
Last Updated: July 7, 2026
Introduction to Gluconeogenesis
Gluconeogenesis is the biosynthesis of new glucose, (i.e. not glucose from glycogen). This process is frequently referred to as endogenous glucose production (EGP). The production of glucose from other carbon skeletons is necessary since the testes, erythrocytes, and kidney medulla exclusively utilize glucose for ATP production.
The brain also utilizes large amounts of the daily glucose consumed or produced via gluconeogenesis. However, in addition to glucose, the brain can derive energy from ketone bodies which are converted to acetyl-CoA and shunted into the TCA cycle.
The primary carbon skeletons used for gluconeogenesis are derived from pyruvate, lactate, glycerol, and the amino acids, alanine and glutamine. The liver is the major site of gluconeogenesis, however, as discussed below, the kidney and the small intestine also have important roles to play in this pathway.
Pathway of Hepatic Gluconeogenesis
Synthesis of glucose from three and four carbon precursors is essentially a reversal of glycolysis. Gluconeogenesis from two moles of pyruvate to two moles of 1,3-bisphosphoglycerate consumes six moles of ATP. This makes the process of gluconeogenesis very costly from an energy standpoint considering that glucose oxidation to two moles of pyruvate yields two moles of ATP.
The relevant features of the pathway of gluconeogenesis, that take place in the liver, are diagrammed below:
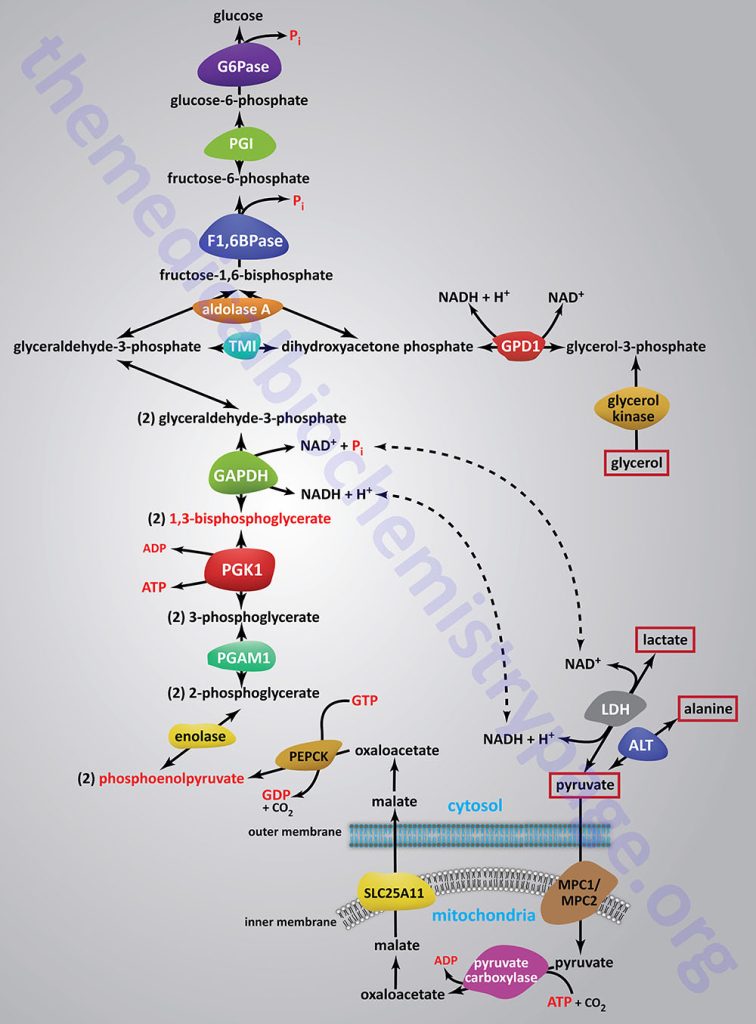
The three reactions of glycolysis that proceed with a large negative free energy change are bypassed during gluconeogenesis by using different enzymes. These three reactions are catalyzed by pyruvate kinase, phosphofructokinase-1 (PFK-1: 6-phosphofructo-1-kinase) and hexokinase/glucokinase. In the liver, intestine, or kidney cortex, the glucose-6-phosphate (G6P) produced by gluconeogenesis can be dephosphorylated by glucose-6-phosphatase and released as free glucose.
Bypass 1: Pyruvate to Phosphoenolpyruvate (PEP)
Conversion of pyruvate to PEP requires the action of two enzymes: pyruvate carboxylase (PC) and phosphoenolpyruvate carboxykinase (PEPCK).
Pyruvate Carboxylase Reaction
The first reaction of bypass 1 utilizes the ATP and biotin-requiring enzyme pyruvate carboxylase, (PC). PC is referred to as an ABC enzyme due to the role of ATP, Biotin, and CO2 in its catalytic activities. The CO2 utilized in the PC reaction is in the form of bicarbonate (HCO3–) . As the name of the enzyme implies, pyruvate is carboxylated to form oxaloacetic acid (OAA; oxaloacetate).
PC is a somewhat unique enzyme in that it is one of only two metabolically important enzymes that requires an obligate activator. In the absence of its obligate activator, acetyl-CoA, PC is essentially inactive. The primary source of the acetyl-CoA required by PC comes from the oxidation of fatty acids which are being delivered to the liver after release from adipose tissue in response to fasting or stress. Another critical enzyme that functions only in the presence of an obligate activator is carbamoyl phosphate synthetase 1 (CPS1) of the urea cycle. Given the low to no activity of PC in the absence of acetyl-CoA the enzyme can be considered rate limiting for hepatic gluconeogenesis. Despite this, the commonly stated rate-limiting enzyme of gluconeogenesis is fructose-1,6-bisphosphatase, F1,6BPase.
Like the other biotin-dependent carboxylating enzymes in mammals, PC is multi-functional and contains three distinct enzymatic domains. These three d0mains, arranged from the N-terminal to the C-terminal of the proteins are the biotin carboxylase (BC) domain, the carboxyltransferase (CT) domain, and the biotin carboxyl carrier protein (BCCP) domain. Functional PC is composed of four identical subunits generating an α4 homotetrameric enzyme.
The reaction catalyzed by PC occurs in a two-step process. The first partial reaction involves the fixation of CO2 to biotin that involves the BC and BCCP domains. During this initial stage of the reaction, biotin is moved to interact with the BC domain forming carboxybiotin. The carboxybiotin is brought into contact with the carboxyltransferase domain resulting in the formation of carboxylated biotin. This biotin carboxylase reaction involves a carboxyphosphate intermediate formed directly from ATP and bicarbonate. During the second step of the overall PC reaction, carboxybiotin is decarboxylated and pyruvate is concurrently carboxylated forming oxaloacetate.
Regulation of Pyruvate Carboxylase Gene Expression
The human PC gene is located on chromosome 11q13.2 and contains 31 exons that generate three mRNAs through the use of two distinct promoters and via alternatively splicing from three 5′-untranslated exons identified as UE1/2, UE3, and UE4. Despite the use of different promoters and the alternative splicing, all three PC mRNAs encode the same 1178 amino acid precursor protein.
The two promoter elements in the PC gene are identified as P1 (proximal) and P2 (distal). The P1 promoter is actively utilized in gluconeogenic tissues such as the liver and kidney as well as in lipogenic tissues such as the liver and adipose tissue. The P2 promoter is active in promoting anaplerosis, particularly in pancreatic β-cells.
The P1 promoter possesses both a cAMP-responsive element (CRE) and a peroxisome proliferator activated receptor response element (PPRE). The P2 promoter lacks a TATA box but contains a GC box and two CCAAT boxes. Sequences that are known to bind several different transcription factors are associated with the P2 promoter. These include binding sites for Sp1, HNF3β, and FOXA2.
Pyruvate Carboxylase-Mediated TCA Cycle Anaplerosis
Although the major function of PC is to drive precursor carbon atoms (from pyruvate, lactate, and alanine) into the generation of endogenous glucose, the production of oxaloacetate is also an important anaplerotic reaction since it can be used to fill-up the TCA cycle. The function of PC in TCA cycle anaplerosis is a part of a metabolic pathway referred to as the PEP cycle.
Indeed, within the brain the primary function of PC is to ensure that glial cells have sufficient oxaloacetate to drive the TCA cycle. In these cells, in addition to energy generation, the TCA cycle is vital to the continued generation of 2-oxoglutarate (α-ketoglutarate) which can be siphoned off the TCA cycle and utilized for the synthesis of glutamate as a critical excitatory neurotransmitter.
Within the liver, the role of PC in TCA cycle anaplerosis is critical for hepatoprotection from oxidative stress and for providing important intermediates required for the maintenance of overall hepatocyte biomass.
Clinical Significance of Pyruvate Carboxylase Mutations
Mutations in the PC gene are the cause of the various forms of pyruvate carboxylase deficiency. The principal pathology of pyruvate carboxylase deficiency includes metabolic acidosis due to elevated lactate, failure to thrive, developmental delay, and recurrent seizures.
Phosphoenolpyruvate Carboxykinase Reaction
The second enzyme in the gluconeogenic conversion of pyruvate to phosphoenolpyruvate (PEP) is PEP carboxykinase (PEPCK). PEPCK requires GTP in the decarboxylation of OAA to yield PEP. Since PC incorporated CO2 into pyruvate and it is subsequently released in the PEPCK reaction, no net fixation of carbon occurs. Humans express two forms of PEPCK, a mitochondrial and a cytosolic PEPCK (designated PEPCK-m and PEPCK-c, respectively). Thus, the second reaction can occur in either cellular compartment. The amount of PEPCK-c versus PEPCK-m is different dependent upon the tissue but in the liver the distribution is essentially 50% for each form.
PEPCK-c is encoded by the PCK1 gene. The PCK1 gene is located on chromosome 20q13.31 and is composed of 10 exons encoding a protein of 622 amino acids. The PCK1 gene is expressed predominantly in the liver and kidney with slightly lower levels of expression in the small intestine as would be expected for a gluconeogenic enzyme.
PEPCK-m is encoded by the PCK2 gene. The PCK2 gene is located on chromosome 14q11.2–q12 and is composed of 12 exons that generate four alternatively spliced mRNAs that collectively encode three distinct protein isoforms. The PCK2 gene is primarily expressed in the liver, kidney, and small intestine as would be expected for a gluconeogenic enzyme. The liver expresses the PCK1 and PCK2 genes at essentially equivalent levels.
Mutations in the either the PCK1 or PCK2 gene results in significant impairment of gluconeogenesis.
Transcription of the PCK1 and PCK2 genes has been shown to be regulated by insulin, glucagon, and glucocorticoids. Regulation of gluconeogenic gene expression by glucagon is exerted via the activation of the transcription factor, CREB (cAMP response element-binding protein; encoded by the CREB1 gene).
Humans express seven genes in the CREB subfamily of the basic leucine zipper (bZIP) family of transcription factors. The seven CREB genes are identified as CREB1, CREB3, CREB5, and CREB3-like 1, 2, 3, and 4 (CREB3L1, CREB3L2, CREB3L3, and CREB3L4). The CREB3L3 encoded protein is commonly identified as CREBH. The CREB1 encoded proteins are most closely related in structure and function to two additional transcription factors called cAMP response element modulator (CREM) and activating transcription factor 1 (ATF-1). Another member of the activating transcription factor (ATF) family, ATF-4, was originally identified as CREB2.
When glucagon binds its receptor the result is activation of adenylate cyclase with resultant increases in cAMP production. The increased cAMP in turn activates PKA which, among numerous substrates, phosphorylates CREB. Active PKA migrates to the nucleus where it phosphorylates CREB which then binds to a cAMP-response element (CRE) in the PCK1, PCK2, and G6PC1 (glucose-6-phosphatase) genes activating their rate of transcription. The stress hormone, cortisol, exerts a very similar effect on gluconeogenic gene expression via binding of the cortisol-activated glucocorticoid receptor to a glucocorticoid-response element (GRE) in the PCK1 and PCK2 genes.
In addition to its role in gluconeogenesis, the PEPCK-c enzyme can participate as a contributor to TCA cycle anaplerosis and to the process referred to as glyceroneogenesis. PEPCK-c functions in these multiple roles via its ability to catalyze the reversible interconversions of OAA and PEP. The reversible activity of PEPCK-c is, in part, controlled by its state of post-translational modification.
Role of The PEP Cycle in Insulin Secretion
The PEP cycle refers to the process whereby cytosolic PEP is used to replenish the mitochondrial TCA cycle, reactions that constitute anaplerosis. Since the NADH and FADH2 generated via the TCA cycle are major contributors to mitochondrial oxidative phosphorylation and ATP production, the PEP cycle is a critical process of overall cellular energy generation.
PEP is a substrate for pyruvate kinase (PK) which converts it to pyruvate. The pyruvate can be transported into the mitochondria where it will be oxidatively decarboxylated to acetyl-CoA via the actions of the PDHc. The acetyl-CoA product of the PDHc can then be completely oxidized via the TCA cycle.
Depending on the energy charge and the mitochondrial level of acetyl-CoA, the mitochondrial pyruvate can be carboxylated by pyruvate carboxylase (PC) generating oxaloacetate (OAA). The mitochondrial form of PEPCK (PEPCK-m) can then decarboxylate OAA yielding PEP which can be transported back to the cytosol, effectively completing one turn of the PEP cycle. The transport of mitochondrial PEP to the cytosol is the function of the SLC family transporter encoded by the SLC25A35 gene.
Under conditions of enhanced PK activity, as can be exerted through pharmacological means, the rate of the PEP cycle increases and the resulting increase in ATP generation, specifically in pancreatic β-cells, results in enhanced insulin secretion. Mitochondrial PEPCK (PEPCK-m), which is encoded by the PCK2 gene, plays a pivotal role in the pancreas by regulating the level of glucose-stimulated insulin secretion (GSIS). This was demonstrated in PCK2 knockout mice that manifest with impaired pancreatic β-cell function resulting in impaired insulin secretion.
Transport of Mitochondrial OAA to Cytosol
For gluconeogenesis to proceed, the OAA produced by PC needs to be transported to the cytosol. However, no transport mechanism exist for its direct transfer and OAA will not freely diffuse out of the mitochondria. Mitochondrial OAA can become cytosolic via three pathways: (1) conversion to PEP as indicated above through the action of the mitochondrial PEPCK; (2) transamination to aspartate; or (3) reduction to malate, all of which are transported to the cytosol.
Transport of mitochondrial PEP to the cytosol is carried out by the tricarboxylate transporter encoded by the SLC25A1 gene. The SLC25A1 encoded transporter is also identified as the mitochondrial citrate carrier (CiC). The transport of malate to the cytosol is carried out by the transporter encoded by the SLC25A11 gene. The transport of aspartate to the cytosol is carried out by either of two transporters, one is encoded by the SLC25A12 gene and the other is encoded by the SLC25A13 gene.
In the context of the transamination of OAA to aspartate and the reduction of OAA to malate, there is a need for adequate levels of the other intermediates of the malate-aspartate shuttle to ensure these latter two reactions can continue.
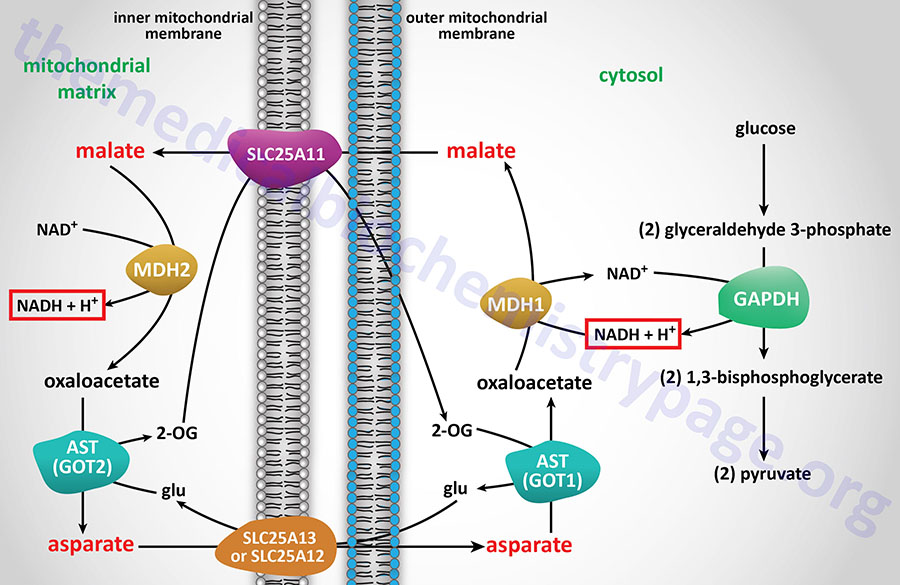
If OAA is converted to PEP by mitochondrial PEPCK-m, the PEP is transported to the cytosol via the SLC family transporter encoded by the SLC25A35 gene. The fate of this mitochondrial PEP once transported to the cytosol depends in part on the tissue as well as the energy state of the mitochondria from which it was transported. Within the liver this mitochondrial PEP when transported to the cytosol can serve as a direct substrate for gluconeogenesis and nothing further is required.
Within adipocytes, hepatocytes, and other lipogenic tissues, mitochondrial PEP transported to the cytosol via the SLC25A35 transporter can ultimately serve as a substrate for glycerol-3-phopshate synthesis and thus, as the backbone for triglyceride synthesis.
Transamination of OAA to aspartate allows the aspartate to be transported to the cytosol where the reverse transamination occurs yielding cytosolic OAA. This transamination reaction requires continuous transport of glutamate into, and 2-oxoglutatrate (α-ketoglutarate) out of, the mitochondrion. Therefore, this process is limited by the availability of these other substrates. Either of these latter two reactions will predominate when the substrate for gluconeogenesis is lactate. Whether mitochondrial decarboxylation or transamination occurs is a function of the availability of PEPCK or transamination intermediates.
Mitochondrial OAA can also be reduced to malate in a reversal of the TCA cycle reaction catalyzed by malate dehydrogenase (MDH). The reduction of OAA to malate requires NADH, which will be accumulating in the mitochondrion as the energy charge increases, primarily as a result of fatty acid oxidation. The increased energy charge will allow hepatocytes to carry out the ATP costly process of gluconeogenesis. The resultant malate is transported to the cytosol where it is oxidized to OAA by cytosolic MDH which requires NAD+ and yields NADH. The NADH produced during the cytosolic oxidation of malate to OAA is utilized during the glyceraldehyde-3-phosphate dehydrogenase reaction of gluconeogenesis. The coupling of these two oxidation-reduction reactions is required to keep gluconeogenesis functional when pyruvate is the principal source of carbon atoms.
The conversion of OAA to malate predominates when pyruvate (derived from glycolysis or amino acid catabolism) is the source of carbon atoms for gluconeogenesis. When in the cytoplasm, OAA is converted to PEP by the cytosolic version of PEPCK. Hormonal signals control the level of PEPCK protein as a means to regulate the flux through gluconeogenesis (see Regulation of Gluconeogenesis below).
The net result of the PC and PEPCK reactions is:
Pyruvate + ATP + GTP + H2O → PEP + ADP + GDP + Pi + 2H+
Role of Cytoplasmic PEPCK in Cataplerosis
As described in the PEP Cycle section above, mitochondrial OAA can be transaminated to aspartate by the mitochondrial aspartate transaminase, AST (which is encoded by the GOT2 gene). The aspartate can then be transported to the cytosol by either of the aspartate-glutamate carriers, encoded by the SLC25A12 and SLC25A13 genes. In the cytosol, the cytosolic AST, which is encoded by the GOT1 gene, can deaminate aspartate back to OAA.
The cytosolic OAA is decarboxylated to PEP via the cytosolic version of PEPCK (encoded by the PCK1 gene). The series of reactions converting mitochondrial OAA to aspartate and cytosolic aspartate to OAA, which constitutes reactions of the malate-aspartate shuttle, allow for the generation of pyruvate in what is referred to as pyruvate cycling. Cytoplasmic PEP generated through the action of the PCK1 encoded PEPCK can be used as a substrate for pyruvate kinase (PK) yielding pyruvate. The pyruvate can re-enter the mitochondria and be oxidized by the PDHc yielding acetyl-CoA and OAA, both of which represent anaplerotic substrates for the TCA cycle.
The reactions of the malate-aspartate shuttle allow for the diversion of the carbons of mitochondrial OAA, not only for glucose production via gluconeogenesis but also to other processes such as glycogen synthesis and glyceroneogenesis. The diversion of potential TCA cycle intermediates to these other pathways is referred to as a series of cataplerotic reactions. In the context of pyruvate cycling, the PCK1 catalyzed reaction represents one of the major contributors to cataplerosis. Indeed, in circumstances associated with reduced PCK1 activity there is an accumulation of TCA cycle intermediates.
Bypass 2: Fructose-1,6-bisphosphate to Fructose-6-phosphate
Fructose-1,6-bisphosphate (F1,6BP) conversion to fructose-6-phosphate (F6P) is the reverse of the rate-limiting step of glycolysis. The reaction, a simple hydrolysis, is catalyzed by fructose-1,6-bisphosphatase (F1,6BPase). The existence of two distinct forms of F1,6BPase was recognized by comparison of the kinetic and regulatory properties of the purified liver and muscle enzymes. In addition, in patients with an inborn error in the gene encoding the liver F1,6BPase isoform, there is no reduction in skeletal muscle F1,6BPase activity. This led to the characterization of two F1,6BPase genes in the human genome. One expresses a liver version of the enzyme (FBP1 gene) and the other expresses a muscle version of the enzyme (FBP2 gene).
The FBP1 gene is located on chromosome 9q22.32 and is composed of 8 exons that generate two alternatively spliced mRNAs, both of which encode the same 338 amino acid protein.
The FBP2 gene is located at the same chromosomal location as the FBP1 gene but is composed of 7 exons that encode a protein of 339 amino acids.
The liver and muscle F1,6BPase enzymes share 77% amino acid sequence identity. Despite being originally identified in skeletal muscle, expression of the FBP2 gene is nearly exclusive to the gastrointestinal system.
Like the regulation of glycolysis occurring at the PFK-1 reaction, the F1,6BPase reaction is a major point of control of gluconeogenesis as discussed in detail below.
Role of Nuclear Fructose-1,6-Bisphosphatase in Cancer Proliferation
In response to stress, particularly to glucose deprivation, the eIF2α kinase, PERK, phosphorylates fructose-1,6-bisphosphatase 1 (FBP1). The phosphorylation of FBP1 causes the normal tetrameric enzyme to dissociate into monomers. Monomeric FBP1 proteins have an exposed nuclear localization signal (NLS) allowing the enzyme to be transported into the nucleus. Within the nucleus FBP1 binds to the transcription factor, peroxisome proliferator-activated receptor alpha (PPARα). In a complex with PPARα, FBP1 dephosphorylates histone H3 (phosphorylated on threonine 11: H3T11) in the promoter region of several genes encoding metabolic enzymes. The dephosphorylation of H3T11 results in suppression of PPARα-mediated activation of genes whose encoded enzymes function in fatty acid β-oxidation and amino acid oxidation such as the ACAA2 (acetyl-CoA acyltransferase 2) and ACAD8 (isobutyryl-CoA dehydrogenase) genes, respectively.
In certain forms of cancer, such as in hepatocellular carcinomas, there is enhanced expression of the enzyme, UDP-N-acetylglucosamine:polypeptide β-N-acetylglucosaminyltransferase (more commonly just O-GlcNAc transferase, OGT) that is responsible for the O-GlcNAcylation of numerous nuclear and cytoplasmic proteins. OGT is known to O-GlcNAcylate FBP1 which results in inhibition of the phosphorylation of FBP1 by PERK. In cancer cells the result is that there is enhanced PPARα-mediated fatty acid β-oxidation providing increased energy needed for tumor cell proliferation.
Clinical significance in the expression of the FBP2 gene has been associated with the development of soft tissue sarcomas. Expression of the gene is reduced in many different types of soft tissue sarcomas and ectopic expression has been shown to repress the progression of these types of cancer. The effect of FBP2 expression on sarcoma progression is the result of both cytoplasm-localized and nuclear-localized enzyme. Within the cytosol the activity of FBP2 reduces the elevated levels of glucose typical of the Warburg effect in numerous cancers. Within the nucleus FBP2 acts as a transcriptional co-repressor with MYC to suppress the expression of the TFAM (mitochondrial transcription factor A) gene thereby reducing mitochondrial biogenesis.
Role of Fructose-1,6-Bisphosphatase in Regulation of Insulin Responses
In response to the effects of insulin in the liver, gluconeogenesis is inhibited, and glucose is driven into glycogen synthesis and fatty acid synthesis. In the absence of finely regulated metabolism, the actions of insulin could lead to potentially severe hypoglycemia and hepatosteatosis. When insulin activates its receptor on hepatocytes, the primary signal transduction molecules that are activated are AKT2/PKBβ and PI3K.
The role of F1,6BPase in the prevention of the potential for hypoglycemia and hepatosteatosis by insulin has been determined, in part, by studying the rare inborn error of metabolism that results from mutations in the FBP1 gene and from studying mice in which the FBP1 gene was knocked out specifically in the liver. The fasting pathology seen in FBP1-deficient infants, that includes hepatomegaly, hepatosteatosis, and hyperlipidemia, are also seen in fasting mice lacking the liver FBP1 gene.
The mechanism by which F1,6BPase plays a role in the prevention of elevated responses to the effects of insulin involves the formation of a multiprotein complex with F1,6BPase serving as the nucleation center. The complex contains F1,6BPase, the ALDOB encoded enzyme of hepatic glycolysis (fructose-1,6-bisphosphate aldolase), the catalytic subunit of protein phosphatase 2A, and AKT2/PKBβ. PP2A is known to dephosphorylate AKT2 which reduces its kinase activity. Since AKT2/PKBβ is a critical kinase in the insulin signaling cascade, the effect of sequestering AKT2/PKBβ and its dephosphorylation, reduces the effects of insulin in the liver.
Bypass 3: Glucose-6-phosphate (G6P) to Glucose (or Glycogen)
Glucose-6-phosphate is converted to free glucose through the action of enzymes of the glucose-6-phosphatase (G6Pase) family. The G6Pase reaction is also a simple hydrolysis reaction like that of F1,6BPase. Since the brain and skeletal muscle, as well as most non-hepatic tissues, lack G6Pase activity, any gluconeogenesis that might occur in these tissues is not utilized for blood glucose supply. In the kidney, muscle and especially the liver, G6P be shunted toward glycogen if blood glucose levels are adequate. The reactions necessary for glycogen synthesis are an alternate bypass series of reactions.
The glucose-6-phosphatase activities are membrane-associated multi-subunit complexes associated with the membranes of the endoplasmic reticulum, ER. The complexes are composed of a catalytic subunit and transporter proteins for the transport of glucose-6-phosphate, inorganic phosphate, and glucose across the membranes of the ER. The catalytic activity of G6Pases resides in a domain of the enzyme that is within the lumen of the ER, thus glucose-6-phosphate must first be transported into the ER for the phosphate to be removed.
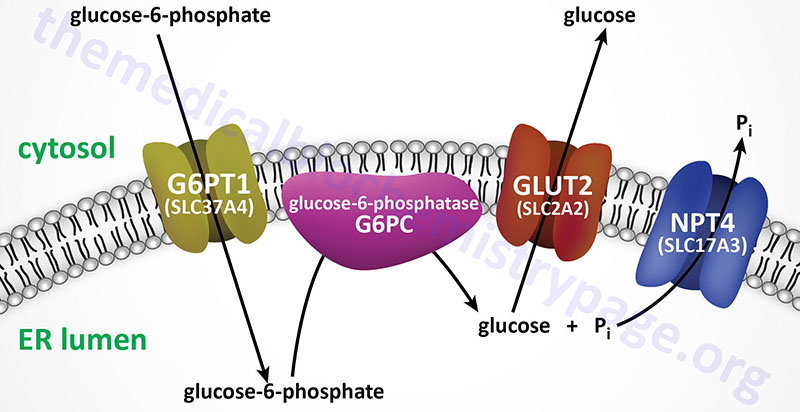
Humans express three distinct genes of the glucose-6-phosphatase family identified as G6PC1 (also identified as G6PC), G6PC2, and G6PC3. The G6PC1 gene encodes the predominantly expressed functional phosphatase form of the glucose-6-phosphatase.
The G6PC1 gene is located on chromosome 17q21.31 and is composed of 5 exons that generate two alternatively spliced mRNAs encoding proteins of 357 amino acids (isoform 1) and 176 amino acids (isoform 2). Only three human tissues express the G6PC1 gene, liver, kidney, and small intestine. Likewise, these are the only tissues that can contribute to endogenous glucose production. Defects in the G6PC1 gene are associated with the glycogen storage disease known as von Gierke disease (glycogen storage disease type Ia).
The G6PC2 gene is located on chromosome 2q31.1 and is composed of 5 exons that generate two alternatively spliced mRNAs, both of which encode distinct protein isoforms. The G6PC2 gene is expressed in pancreatic islets but the encoded protein does not possess glucose-6-phosphatase activity.
The G6PC3 gene is located on chromosome 17q21.31 and is composed of 8 exons that generate six alternatively spliced mRNAs that collectively encode three distinct protein isoforms. The G6PC3 gene encoded proteins are not involved in endogenous free glucose production but are believed to have functions in the activities of neutrophils. Although the G6PC3 encoded proteins can hydrolyze phosphate from glucose-6-phosphate (G6P) in vitro, the enzymes have a high Km for G6P and thus, are likely to catalyze hydrolysis of other substrates in vivo.
The ER membrane-localized glucose-6-phosphate transporter is encoded by the SLC37A4 gene with the encoded protein being identified as G6PT1. The SLC37A4 gene is located on chromosome 11q23.3 and is composed of 12 exons that generate five alternatively spliced mRNAs that collectively encode three distinct protein isoforms.
The ER membrane-localized phosphate transporter is encoded by the SLC17A3 gene and the encoded protein is identified as NPT4 (Na+-phosphate transporter 4). The SLC17A3 gene is located on chromosome 6p22.2 and is composed of 13 exons that generate two alternatively spliced mRNAs. One of the SLC17A3 derived mRNAs encodes a 498 amino acid transporter that is localized to the apical membrane of epithelial cells of the proximal tubule of the kidney. The other SLC17A3 derived mRNA encodes a 420 amino acid transporter that is localized to the ER membrane.
The transport of free glucose, from the lumen of the ER to the cytosol, most likely occurs through the actions of plasma membrane localized GLUT transporters (most likely GLUT2 in the liver) as they are transiting the ER on their way to the plasma membrane.
Phosphorolysis of glycogen is carried out by glycogen phosphorylase, whereas, glycogen synthesis is catalyzed by glycogen synthase. The G6P produced from gluconeogenesis could be used as a substrate for the synthesis of glycogen. If this were to occur the G6P is converted to glucose-1-phosphate (G1P) by phosphoglucomutase (PGM). G1P is then converted to UDP-glucose (the substrate for glycogen synthase) by UDP-glucose pyrophosphorylase, a reaction requiring hydrolysis of UTP. However, given that hepatic gluconeogenesis is stimulated by glucagon and epinephrine, hormones that also result in inhibition of glycogen synthesis, it is unlikely that any G6P produced as a result of gluconeogenesis would end up in glycogen.
Substrates for Gluconeogenesis
Lactate
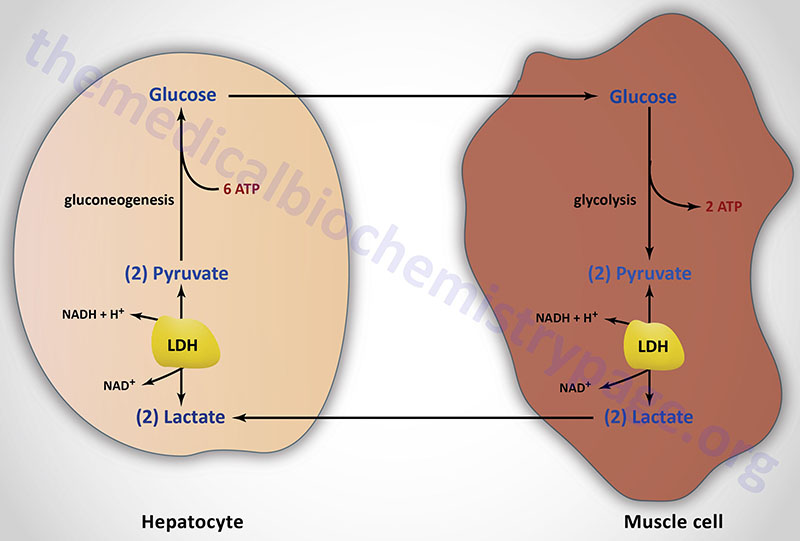
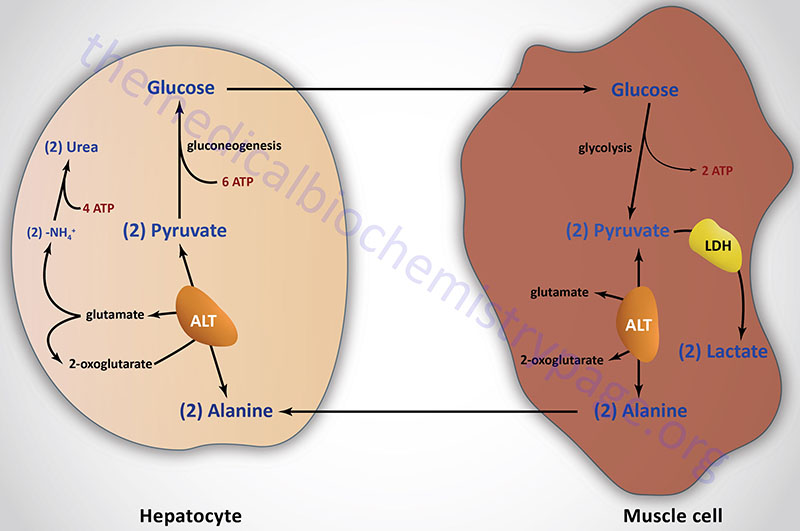
Lactate is a predominate source of carbon atoms for glucose synthesis via hepatic gluconeogenesis. During anaerobic glycolysis in erythrocytes and skeletal muscle, pyruvate is reduced to lactate by the lactate dehydrogenase (LDH) encoded by the LDHA gene. This reaction serves two critical functions during anaerobic glycolysis. First, in the direction of lactate formation the LDH reaction requires NADH and yields NAD+ which is then available for use by the glyceraldehyde-3-phosphate dehydrogenase reaction of glycolysis. These two reaction are, therefore, intimately coupled during anaerobic glycolysis. Secondly, the lactate produced by the LDH reaction is released to the blood stream and transported to the liver where it is converted to glucose. The glucose is then returned to the blood for use by muscle as an energy source and to replenish glycogen stores. This cycle is termed the Cori cycle.
Pyruvate
Pyruvate, generated in muscle and other peripheral tissues, can be transaminated to alanine via the alanine transaminase (ALT) reaction. The alanine is then delivered to the liver for use in gluconeogenesis. The transamination reaction requires an α-amino acid as donor of the amino group, generating an α-keto acid in the process. This pathway is termed the glucose-alanine cycle. Although the majority of amino acids are degraded in the liver some are deaminated in muscle. The glucose-alanine cycle is, therefore, an indirect mechanism for muscle to eliminate nitrogen while replenishing its energy supply. However, the major function of the glucose-alanine cycle is to allow non-hepatic tissues to deliver the amino portion of catabolized amino acids to the liver for excretion as urea. Within the liver the alanine is deaminated by ALT back to pyruvate and used as a gluconeogenic substrate (if that is the hepatic requirement) or oxidized in the TCA cycle. The amino nitrogen is converted to urea in the urea cycle and excreted by the kidneys.
Amino Acids
All of the amino acids present in proteins, excepting leucine and lysine, can be degraded to TCA cycle intermediates as discussed in the metabolism of amino acids page. This allows the carbon skeletons of the amino acids to be converted to those in oxaloacetate and subsequently into pyruvate. The pyruvate thus formed can be utilized by the gluconeogenic pathway. The major amino acids utilized for gluconeogenesis are alanine and glutamine.
When glycogen stores are depleted, in muscle during exertion and liver during fasting, catabolism of muscle proteins to amino acids contributes the major source of carbon for maintenance of blood glucose levels. Of all the amino acids utilized for gluconeogenesis, glutamine is the most important as this amino acid is critical for glucose production by the kidneys and small intestine.
Glutamine
Glutamine is the sole source of carbon atoms for the gluconeogenesis pathway carried out in the kidney and the small intestine. In these two tissues, glutamine is first deaminated to glutamate via the action of glutaminase. The glutamate is then further deaminated, via the action of the enzyme glutamate dehydrogenase, yielding 2-oxoglutarate (α-ketoglutarate). The 2-oxoglutarate can then enter the TCA cycle where it is eventually converted to malate.
As described earlier, malate can be transported out of the mitochondria and oxidized to oxaloacetate via the action of cytoplasmic malate dehydrogenase. The oxaloacetate is then converted to PEP via the action of the cytoplasmic version of phosphoenolpyruvate carboxykinase (PEPCK-c). Alternatively, malate can be oxidized to oxaloacetate within the mitochondria then the action of mitochondrial PEPCK (PEPCK-m) can convert the oxaloacetate to PEP. If this pathway is utilized the PEP is transported to the cytosol for gluconeogenesis.
Glycerol
Oxidation of fatty acids yields enormous amounts of energy on a molar basis, however, the carbons of the fatty acids cannot be utilized for net synthesis of glucose. The two carbon unit of acetyl-CoA derived from β-oxidation of fatty acids can be incorporated into the TCA cycle, however, during the TCA cycle two carbons are lost as CO2. Thus, explaining why fatty acids do not undergo net conversion to carbohydrate.
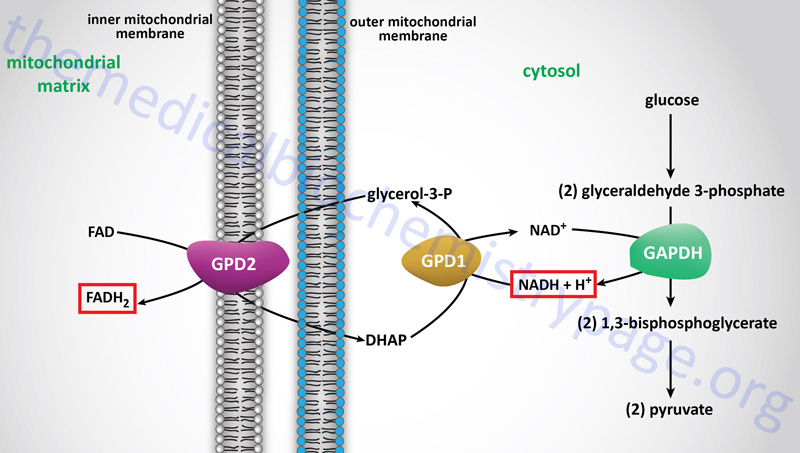
However, the glycerol backbone that is released from adipocytes, following hormone-induced triglyceride breakdown, can be used for gluconeogenesis. This requires phosphorylation of the glycerol to glycerol-3-phosphate by glycerol kinase within hepatocytes. Following formation of glycerol-3-phosphate it is oxidized to dihydroxyacetone phosphate (DHAP) by cytosolic glycerol-3-phosphate dehydrogenase 1 (GPD1). The glycerol backbone of adipose tissue stored triglycerides is ensured of being used as a gluconeogenic substrate by the liver since adipocytes lack glycerol kinase. In fact adipocytes require a basal level of glycolysis in order to provide them with DHAP as an intermediate in the synthesis of triglycerides. The GPD1 reaction is the same as that used in the transport of cytosolic reducing equivalents into the mitochondrion for use in oxidative phosphorylation. This transport pathway is called the glycerol phosphate shuttle.
Propionate
Oxidation of fatty acids with an odd number of carbon atoms and the oxidation of some amino acids generates as the terminal oxidation product, propionyl-CoA. In addition, there is another source of propionyl-CoA that is exclusive to the liver which is the pathway of bile acid synthesis from cholesterol.
Propionyl-CoA is converted to the TCA cycle intermediate, succinyl-CoA. This conversion is carried out by the ATP-requiring enzyme, propionyl-CoA carboxylase then methylmalonyl-CoA epimerase and finally the vitamin B12 requiring enzyme, methylmalonyl-CoA mutase. The utilization of propionate in gluconeogenesis only has quantitative significance in ruminants.
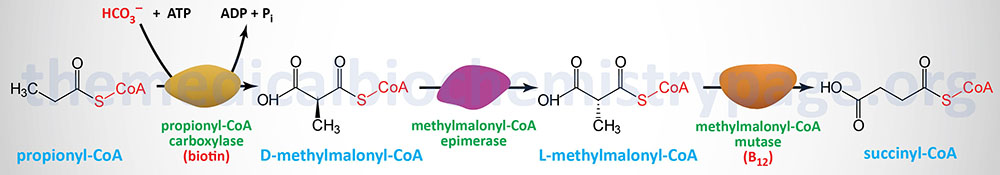
Propionyl-CoA carboxylase functions as a heterododecameric enzyme (subunit composition: α6β6) and the two different subunits are encoded by the PCCA and PCCB genes, respectively.
The PCCA gene is located on chromosome 13q32.3 and is composed of 32 exons that generate eleven alternatively spliced mRNAs, each of which encode a unique protein isoform.
The PCCB gene is located on chromosome 3q22.3 and is composed of 17 exons that generate two alternatively spliced mRNAs encoding proteins of 539 amino acids (isoform 1) and 559 amino acids (isoform 2).
Mutations in either the PCCA or PCCB gene are associated with propionic acidemia and severe ketoacidosis. The original identification of a child suffering from propionyl-CoA deficiency was in 1961. This child suffered frequent episodes of severe ketoacidosis, all of which were precipitated by protein ingestion. Blood and urine analysis demonstrated marked elevations in glycine levels. These initial laboratory studies lead to the disorder being called ketotic hyperglycinemia. However, there is no defect in glycine metabolism with inherited mutations in PCCA or PCCB. The clinical hallmark of the disease is severe ketoacidosis of an episodic nature, primarily induced by the consumption of protein.
Methylmalonyl-CoA epimerase is encoded by the MCEE gene located on chromosome 2p13.3 and is composed of 4 exons that encode a 176 amino acid precursor protein.
Methylmalonyl-CoA mutase is encoded by the MMUT gene located on chromosome 6p12.3 and is composed of 13 exons that encode a precursor protein of 750 amino acids. Mutations in the MMUT gene are one cause of the methylmalonic acidemias.
Regulation of Gluconeogenesis
Control of gluconeogenesis, particularly hepatic gluconeogenesis, is exerted by numerous distinct and interconnected processes. These processes include hormone secretion, regulation of gene expression, and posttranslational modification. The major hormones that regulate hepatic gluconeogenesis include insulin which inhibits the process, and glucagon, epinephrine, and cortisol, all of which stimulate the process.
Insulin Regulation of Gluconeogenesis
Insulin regulates hepatic gluconeogenesis as a result of its receptor activating phosphatidylinositol-3 kinase (PI3K) and AKT/PKB. Humans express three forms of AKT/PKB with AKT2/PKBβ being the predominant form regulated by insulin in hepatocytes. Insulin is released from pancreatic β-cells in response to the well fed state. When insulin binds its receptor on hepatocytes it triggers a signaling cascade involving PI3K and AKT2. PI3K phosphorylates phosphatidylinositol-4,5-bisphosphate (PIP2; PtdIns-4,5-P2) generating the lipid second messenger phosphatidylinositol-3,4,5-trisphosphate (PIP3; PtdIns-3,4,5-P2). PIP3 recruits AKT2 to the plasma membrane where it is activated by being phosphorylated. Activated AKT2 then phosphorylates numerous substrates including the transcription factors FOXO1 and CRTC2 (CREB regulated transcription coactivator 2).
As indicated below, the phosphorylation of these transcription factors leads to their exclusion from the nucleus. Insulin signaling also activates protein phosphatase 1 (PP1) and the phosphodiesterase, PDE3B (also called adipocyte cAMP phosphodiesterase). The activation of these two enzymes results in reduced levels of active PKA and reduced levels of phosphorylation of enzymes that regulate the activity level of gluconeogenesis, such as PFK2 (see next section).
Glucagon and Epinephrine Regulation of Gluconeogenesis
Both glucagon and epinephrine stimulate hepatic gluconeogenesis. Hepatocytes express both α1– and β2-adrenergic receptors to which epinephrine binds. The α1-adrenergic receptors are coupled to a Gq-type G-protein whereas the β2-adrenergic receptors and the glucagon receptors are coupled to Gs-type G-proteins. It is important to note that within hepatocytes it has been demonstrated that the glucagon receptor is also coupled to a Gq-type G-protein.
Activation of the receptor-associated Gs-type G-proteins leads to activation adenylate cyclase and the production of cAMP. Increased levels of cAMP in turn activates PKA. PKA phosphorylates numerous proteins (enzymes and transcription factors) that are involved in the regulation of gluconeogenesis or are directly involved in the pathway as described in more detail below.
Activation of Gq-type G-proteins leads to activation of phospholipase C-β (PLCβ) resulting in increased production of diacylglycerol (DAG) and inositol trisphosphate (IP3; Ins-1,4,5-P3) from membrane phosphatidylinositol-4.5-bisphosphate (PIP2; PtdIns-4,5-P2). The released IP3 binds to specific receptors on the ER membrane which, when activated, leads to the mobilization of stored Ca2+ into the cytosol. Of significance to hepatic glucose homeostasis, the release of stored intracellular Ca2+ results in binding to the calmodulin subunits of several kinases such as Ca2+/calmodulin-dependent protein kinase II (CaMKII).
Increased CaMKII activity leads to phosphorylation of glycogen synthase and consequent inhibition of the activity of the enzyme. This contributes to the ability of hepatocytes to release glucose stored in glycogen. CaMKII also phosphorylates the lipolytic enzyme, ATGL leading to increased triglyceride breakdown in hepatocytes. The release of fatty acids from triglycerides leads to increased fatty acid oxidation which increases the concentration of acetyl-CoA. Acetyl-CoA is the activator of pyruvate carboxylase which ultimately leads to increased gluconeogenesis.
Within the liver, glucagon activation of its receptor also results in increased amino acid transport into hepatocytes and increased amino acid metabolism as a means to provide the carbon skeletons necessary for glucose synthesis via gluconeogenesis. The increase in amino acid metabolism also results in an increase in urea production.
The uptake and oxidation of free fatty acids from the blood provides some of the energy required by hepatocytes to carry out gluconeogenesis. However, glucagon effects are also exerted on hepatic lipid metabolism such that fatty acid synthesis is inhibited and fatty acid oxidation is enhanced. One primary site of action of glucagon on fatty acid metabolism is via PKA-mediated phosphorylation and inhibition of the rate-limiting enzyme of fatty acid synthesis, acetyl-CoA carboxylase 1 (ACC1). The inhibition of ACC1 results in direct inhibition of fatty acid synthesis but also results in decreased production of its product, malonyl-CoA, which is an inhibitor of the outer mitochondrial membrane-localized fatty acid transporter, carnitine palmitoyltransferase 1 (CPT1). The de-repression of CPT1 allows higher rates of fatty acyl-CoA import into the mitochondria allowing for increased β-oxidation.
In addition to direct changes in metabolic pathway activity, exerted primarily via PKA-mediated phosphorylation events, the actions of PKA and CaMKII lead to changes in gene expression in these same cells. PKA phosphorylates numerous substrates including the transcription factor, cAMP response element-binding protein, CREB. Active PKA migrates to the nucleus where it phosphorylates CREB bound to cAMP-response elements (CRE) resulting in altered transcription. Important CREB target genes are those encoding the gluconeogenic enzymes, phosphoenolpyruvate carboxykinase, (PEPCK) and glucose-6-phosphatase.
As indicated, glucagon binds to its receptor in the liver and kidneys and to a lesser extent, in heart, endocrine pancreas, adrenal glands, spleen, adipose tissue, and cerebral cortex. Glucagon actions have been shown to result in increased activation of hormone-sensitive lipase, HSL in adipose tissue but there is controversy as to whether or not the effects in adipose tissue are direct or indirect. Nonetheless, the activation of HSL leads to increased release of fatty acids stored in the triglycerides in adipose tissue. The released fatty acids enter the circulation, are bound by albumin and transported to various tissues for oxidation. In the liver the oxidation of fatty acids is necessary to provide the energy needed for gluconeogenesis which is activated in liver in response to glucagon. Within the endocrine pancreas, the glucagon receptor is found on the β-cells that secrete insulin. The effect of glucagon on these cells is to stimulate insulin release so that there results a fine regulatory control over the overall level of circulating glucose.
Cortisol Regulation of Gluconeogenesis
Cortisol, an adrenal cortical glucocorticoid, exerts its effects as a result of binding to its receptor (glucocorticoid receptor, GR) which is a member of the nuclear receptor family of ligand-activated transcription factors. Cortisol activated GR binds to regulatory sequences in several genes encoding enzyme/proteins involved gluconeogenesis. These genes include those encoding pyruvate carboxylase (PC), cytosolic PEPCK (PCK1), fructose-1,6-bisphosphatase (FBP1), glucose 6-phosphatase (G6PC1), PFK2 (PFKFB1), and the glucose-6-phosphate transporter (SLC37A4). Details of transcriptional regulation of gluconeogenesis are covered below.
In addition to transcriptional effects in the liver that regulate hepatic gluconeogenesis, cortisol affects hepatic gluconeogenesis at the level of adipose tissue and skeletal muscle. These effects of cortisol are the result of stimulating the release of hepatic gluconeogenic substrates such as amino acids (skeletal muscle) and glycerol (adipose tissue).
Post-Translational Regulation of Gluconeogenic Enzyme Activity
Obviously the regulation of gluconeogenesis will be in direct contrast to the regulation of glycolysis. In general, negative effectors of glycolysis are positive effectors of gluconeogenesis. Regulation of the activity of PFK-1 and F1,6BPase is the most significant site for controlling the flux toward glucose oxidation or glucose synthesis. As described in control of glycolysis, this is predominantly controlled by fructose-2,6-bisphosphate (F2,6BP) which is a powerful negative allosteric effector of F1,6Bpase activity.
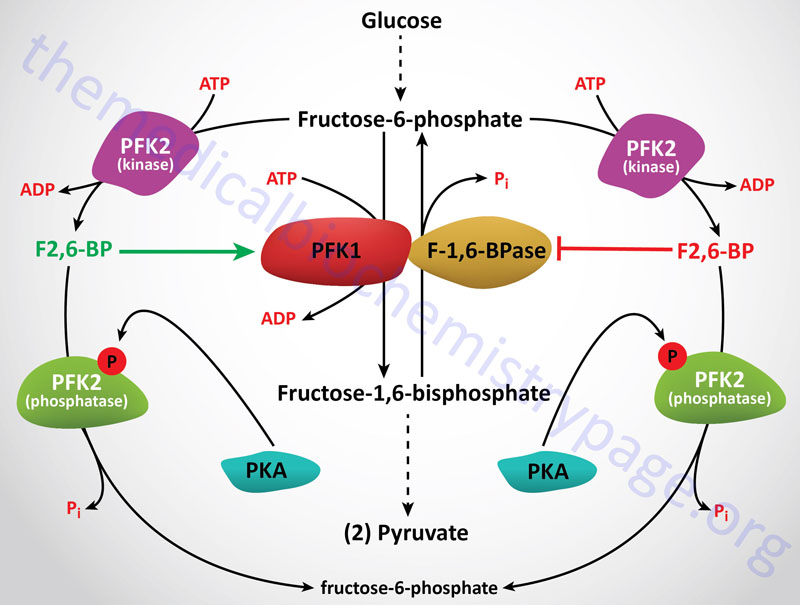
The level of F2,6BP will decline in hepatocytes in response to glucagon stimulation as well as stimulation by catecholamines. Each of these signals is elicited through activation of cAMP-dependent protein kinase (PKA). One substrate for PKA is PFK-2, the bifunctional enzyme responsible for the synthesis and hydrolysis of F2,6BP. When PFK-2 is phosphorylated by PKA it acts as a phosphatase leading to the dephosphorylation of F2,6BP with a concomitant increase in F1,6Bpase activity and a decrease in PFK-1 activity. Secondarily, F1,6Bpase activity is regulated by the ATP/ADP ratio. When this is high, gluconeogenesis can proceed maximally.
Gluconeogenesis is also controlled at the level of the pyruvate to PEP bypass. The hepatic signals elicited by glucagon or epinephrine lead to phosphorylation and inactivation of pyruvate kinase (PK) which will allow for an increase in the flux through gluconeogenesis. PK is also allosterically inhibited by ATP and alanine. The former signals adequate energy and the latter that sufficient substrates for gluconeogenesis are available. Conversely, a reduction in energy levels, as evidenced by increasing concentrations of ADP, lead to inhibition of both PC and PEPCK. Activation of PC occurs through interaction with acetyl-CoA. Indeed, PC is catalytically inactive in the absence of acetyl-CoA. This fact defines the role of acetyl-CoA as an obligate activator of PC. These regulations occur on a short time scale, whereas long-term regulation can be effected at the level of PEPCK. The amount of this enzyme increases in response to prolonged glucagon stimulation. This situation would occur in a starving individual or someone with an inadequate diet.
Regulation of PEPCK by Post-Translational Modification
The function of PEPCK is, in part, controlled by its state of lysine acetylation. Under conditions of high glucose the cytosolic PEPCK enzyme (PEPCK-c encoded by the PCK1 gene) is acetylated by the p300/CBP acetyltransferase. Under conditions of low glucose (low energy) the PEPCK-c enzyme is deacetylated by the SIRT1 deacetylase. When PEPCK-c is acetylated it functions as an anaplerotic enzyme converting PEP to OAA which allows maximal TCA cycle activity. The cytosolic OAA can be oxidized to malate which can be transported to the mitochondria, in a reversal of the malate-aspartate shuttle, for incorporation into the TCA cycle. When PEPCK-c is deacetylated it functions to convert OAA to PEP driving gluconeogenesis.
The post-translational modification referred to as Neddylation is a mechanism that adds the protein NEDD8, a ubiquitin-like protein, to target lysine residues in numerous proteins. The primary targets for NEDDylation are the cullin proteins that serve as molecular scaffolds for the RING domain family of E3 ubiquitin ligases which together form the CRL family of E3 ligase complexes.
Recent evidence has demonstrated the the cytosolic PEPCK (PEPCK-c) is a target for NEDDylation. There are three lysine residues in the PEPCK-c protein, K278, K342, and K387, that have been found to be NEDDylated. The extent of PEPCK NEDDylation in the liver is regulated by nutrient status, being highest in the fasted state. The NEDDylation of PEPCK-c is required for it gluconeogenic activity indicating that this post-translational modification is of critical significance to overall hepatic glucose output, particularly in the fasted, nutrient deprived state.
Role of AMPK in Regulation of Gluconeogenesis
The master metabolic regulating kinase, AMP-activated protein kinase (AMPK), is a significant regulator of hepatic gluconeogenesis. AMPK is activated when cellular energy levels are low which results in increased concentrations of AMP. Activated AMPK phosphorylates numerous substrates that result in a switching off ATP-consuming anabolic pathways and switching on ATP-generating catabolic pathways. One target for AMPK phosphorylation is the transcription factor, CREB regulated transcription coactivator 2 (CRTC2). Phosphorylated CRTC2 is sequestered in the cytosol resulting in reduced transcription of several gluconeogenic genes as well as the gene encoding the transcriptional coactivator, PGC-1α as discussed below. AMPK also phosphorylates class II histone deacetylases resulting in their exclusion from the nucleus and a consequent reduction the expression of gluconeogenic genes.
Transcriptional Regulation of Gluconeogenesis
Within the liver the actions of glucagon and epinephrine (via β2-adrenergic receptors) result in increased levels of cAMP and subsequent activation of gluconeogenesis. Insulin action in the liver exerts the opposite effect, an inhibition of gluconeogenesis. The mechanisms by which insulin turns off gluconeogenesis are complex and include a reduction in the level of cAMP via the insulin-mediated activation of phosphodiesterase (PDE3B) which hydrolyzes cAMP to AMP. In addition, insulin activation of protein phosphatase 1 (PP-1) results in removal of phosphate from enzymes that promote gluconeogenesis over glycolysis such as PFK2 and pyruvate kinase.
The primary transcriptional control of gluconeogenesis occurs at the level of the genes encoding pyruvate carboxylase (PC), phosphoenolpyruvate carboxykinase (PEPCK), fructose-1,6-bisphosphatase (F1,6BPase), and glucose 6-phosphatase (G6Pase; specifically the G6PC1 gene).
At the level of the regulation of genes involved in gluconeogenesis, cAMP activates PKA which then phosphorylates numerous substrates. One of the PKA substrates is the transcription factor CREB1 [cAMP response element (CRE)-binding protein 1] which it phosphorylates at Ser133. As indicated above, humans express seven genes in the CREB subfamily of the basic leucine zipper (bZIP) family of transcription factors. The seven CREB genes are identified as CREB1, CREB3, CREB5, and CREB3-like 1, 2, 3, and 4 (CREB3L1, CREB3L2, CREB3L3, and CREB3L4). The CREB3L3 encoded protein is commonly identified as CREBH. The CREB1 encoded proteins are most closely related in structure and function to two additional transcription factors called cAMP response element modulator (CREM) and activating transcription factor 1 (ATF-1). Another member of the activating transcription factor (ATF) family, ATF-4, was originally identified as CREB2.
When phosphorylated, CREB1 binds to the cAMP response element (CRE) of target genes resulting in the recruitment of the coactivators CRTC2 (CREB regulated transcription coactivator 2) and p300/CBP. The p300/CBP complex activates gene expression through its intrinsic histone acetyltransferase activity and through recruitment of other coactivator molecules. The coactivator CBP is a target of insulin-dependent phosphorylation at Ser436. This residue, which is adjacent to the CREB-binding domain (CREB-BD), is phosphorylated by a phosphatidylinositol-3-kinase (PI3K)−dependent insulin signaling pathway, and is not conserved in the related cofactor p300.
The transcriptional coactivator PGC-1α (peroxisome proliferator-activated receptor-γ coactivator-1α) also functions as a central regulator of gluconeogenesis by binding to several transcription factors, including the glucocorticoid receptor, hepatic nuclear factor 4α (HNF4α), and FOXO1 (a forkhead transcription factor family member) to the promoter regions of several genes encoding gluconeogenic enzymes.
Regulation of hepatic gluconeogenic gene expression, via phosphorylation of transcription factors, is a major mechanism of control. Phosphorylation of both CRTC2 and FOXO1 represses their ability to activate gluconeogenic gene expression. Insulin signaling antagonizes gluconeogenesis through stimulation of the phosphorylation of FOXO1 which results in its subsequent exclusion from the nucleus. Given that FOXO1 regulates not only the expression of gluconeogenic genes but also it own gene, its exclusion from the nucleus leads to enhanced repression of gluconeogenesis.
The phosphorylation of CRTC2 also results in its exclusion from the nucleus. In the case of CRTC2, when it is excluded from the nucleus it is targeted for ubiquitylation and proteasomal degradation. The major kinase regulating the nuclear exclusion of both CRTC2 and FOXO1 is nemo-like kinase (NLK) although AMPK has also been shown to phosphorylate CRTC2. Expression of PGC-1α is also regulated by CREB through a well-defined CRE present in the promoter region of the gene (PPARGC1) encoding PGC-1α.
The clinical significance of the role of NLK in the regulation of hepatic gluconeogenesis can be appreciated from the fact that expression of the NLK gene is downregulated in individuals with diabetes. In animal models of type 2 diabetes the expression of NLK is also repressed. Forced expression of the NLK gene in these animal models results in significant reductions in plasma glucose levels and increased glucose tolerance.
The transcription factor identified as p52 has been shown to be involved in the regulation of hepatic gluconeogenesis. The p52 protein is derived from the mRNA transcribed from the NFKB2 gene which encodes a subunit of the NFκB (nuclear factor-kappa-B) complex. The full-length p100 protein encoded by the NFKB2-derived mRNA is post-translationally processed into the active p52 form. Depending upon the proteins to which p52 dimerizes it can function as a transcriptional activator or repressor. One important gene whose transcription is repressed by p52 is PDE4B which encodes a cAMP-specific phosphodiesterase. The inhibition of PDE4B expression in hepatocytes leads to cAMP accumulation which in turn leads to enhancement of glucagon responses. The diabetes drug metformin exerts a portion of its effect on hepatic gluconeogenesis by inhibiting the activation of p52.
Role of Malonyl-CoA in Regulation of Hepatic Gluconeogenesis
Malonyl-CoA represents a critical intermediate in the process of de novo fatty acid synthesis. Synthesis of malonyl-CoA is catalyzed by either of two related biotin-dependent enzymes, acetyl-CoA carboxylase 1 (ACC1; encoded by the ACACA gene) and acetyl-CoA carboxylase 2 (ACC2; encoded by the ACACB gene). The ACC1 enzyme is localized to the cytosol and constitutes the rate-limiting enzyme of fatty acid synthesis. The ACC2 enzyme is localized to the outer mitochondrial membrane where the malonyl-CoA that is generated by the enzyme can directly inhibit the activity of the fatty acid transport enzyme, carnitine palmitoyltransferase 1, CPT1. The activity of ACC2 is, therefore, critical for the prevention of the oxidation of newly synthesized palmitic acid, the end-product of de novo fatty acid synthesis.
Increased fatty acid oxidation, as occurs in the liver in response to fasting, contributes to enhanced gluconeogenesis by providing the acetyl-CoA required for activation of pyruvate carboxylase. The conversion of pyruvate to phosphoenolpyruvate (PEP), catalyzed by pyruvate carboxylase, not only contributes to enhanced gluconeogenesis but also provides anaplerotic substrates for the TCA cycle contributing to overall energy production, a consequence required to drive gluconeogenesis.
As indicated above, fasting decreases the activity of ACC1 and ACC2 as a result of their phosphorylation by PKA, which is activated in response to the activation of the glucagon receptor, and by AMPK which is activated as the ATP/ADP ratio decreases. These regulatory effects are reversed in response to food intake as a result of the actions of insulin receptor signaling.
Malonyl-CoA synthesis therefore, represents an acute process that controls the overall rate of gluconeogenesis, as well as and TCA cycle activity, by controlling the delivery of fatty acids into the mitochondria for oxidation. More broadly, by controlling the rate of fatty acid oxidation, malonyl-CoA not only regulates hepatic gluconeogenesis but also results in the regulation of the redox state of hepatocytes.
Role of Intestinal Gluconeogenesis and Control of Feeding Behaviors
The gut, in particular the small intestine, plays a critical role in the uptake and delivery of glucose from the diet. As such, the gut plays a central role in the overall regulation of glucose homeostasis. Glucose uptake from the lumen of the gut and trans-epithelial transport to the portal circulation had been shown to occur via the action of two distinct glucose transporters. First, glucose is taken up from the intestinal lumen through the action of the sodium-dependent glucose transporter-1 (SGLT-1) present in the apical (luminal) membrane . The glucose is then transported into the superior mesenteric blood via the action of the facilitated glucose transporter, GLUT2, present in the basolateral membrane. The superior mesenteric vein terminates by uniting with the splenic vein and then forms the portal vein.
Evidence has also indicated that GLUT2 present in the apical membrane of enterocytes was involved in glucose uptake. However, GLUT2 is not present in the apical membrane in the absence of a glucose load. The mechanism of GLUT2 presentation in the apical membrane involves a glucose-induced translocation of GLUT2 to this membrane. Thus, glucose uptake by the small intestine enhances additional uptake by promoting presentation of an additional transporter in the apical membrane.
The small intestine also utilizes glucose, obtained from the diet or from the blood, for energy production. Recently it was shown that the intestine is able to utilize glutamine for energy with the same efficiency as glucose. Indeed, glutamine has been considered to be a major energy substrate for this organ.
Of additional, significance, and only recently having been determined, is the role of intestinal gluconeogenesis in overall endogenous glucose production (EGP). A little over 10 years ago, molecular analysis allowed for the characterization of the expression of glucose-6-phosphatase (G6Pase) within enterocytes of the small intestine. Expression of G6Pase thus, confers upon the intestine, the ability to carry out gluconeogenesis. Now it is known that glutamine serves as the major precursor of glucose formed within the small intestine. One major source of intestinal glutamine is the transamination reactions occurring in other tissues as a mechanism to ultimately deliver waste nitrogen, derived from nucleotide and amino acid catabolism, to the liver for incorporation into urea. Glutamine is also derived from protein digestion in the small intestines.
The genes for both G6Pase and the cytosolic form of phosphoenolpyruvate carboxykinase (PEPCK-c) are controlled by insulin in the small intestine similarly to the regulation of these genes in the liver. The presence of G6Pase within the small intestine also plays a role in the export of glucose to the portal circulation. This can either be dietary glucose, glucose released from intestinal glycogen stores, or glucose produced via gluconeogenesis. This glucose export mechanism is dependent on the previous phosphorylation of glucose by hexokinases followed by G6Pase-mediated dephosphorylation. Within the small intestines the major hexokinases are hexokinase I and II, but small amounts of hexokinase III and IV (more commonly called glucokinase) are also present.
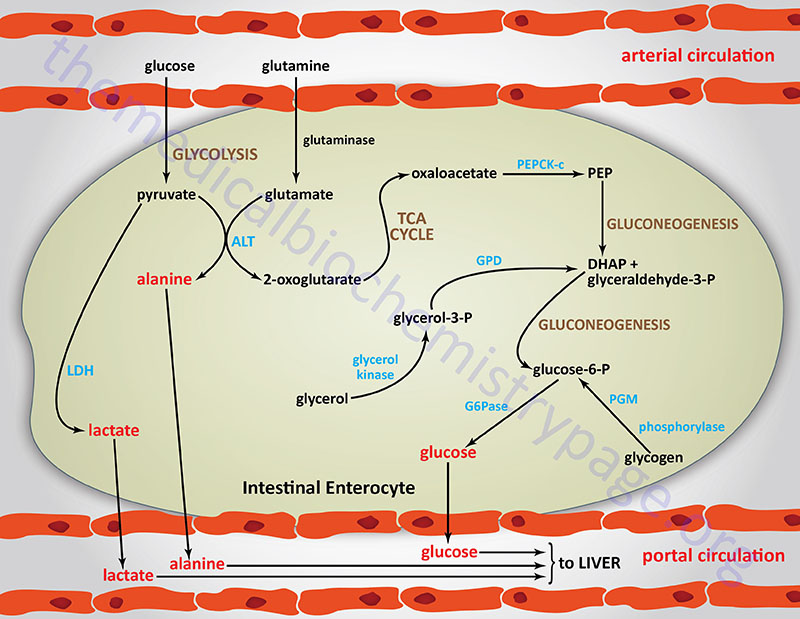
The importance of intestinal gluconeogenesis, to overall EGP, has been demonstrated both in experimental animals (mice with specific knockout of PEPCK-c in the liver) and in humans in the anhepatic phase during liver transplantation. In mice without hepatic PEPCK-c there is an efficient adaptation to fasting conditions such that blood glucose levels decrease by only 30%. In addition, in these mice, and humans undergoing liver transplant, there occurs a significant increase in plasma glutamine concentration. These observations stressed the likely role of the kidney and/or intestine in glucose production, because glutamine is a major glucose precursor in the kidney and the small intestine, but not in the liver. The role of the intestine in this glucose control was demonstrated by the fact that in these experimental conditions there is no observable difference in glucose concentration between arterial and portal blood.
During periods of fasting the small intestine accounts for approximately 20% of EGP by 48hrs and up to 35% by 72hrs. However, expression of the key gluconeogenic genes, G6Pase and PEPCK-c, is dependent on plasma insulin concentrations, and these do not change throughout these time frames of fasting. Yet expression of both of these genes is seen to increase within intestinal cells between 24 hrs and 48 hrs of the initiation of fasting. The promoter region of the G6PC1 gene, containing the presumed TATA box (TATAAAA, located -31 to -25 bp upstream of the transcription start site), also constitutes a putative binding site for transcription factors of the caudal-related homeobox (CDX) family. In adult mammals, the CDX genes are exclusively expressed in the gut, where they are involved in the differentiation of both the crypt-villus and anteroposterior axis. The TATAAAA element in the G6PC1 gene is indeed responsible for its transactivation by CDX1 (but not CDX2) since disruption of the sequence strongly blunts basal transcription and CDX1 transactivation of the gene.
Intestinal Gluconeogenesis and Feeding Behaviors
That intestinal gluconeogenesis is indeed crucial in the control of food intake by dietary protein was established with the use mice in which expression of the G6Pase gene was specifically and conditionally abolished in the intestine. When these mice are fed a protein-rich, carbohydrate-free diet they do not exhibit a decrease in their level of food intake such as is seen in control mice on the same diet. The same loss of satiety induction by protein-rich diets or portal glucose infusion is seen in animals whose portal vein afferent nerve connections are chemically or surgically destroyed. Afferent nerves send signals from body locations to the brain. These types of studies demonstrate that portal sensing of intestinal gluconeogenesis is a key mechanism in the satiety effect induced by dietary protein.
Role of Renal Gluconeogenesis
Although the liver has the critical role of maintaining blood glucose homeostasis and therefore, is the major site of gluconeogenesis, the kidney plays an important role. During periods of severe hypoglycemia that occur under conditions of hepatic failure, the kidney can provide glucose to the blood via renal gluconeogenesis. In the renal cortex, glutamine is the preferred substance for gluconeogenesis.
Glutamine is produced in high amounts by skeletal muscle during periods of fasting as a means to export the waste nitrogen resulting from amino acid catabolism. Through the actions of transaminases, a mole of waste ammonia is transferred to 2-oxoglutarate (α-ketoglutarate) via the glutamate dehydrogenase catalyzed reaction yielding glutamate. Glutamate is then a substrate for glutamine synthetase which incorporates another mole of waste ammonia generating glutamine (go to the Nitrogen Metabolism and the Urea Cycle page for more details). The glutamine is then transported to the kidneys where the reverse reactions occur liberating the ammonia and producing 2-oxoglutarate which can enter the TCA cycle and the carbon atoms diverted to gluconeogenesis via oxaloacetate. This process serves two important functions. The ammonia (NH3) that is liberated spontaneously ionizes to ammonium ion (NH4+) and is excreted in the urine effectively buffering the acids in the urine. In addition, the glucose that is produced via gluconeogenesis can provide the brain with critically needed energy.
Regulation of Blood Glucose Levels
If for no other reason, it is because of the demands of the brain for oxidizable glucose that the human body exquisitely regulates the level of glucose circulating in the blood. This level is maintained in the range of 5mM (90mg/dL) during normal between meal fasting.
Nearly all carbohydrates ingested in the diet are converted to glucose following transport to the liver. Catabolism of dietary or cellular proteins generates carbon atoms that can be utilized for glucose synthesis via gluconeogenesis. Additionally, other tissues besides the liver that incompletely oxidize glucose (predominantly skeletal muscle and erythrocytes) provide lactate that can be converted to glucose via gluconeogenesis.
Maintenance of blood glucose homeostasis is of paramount importance to the survival of the human organism. The predominant tissue responding to signals that indicate reduced or elevated blood glucose levels is the liver. Indeed, one of the most important functions of the liver is to produce glucose for the circulation. Both elevated and reduced levels of blood glucose trigger hormonal responses to initiate pathways designed to restore glucose homeostasis. Low blood glucose triggers release of glucagon from pancreatic α-cells. High blood glucose triggers release of insulin from pancreatic β-cells and inhibition of glucagon release from α-cells.
Additional hormonal signals, such as via ACTH and growth hormone, released from the pituitary, act to increase blood glucose by inhibiting its uptake by extrahepatic tissues such as adipose tissue and skeletal muscle. Glucocorticoids also act to increase blood glucose levels by inhibiting glucose uptake (also primarily at the level of adipose tissue and skeletal muscle) and by stimulation of gluconeogenesis. Cortisol, the major glucocorticoid released from the adrenal cortex, is secreted in response to the increase in circulating ACTH. Within the liver, cortisol binding to the glucocorticoid receptor (GR), results in transcriptional activation of the PEPCK gene, thereby, resulting in increased rates of gluconeogenesis and glucose output to the blood.
The adrenal medullary hormone, epinephrine, stimulates production of glucose by activating hepatic glycogenolysis and gluconeogenesis. These effects are exerted via the presence of α1 and β2 adrenergic receptor subtypes on hepatocytes. Epinephrine also exerts an effect on skeletal muscle glycogenolysis in response to stressful stimuli. Within skeletal muscle, epinephrine exerts its effects primarily through activation of the β2 adrenergic receptor but a small percentage of the total adrenergic receptor subtypes in skeletal muscle includes the β1 subtype (7–10% of the total).
The significance of adrenergic (epinephrine primarily) receptor function to the control of blood glucose can be seen by the consequences of several identified receptor mutations. For example, mutations in either the β1 or the β3 adrenergic receptors are highly correlated to insulin resistance associated with type 2 diabetes. In addition, mutations in all three β-adrenergic receptors are associated with hyperlipidemia (which exacerbates the hyperglycemia of diabetes) as well as the associated pathophysiology of the metabolic syndrome. Mutations in all three β-receptors are also associated with increased risk for obesity.
Glucagon binding to its receptors on the surface of liver cells triggers an increase in cAMP production leading to an increased rate of glycogenolysis by activating glycogen phosphorylase via the PKA-mediated cascade. This is the same response hepatocytes have to epinephrine binding to the β2-adrenergic receptors on hepatocytes. The resultant increased levels of G6P in hepatocytes is hydrolyzed to free glucose, by glucose-6-phosphatase, which then diffuses to the blood. The glucose enters extrahepatic cells where it is re-phosphorylated by hexokinase. Since all tissues, excluding liver, kidney, and small intestine, lack glucose-6-phosphatase, the glucose-6-phosphate product of hexokinase is retained and oxidized by these tissues.
In opposition to the cellular responses to glucagon, cortisol, and epinephrine, insulin stimulates extrahepatic uptake of glucose from the blood and inhibits glycogenolysis in extrahepatic cells and conversely stimulates glycogen synthesis. As the glucose enters hepatocytes it binds to and inhibits glycogen phosphorylase activity. The binding of free glucose stimulates the dephosphorylation of phosphorylase thereby, inactivating it. Why is it that the glucose that enters hepatocytes is not immediately phosphorylated and oxidized? Hepatocytes express the isoform of hexokinase called glucokinase. Glucokinase has a much lower affinity for glucose than does hexokinase. Therefore, it is not fully active at the physiological ranges of blood glucose. Additionally, glucokinase is not inhibited by its product G6P, whereas, hexokinase is inhibited by G6P.
Hepatocytes, unlike most other cells, are essentially freely permeable to glucose and are, therefore, not directly affected by the action of insulin at the level of increased glucose uptake. When blood glucose levels are low, the liver does not compete with other tissues for glucose since the extrahepatic uptake of glucose is stimulated in response to insulin. Conversely, when blood glucose levels are high extrahepatic needs are satisfied and the liver takes up glucose for conversion into glycogen for future needs. Under conditions of high blood glucose, liver glucose levels will be high and the activity of glucokinase will be elevated. The G6P produced by glucokinase is rapidly converted to G1P by phosphoglucomutase, where it can then be incorporated into glycogen.
Inherited Disorders of Gluconeogenesis
Inherited mutations in several genes are associated with defective gluconeogenesis and result in the common pathology of hypoglycemia. Inherited mutations that are associated with disorders of gluconeogenesis include those in the genes encoding fructose-1,6-bisphosphatase (FBP1), pyruvate carboxylase (PC), and phosphoenolpyruvate carboxykinase (PCK1 and PCK2). The consequences of mutations in the FBP1 gene are described in the Fructose Metabolism Disorders page. The consequences of mutations in the PC gene are described in the Pyruvate Carboxylase Deficiency page.
Mutations in both genes (PCK1 and PCK2) encoding PEPCK enzymes have been identified and are associated with defective gluconeogenesis. Inheritance of either form of PEPCK deficiency is autosomal recessive. In addition to severe fasting hypoglycemia, mutations in PCK1 are associated with ketonuria, dicarboxylic aciduria, and urea cycle dysfunction. Mutations in the PCK2 gene most often lead to fatal liver failure associated with massive fatty infiltration. The fat infiltration is observed in the kidneys of PCK2 deficient patients as well.