the serum and are referred to as the secreted forms. Some individuals produce the glycoprotein forms of the ABO antigens while others do not. This property distinguishes secretors from non-secretors, a property that has forensic importance such as in cases of rape.
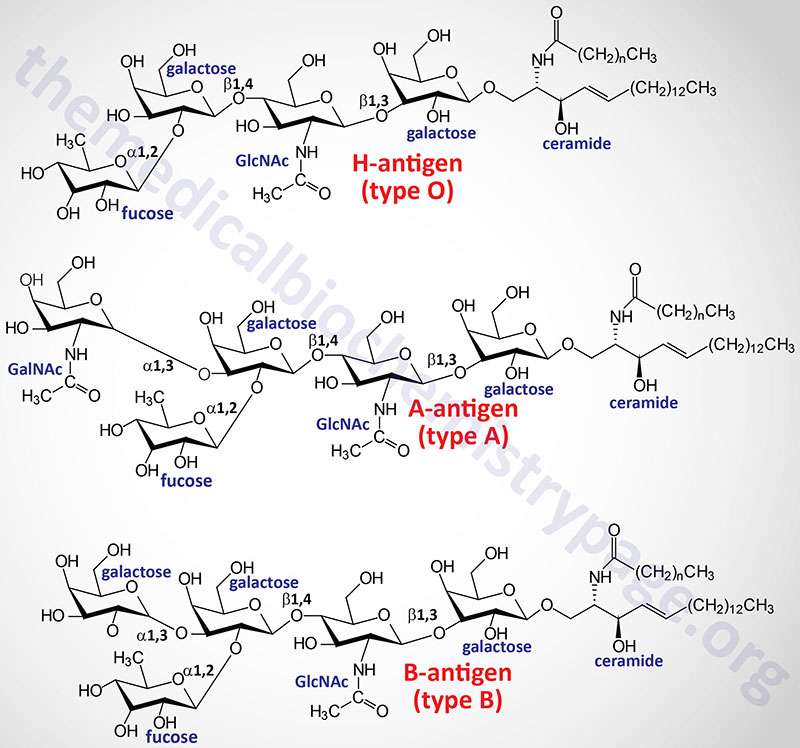
2. The truncation of erythrocyte surface glycoproteins leads to cell clumping, as in congenital dyserythropoietic anemia type II. Also referred to as HEMPAS (hereditary erythroblastic multinuclearity with positive acidified-serum test).
3. Several viruses, bacteria and parasites have exploited the presence of cell-surface carbohydrates, principally associated with protein (glycoproteins), using them as portals of entry into the cell.
- Human immunodeficiency virus (HIV), the causative agent of AIDS, gains entry into cells of the immune system by attaching to a class of cellular receptors known as the chemokine receptors, most notably CXCR4 and CCR5.
- Members of the poxvirus family of viruses gain entry into cells, most frequently migratory leukocytes, by attaching to chemokine receptors including CCR1, CCR5 and CXCR4.
- Dystroglycan (DG) is a component of the dystrophin-glycoprotein complex. It is a laminin receptor encoded by a single gene and cleaved by post-translational processing into two proteins, peripheral membrane α-DG and transmembrane β-DG. α-DG interacts with laminin-2 in the basal lamina and β-DG binds to dystrophin containing cytoskeletal proteins in muscle and peripheral nerves. DG is involved in agrin- and laminin-induced acetylcholine receptor clustering at neuromuscular junctions, morphogenesis, early development, and the pathogenesis of muscular dystrophies. Evidence has shown that α-DG present on Schwann cell membranes is the receptor for Mycobacterium leprae and also serves as the receptor for the arenavirus class of pathogens. Arenaviruses cause hemorrhagic fever in humans. Lymphocytic choriomeningitis virus (LCMV), Lassa fever virus (LFV), Oliveros and Mobala (all members of the arenavirus family) all bind to α-DG. The specificity of this interaction was demonstrated by the resistance to LCMV infection of cells harboring a null mutation in DG.
- Rhinoviruses utilize attachment to ICAM-1 (inter-cellular adhesion molecule-1) to gain entry into cells.
- The pathogenic human parvovirus, B19, attaches to the erythrocyte-specific cell-surface globoside identified as erythrocyte P antigen to infect erythrocytes.
- The malarial parasite Plasmodium vivax, binds to the erythrocyte chemokine receptor known as the Duffy blood group antigen (also known as the erythrocyte receptor for interleukin-8) to infect erythrocytes.
- The MNS blood group system is a well-characterized set of erythrocyte surface antigens that represent the variable carbohydrate modifications of the trans-membrane glycoprotein, glycophorin. Glycophorin is the cellular receptor for influenza virus as well as the receptor for erythrocyte invasion by the malarial parasite Plasmodium falciparum.
- Helicobacter pylori is the bacterium responsible for chronic active gastritis and gastric and duodenal ulcers; it is also the causative agent for one of the most common forms of cancer in humans, adenocarcinoma. This bacterium attaches to the Lewis blood group antigen on the surfaces of gastric mucous cells.
- Rabies virus binds to cells through interactions with neural cell adhesion molecule (N-CAM).
- Human herpesvirus 6 (HHV-6) infection occurs in virtually all persons within the first 2 years of life and persists the entire lifetime. In immunocompromised patients HHV-6 causes opportunistic infections and is the causative agent of exanthema subitum. HHV-6 has been linked to multiple sclerosis and to the progression of AIDS. The cellular receptor for HHV-6 is the cell-surface type-I glycoprotein, CD46.
4. Defects in the proper targeting of glycoproteins to the lysosomes can also lead to clinical complications. Deficiencies in the enzyme responsible for the transfer of GlcNAc-1-P to Man residues (GlcNAc phosphotransferase) in lysosomal enzymes leads to the formation of dense inclusion bodies formation in the fibroblasts. Two disorders related to deficiencies in the targeting of lysosomal enzymes are termed I-cell disease (mucolipidosis II) and pseudo-Hurler polydystrophy (mucolipidosis III, also called mucolipidosis-HI). I-cell disease is characterized by severe psychomotor impairment, skeletal abnormalities, coarse facial features, painful restricted joint movement, and early mortality. Pseudo-Hurler polydystrophy is less severe; it progresses more slowly, and afflicted individuals live to adulthood.
5. Paroxysmal nocturnal hemoglobinuria (PNH) is an acquired (due to somatic mutations in bone marrow cells) hematopoietic disorder where the cells lack GPI-anchored proteins. This disorder is characterized by intravascular hemolysis of erythrocytes by their own complement system proteins. This phenotype is principally due to the lack of the complement inhibitor proteins CD55 and CD59, which are GPI-anchored, on the surface of erythrocytes. PNH has been shown to be the result of somatic mutations in the PIGA gene whose encoded protein catalyzes the first reaction in the synthesis of the GPI anchor. The PIGA gene is located on the X chromosome (Xp22.2). Due to X chromosome inactivation, females harbor only one functional copy of the PIGA gene which explains the equal distribution of PNH in males and females.
Clinical Significance of Defective Glycoprotein Degradation
The proper degradation of glycoproteins has important medical relevance. Degradation occurs within lysosomes and requires specific lysosomal hydrolases, termed glycosidases. Exoglycosidases remove sugars sequentially from the non-reducing end and exhibit restricted substrate specificities. In contrast, endoglycosidases cleave carbohydrate linkages from within and exhibit broader substrate specificities.
Several inherited disorders involving the abnormal storage of glycoprotein degradation products have been identified in humans. These disorders result from defects in the genes encoding specific glycosidases, leading to incomplete degradation and subsequent over-accumulation of partially degraded glycoproteins. As a general class, such disorders are known as lysosomal storage diseases (lysosomal storage disorders).
Numerous proteins and sphingolipids harbor similar carbohydrate modifications. The enzymes that remove these sugar residues are the same for both glycoproteins and glycolipids and as such there is often overlapping phenotypes in diseases that were originally identified as being caused by defects in glycoprotein degradation or glycolipid degradation.
The following Figure shows the locations of the actions of several glycosidases involved in glycoprotein metabolism for a typical complex-type glycoprotein modification. The structures of the carbohydrates in a typical complex oligosaccharide cluster are included (see Figure above of the three major classes of glycoprotein). The bonds are indicated for each linkage by the solid line between the representative symbols. Enzyme names are in green and diseases associated with defects in the indicated enzymes are in blue. Note that as indicated in the above paragraph, many lysosomal storage disorders (e.g. Tay-Sachs) resulting from defective enzymes that metabolize both glycolipids and glycoproteins are defined by one or the other defective pathway (see the Sphingolipid Metabolism and the Ceramides page for more information).
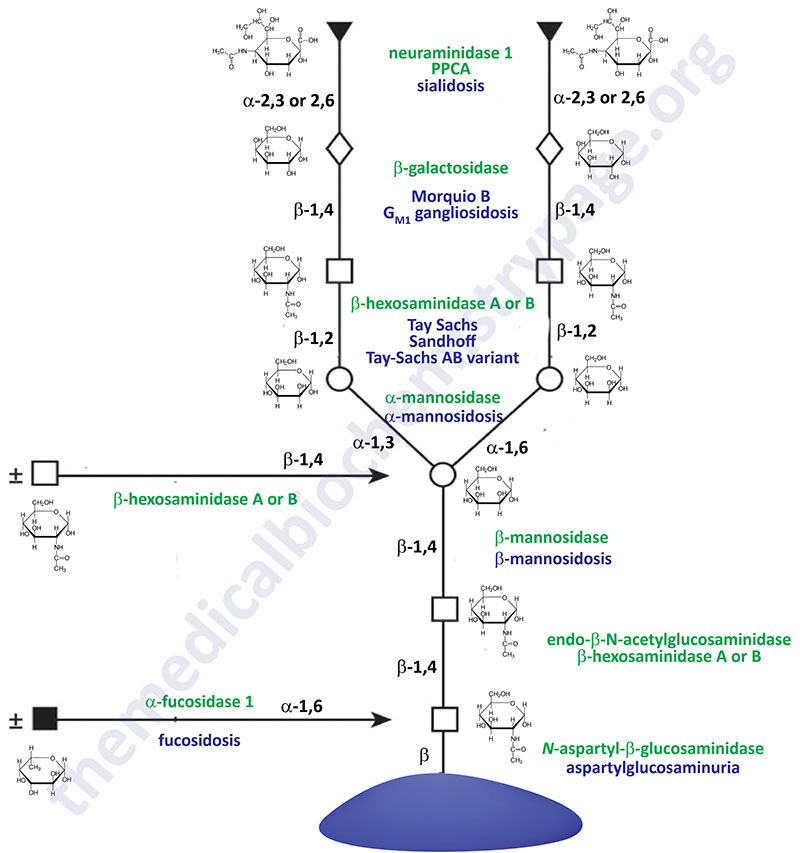
Table of Diseases Resulting from Defects in Glycoprotein Degradation
| Disease | Enzyme Deficiency | Symptoms/Comments |
| Aspartylglucosaminuria | aspartylglucosaminidase (N-aspartyl-β-glucosaminidase) | progressive intellectual impairment, delayed speech and motor development, coarse facial features |
| β-Mannosidosis | β-mannosidase | the defective β-mannosidase associated with this disease is encoded by the mannosidase beta gene (MANBA); primarily neurological defects, speech impairment |
| α-Mannosidosis | α-mannosidase | the defective α-mannosidase associated with this disease is encoded by the mannosidase alpha class 2B, member 1 gene (MAN2B1); symptoms include intellectual impairment, dystosis multiplex, hepatosplenomegaly, hearing loss, delayed speech |
| GM1 Gangliosidosis | β-galactosidase (galactosidase beta 1) | also identified as a glycosphingolipid storage disease or lysosomal storage disease |
| Sandhoff disease | β-hexosaminidases A and B | also identified as a glycosphingolipid storage disease or lysosomal storage disease |
| Sialidosis (also identified as Mucolipidosis I) | neuraminidase 1 (sialidase) | sialidase is lysosome-localized hydrolase; type 1 sialidosis is also known as cherry-red spot myoclonus syndrome; type 2 is also identified as mucolipidosis I (ML-I); neuraminidase 1 requires the accessory protein: protective protein/cathepsin A (PPCA) which is encoded by the CTSA gene; type I associated with myoclonus, gait disturbances, reduced visual acuity with cherry-red macula; type II is associated hydrops fetalis, hepatosplenomegaly, dysostosis multiplex, coarse facial features, myoclonus, ataxia, tremor and angiokeratoma |
| Fucosidosis | α-fucosidase 1 | two primary types: type 1 and type 2; type 1 is more severe and manifest in infancy; progressive motor and mental deterioration, growth impairment, coarse facial features, recurrent sinus and pulmonary infections |
Carbohydrate Recognition: Lectins
The ability of certain proteins to recognize and bind to specific carbohydrate structures is of critical importance in overall cellular homeostasis. Proteins that recognize specific types of carbohydrates displayed on other proteins and/or lipids are referred to as lectins. All lectins contain a carbohydrate recognition domain, CRD. The lectin family of proteins does not include the immunoglobulins which by themselves constitute a specialized class of carbohydrate recognizing proteins. The clinical laboratory definition of lectins describes non-immunoglobulins that are capable of differential agglutination (Latin for “to glue to”, means clumping) of erythrocytes.
Table of Lectin Families and their Carbohydrate Specificities
| Lectin Family | Characteristics | Binding Specificity |
| C-type (7 subfamilies) | require Ca2+ for activity | variable |
| Collectins (C-type subfamily III) | C-type CRD with collagen-like domains | variable, mannose |
| Galectins (S-type lectins) | sulfydryl-dependent or β-galactosidase binding | strict for β-galactosides |
| Siglecs (I-type lectins) | contain immunoglobulin-like domains | sialic acid |
| Phosphomannosyl receptors (P-type lectins) | recognize mannose-6-phosphate | mannose-6-phosphate |
| Pentraxins | quaternary structure composed of 5 identical polypeptides that form a ring with a central hole) | variable and can be non-carbohydrate |
| Calnexin and calreticulin | recognition of properly folded glycoproteins in the ER | glucose |
| Ficolins | contain fibrinogen-like homology domain | variable |
| Tachylectins | distinct CRD but also contain fibrinogen-like domain | GlcNAc/GalNAc |
C-type Lectins
C-type lectins were initially defined as those proteins having a CRD of approximately 120 amino acids with a dependence on Ca2+ ions for binding activity. The C-type lectin family can be further subdivided into at least 7 subfamilies dependent upon the nature of other non-carbohydrate recognition domains as well as overall gene structure.
C-type lectins recognize carbohydrate structures on many different forms of protein, including cell surface proteins and extracellular matrix (ECM) proteins and other ECM structures containing carbohydrate modifications such as proteoglycans, glycosaminoglycans and glycolipids. C-type lectins also exhibit broad specificity in the types of carbohydrates to which they bind including fucose, galactose, glucose, GlcNAc, maltose, mannose and N-acetylmannosamine (ManNAc). One of the most important functions for carbohydrate recognition by C-type lectins is the role that function plays in pathogen recognition by the innate immune system.
Collectins
The collectins are subtype III C-type lectins defined by having an additional collagen-like domain. The presence of the collagen-like domain allows the collectins to form a triple helical structure that creates a multivalent CRD. The major function of the collectins is recognition of carbohydrate structures on microbial surfaces leading to activation of the complement cascade and phagocytosis.
Mannan-binding lectin, MBL (also termed mannose-binding protein) is the best characterized human collectin. Although termed mannose-binding lectin, MBL actually exhibits higher affinity for GlcNAc than for mannose. MBL is synthesized by the liver and secreted into the circulation and is considered one of the acute phase proteins synthesized by the liver. Expression levels of MBL rise in response to inflammatory signals. Upon binding to carbohydrate structures on microbial surfaces, MBL activates the complement system. The mechanism of MBL activation of complement requires the interaction of MBL with a family of serine-proteases termed mannose-binding lectin-associated serine proteases (MASPs). The action of MBL-MASP in the lectin-activated complement cascade is similar to the C1q-antibody complex that activates the serine proteases C1r and C1s.
Selectins
The selectins are subtype IV C-type lectins defined the presence of the C-type lectin CRD, an epidermal growth factor-like domain and a variable number of complement regulatory protein-like repeat domains. The selectins are also members of the cell adhesion family of proteins. Each selectin is tethered to the plasma membrane via a single transmembrane domain.
A major function of the selectins is the recruitment of neutrophils to sites of inflammation. There are three characterized selectins that were originally identified as the adhesion molecules: endothelial-leukocyte adhesion molecule-1 (ELAM-1), murine lymph node homing receptor and platelet granule membrane protein-140. These three proteins are now referred to as E-selectin, L-selectin and P-selectin, respectively.
E-selectin is also synthesized by endothelial cells but is not constitutively present. Expression of E-selectin is regulated by cytokines such as IL-1 and TNF-α.
L-selectin is expressed by most leukocytes and plays a role in the recruitment of these cell types into lymphatic tissue. Constitutively expressed L-selectin is rapidly released from the surface of activated leukocytes. Activation of L-selectin signaling, via ligand-induced cross-linking, results in potentiation of the responses of neutrophils to other stimuli.
P-selectin is constitutively stored in secretory granules of platelets and endothelial cells. When stimulated by the appropriate response (e.g. IL-1 and TNF-α) these cells present P-selectin to the cell surface. Through this mechanism, P-selectin is involved in mediating very early responses of leukocytes to inflammatory signals.
S-type Lectins: Galectins
The S-type lectins were originally so designated because the original family members exhibited a sulfhydryl-dependence on carbohydrate binding. The S-type lectins are now commonly referred to as the galectins. The galectins are all defined by a canonical CRD having an affinity for β-galactosides. To date, 14 galectins have been identified in mammals.
The galectins can be further divided into 3 subfamilies based upon their structures. The prototype subfamily (galectin-1, -2, -5, -7, -10, -11, -13, and -14) is comprised of a single CRD; the tandem-repeat type family (galectin-4, -6, -8, -9, and -12) contains two homologous CRDs and the chimeric type family contain a single CRD combined with a unique amino or carboxy terminus. Due to their multivalent nature, galectins are capable of cross-linking glycoconjugates containing N-acetyllactosamine (LacNAc) structures. Galectin activities are observed both at the cell surface and intracellularly.
One of the most crucial functions of the galectins is in the regulation of inflammatory responses. Galectin-1 is expressed by antigen-stimulated T cells, activated B cells and by macrophages. The action of galectin-1 in these systems is to stimulate apoptosis resulting in cell death. Negative growth effects of galectin-1 are also seen in breast and prostate cancer cell lines.
In addition to galectin-1, galectin-7, -8, -9, and -12 have been shown to exhibit pro-apoptotic activity. In contrast, galectin-3 exhibits anti-apoptotic activities in similar types of cells. Whereas the proapoptotic effects of galectin-1 can be observed by adding the protein to activated T cells, the anti-apoptotic effects of galectin-3 appears to be the result of intracellular activities associated with the protein. Interestingly, galectin-3 has some striking sequence similarities to the anti-apoptotic Bcl-2 protein family. Galectin-1 is a potent inhibitor of activated T cell-mediated inflammatory responses.
Using experimentally induced models of several autoimmune disorders (e.g. myasthenia gravis, multiple sclerosis and rheumatoid arthritis) it has been shown that administration of recombinant galectin-1 can significantly ameliorate the resultant clinical symptoms. Conversely, the presence of autoantibodies to galectin-1 has been identified in several neurodegenerative disorders. Likewise, the levels of autoantibodies to galectin-3 are inversely associated with severity of Crohn disease (inflammatory bowel disease, IBD).
I-type Lectins: Siglecs
I-type lectin is the term collectively given to the family of carbohydrate binding proteins that belong to the immunoglobulin (Ig) superfamily of proteins. The Ig superfamily of proteins all have at least one domain composed of two β-sheets that are disulfide bonded and stacked together. There are at least three subtypes of Ig domain (V-set, C1-set and C2-set) that are defined by the number and arrangement of the β-strands in the domain. All of the I-type lectins are membrane anchored with their CRDs on the outside of the cell.
The I-type lectins are divided into two main subfamilies. One subfamily includes the cell adhesion molecules, L1, N-CAM and intercellular adhesion molecule-1 (ICAM-1) as well as the cell-surface glycoproteins CD83 and CD2. The other subfamily is composed of the siglecs which are so named because of their binding specificity for sialic acid residues. Siglecs are also characterized by the presence of a characteristic V-set Ig domain at their amino termini followed by varying numbers of C2-set domains.
The siglecs are primarily expressed on cells of the hematopoietic system. A macrophage lectin-like adhesion molecule termed sialoadhesin and CD22, another member of the Ig superfamily that shows restricted B-cell expression were the first two members of the siglec group to be characterized. Both sialoadhesin and CD22 were shown to mediate cell-cell interactions through their abilities to recognize sialylated glycoconjugates. Sialoadhesin is now referred to as siglec-1 and CD22 as siglec-2 in reference to their being the first and second member characterized, respectively.
To date, at least 11 siglecs have been characterized. Like the pentraxins, the siglecs, principally the siglec-3 family, are involved in regulatory aspects of innate immunity reactions. All of the siglec-3 family members contain a motif in their intracellular tails termed an immunoreceptor tyrosine-based inhibitory motif (ITIM) indicating these proteins act as inhibitory receptors on hematopoietic cells. Associated with the ITIM is a tyrosine residue that becomes phosphorylated in response to activation of activating receptors. The function of the ITIM is to recruit phosphatases to activated receptor complexes, thereby, deactivating the activating receptors by phosphate removal. The presence of phosphotyrosine in the cytoplasmic tail of siglec-3 also enhances the subsequent interaction with sialylated ligands suggesting that tyrosine phosphorylation plays a regulatory role in ligand binding to siglec-3.
Pentraxins
The pentraxin family of lectins is defined by having primary sequence similarities to major acute phase reactants, C-reactive protein (CRP) and serum amyloid P component (SAP). The pentraxins assume a characteristic multisubunit complex consisting of five to ten identical subunits that form a pentameric ring structure with a central open core, hence the term pentraxin.
The original identity applied to CRP stems from the observation that it was an antibody-like molecule associated with the C polysaccharide component of the pneumococcal bacterium. CRP was originally shown to be a complement-activating precipitin with a high affinity for phosphocholine. Although originally not identified as a carbohydrate-binding protein, CRP was found to exhibit affinity for galactans (galactose-containing oligosaccharides) and phosphogalactose.
Similarly, SAP not only has carbohydrate-binding specificity it also has an affinity for phosphoethanolamine. In general the pentraxins represent a class of molecules that exhibit calcium-dependent binding to a number of substances bearing phosphate esters and carbohydrates. Classes of pentraxins have been divided into those with binding similarities to CRP (i.e. preference for phosphocholines) or SAP (i.e. preference for phosphoethanolamines).
Another defining feature associated with pentraxins is the observation that their expression is markedly increased as a consequence of the activities of immunomodulatory molecules such as IL-1 and TNF-α as well as in response to tissue injury and necrosis. Both IL-1 and IL-6 can dramatically induce the acute phase response of the liver leading to increased CRP synthesis and secretion. A role for the pentraxins in innate immunity functions is suggested by their reactivities with the complement system as well as phagocytic leukocytes. CRP is consistently shown to activate the classic complement pathway and binds to the site of complement-induced cell injury.
Congenital Disorders of Glycosylation (CDG)
Congenital disorders of glycosylation (CDG) represent a constellation of diseases that result from defects in the N-linked glycosylation pathway. These diseases are clustered into two broad categories. Group I CDG diseases are defined by alterations/deficiencies in the synthesis and/or transfer of the dolichol-pyrophosphate oligosaccharide precursor to Asn residues in substrate proteins. Group II CDG diseases are defined as those that result from defects in subsequent N-linked glycan processing. It is important to note that these disorders are only reflective of deficiencies of N-glycosylation and that diseases/disorders are known to result from deficiencies in O-glycosylation, GPI-linkage and the biosynthesis of proteoglycans, all of which involve carbohydrate addition and remodeling in the context of a protein backbone. For more information on both N-linked and O-linked glycosylation defects see the Congenital Disorders of Glycosylation page.
CDG-Ia is the most commonly occurring CDG, with appearance in individuals of European ancestry being highest. Although there is considerable variability in the clinical phenotypes observed in CDG-Ia patients, there is always some level of psychomotor impairment. In addition, children are ataxic and have skeletal abnormalities consisting of long limbs and short torsos. Due to defective synthesis of coagulation factors by the liver (primarily factor XI, antithrombin III, protein C and protein S), patients have severe coagulation defects. Adding to the situation is hepatomegaly with consequent liver dysfunction. CDG-Ia results from mutations in phosphomannomutase 2 (PMM2), the enzyme that is required to convert Man-6-P to Man-1-P used in the generation of GDP-Man. Over 60 mutations in PMM2 have been identified that either decrease enzyme activity or stability.
CDG-IIc is more commonly referred to as leukocyte adhesion deficiency syndrome II (LAD II). LAD II belongs to the class of disorders referred to as primary immunodeficiency syndromes as the symptoms of the disease manifest due to defects in leukocyte function. Symptoms of LAD II are characterized by unique facial features, recurrent infections, persistent leukocytosis, defective neutrophil chemotaxis and severe growth and intellectual impairment. The genetic defect resulting in LAD II is in the pathway of fucose utilization leading to loss of fucosylated glycans on the cell surface.
An additional feature of LAD II is that individuals harbor the rare Bombay (hh) blood type at the ABO locus as well as lack the Lewis blood group antigens. The Bombay blood type is characterized by a deficiency in the H (referred to as the O-type), A and B antigens due to loss of the fucose residue. Each of these blood group antigens contains a Fuc-α-1,2-Gal modification that is the final carbohydrate addition to these antigens. These fucosylation reactions are catalyzed by α-1,2-fucosyltransferase which is encoded by the H and Se loci. The defective neutrophil chemotaxis is due to the loss of a selectin ligand on these cells. This ligand is the sialylated Lewisx antigen, another blood group antigen.
The recurrent infections seen in LAD II patients are the result of the defective neutrophil function. Neutrophils are involved in innate immunity responses to bacterial infection. To carry out their role in host defense mechanisms, neutrophils must adhere to the surface of the endothelium at the site of inflammation which is an event mediated by cell surface adhesion molecules. The selectin family (E-, L-, and P-selectins) of animal lectins are necessary to mediate the initial process of neutrophil adherence to the endothelium. The selectins recognize sialylated fucosylated lactosamines typified by the Lewisx antigen. Once neutrophils adhere and roll along the surface of the endothelium (due to vascular flow), the integrin family of adhesion molecules allow for firm adherence followed by tissue penetration. A related disorder, termed LAD I, is caused by the absence of CD18 which is the β2 subunit of the leukocyte integrin found on the surface of neutrophils and monocytes.
Because there is widespread loss of fucosylated antigens in LAD II patients, each of which can be formed through the actions of several fucosyltransferases, the role of these enzymes in the disease could be ruled out. In addition, normal levels of the α-1,2-, α-1,3- and α-1,4-fucosyltransferases were observed in the serum of LAD II individuals. These observations indicated that the pathways to GDP-fucose synthesis or utilization by the fucosyltransferases in the Golgi must be deficient in LAD II.
Fucose can be converted to GDP-fucose by salvage of free fucose (exogenous or derived through glycoconjugate degradation) or by epimerization of GDP-mannose. In order for GDP-fucose to be utilized by Golgi fucosyltransferases it must first be transported into the Golgi from the cytosol where it is synthesized. Examinations of the enzymes involved in the synthesis and transport of GDP-fucose have been undertaken. While the activity of one of the enzymes of GDP-mannose epimerization (GDP-D-mannose 4,6-dehydratase, GMD) has been shown to be reduced in LAD II patients, it has been determined that the major defect causing LAD II is an impairment in the transport of GDP-fucose into the Golgi. This latter reaction is catalyzed by the GDP-fucose transporter encoded by the FUCT1 gene (also identified as solute carrier family 35, member C1: SCL35C1).