
 deficiency")
Last Updated: July 13, 2026
Introduction to G6PD Deficiency
Deficiencies in glucose-6-phosphate dehydrogenase (G6PDH; encoded by the G6PD gene) are inherited as X-linked recessive disorders. Glucose-6-phosphate dehydrogenase is found in all cells and is responsible for the first reaction of the pentose phosphate pathway in which glucose-6-phosphate is oxidized to 6-phosphogluconolactone with concomitant production of NADPH.
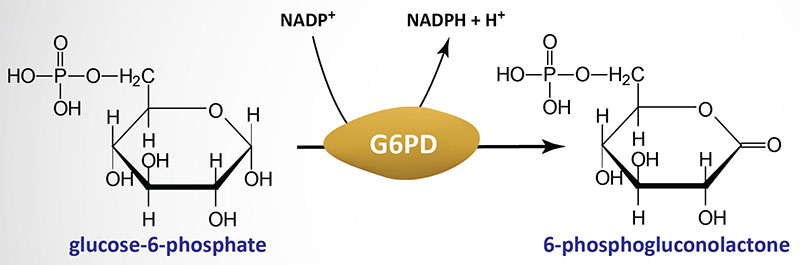
Humans express two genes that generate glucose-6-phosphate dehydrogenase activity, the X-linked G6PD gene and an autosomal gene identified as H6PD (hexose-6-phosphate dehydrogenase/glucose 1-dehydrogenase). Expression of the H6PD gene occurs in all tissues except red blood cells.
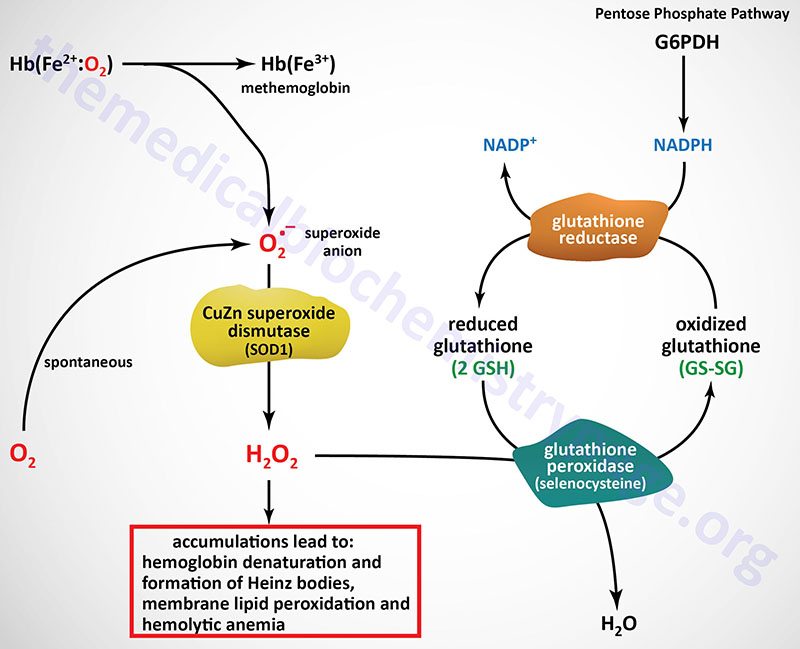
The NADPH produced via the pentose phosphate pathway, as well as through several additional pathways, is required for a variety of reductive biosynthetic reactions as well as for the regeneration of the reduced form of glutathione (GSH) from the oxidized (GSSG) form. GSH is essential for the detoxification of hydrogen peroxide (H2O2) and therefore, cellular defense against the oxidizing effects of H2O2 is absolutely dependent upon the generation of NADPH. GSH converts H2O2 to H2O via the action of glutathione peroxidase and requires one mole of NADPH per mole of H2O2. The critical need for production of NADPH, via the pentose phosphate pathway, is especially true in red blood cells because there are no other NADPH-producing reactions in these cells and they are extremely sensitive to oxidative damage.
Drugs Triggering Hemolytic Anemia in G6PD Deficiency
A deficiency of G6PD is most likely in individuals experiencing episodes of acute hemolytic anemia following an infection, consumption of fava beans, or exposure to medications that are known to be oxidizing. Examples of drugs that are in the latter class include the anti-malarial drugs dapsone and primaquine, as well as rasburicase which is normally used in the treatment tumor lysis syndrome in patients undergoing chemotherapy for hematologic cancers, but has also been used to reduce the hyperuricemia associated with gout. The sulfonamides used to treat bacterial infections can also trigger hemolytic anemia in G6PD deficiency.
Red cell deficiency in G6PD is the basis of favism. Favism is a term relating to a hemolytic anemia that results from the ingestion of the fava bean (Vicia faba) also known as broad beans. With respect to favism, the occurrence of acute hemolysis after ingestion of the fava bean was recognized as far back as the time of the Greek mathematician, Pythagoras. Analysis of extracts from fava beans demonstrated that the toxic compounds are the pyrimidine aglycones, divicine and isouramil, that are derived from the pyrimidine glycosides, vicine and convicine, respectively. Both divicine and convicine rapidly oxidize GSH to GSSG.
Molecular Biology of G6PD Deficiency
The G6PD gene that encodes glucose-6-phosphate dehydrogenase is located on the X chromosome (Xq28) about 1 Mb (million base pairs) from the telomeric end. The gene spans 18 kb and is composed of 14 exons that generate three alternatively spliced mRNAs. These three mRNAs encode two distinct G6PD proteins identified as isoform a (545 amino acids) and isoform b (515 amino acids).
The first exon of the G6PD gene is a non-coding exon with the translational initiation codon present in exon 2. The 545 amino acid isoform a protein is inactive but post-translational processing results in a 515 amino acid functional protein containing an acetylated alanine residue at the N-terminus. Biologically active G6PD is functional as either a homodimer or a homotetramer and both forms co-exist in equal proportions at neutral pH.
Deficiencies in G6PD are the most commonly inherited enzyme deficiencies (enzymopathies) world wide with estimates of greater than 400 million affected individuals. The incidence of deficiency approaches 25% of the population of persons of Mediterranean, tropical African, and tropical and subtropical Asian descent. In fact, the incidence of G6PD deficiency is so high in some populations that the occurrence of homozygous females is not at all rare as would normally be expected for an X-linked disorder.
The inheritance of G6PD deficiency is clinically classified as an X-linked recessive disorder, however, because heterozygous females can develop hemolytic episodes the disorder is not truly recessive in the Mendelian sense. The phenomenon of symptom manifesting heterozygous females is explained by the co-existence of populations of G6PD positive and negative cells in the same female as a result of X-chromosome inactivation.
Deficiency in G6PD represents one of the most genetically heterogeneous disorders. There are over 400 different variants of G6PD defined by their diverse biochemical characteristics. To date a total of 130 different pathogenic variants have been identified in the G6PD gene. Surprisingly, only five in-frame deletions have been identified and no large deletions or insertions have been found. This fact is likely explained by the observation that in mouse models of G6PD deficiency, complete loss of enzyme activity is associated with embryonic lethality.
G6PD deficiency cannot be classified by a single pathogenic variants but is manifest as a consequence of numerous structural allelic variants. In addition, G6PD deficiencies are also classifiable by a variety of physicochemical parameters including chromatographic properties, thermostability, pH dependence, KM for glucose-6-phosphate, and KM for NADP+. G6PD gene variants are currently divided into five classifications dependent upon enzyme activity and clinical manifestations. In addition, the deficiencies are classified as to whether or not they are sporadic or polymorphic.
The five classifications are:
- Class 1: enzyme deficiency with chronic nonspherocytic hemolytic anemia
- Class 2: severe enzyme deficiency, less than 10% of normal activity
- Class 3: moderate to mild enzyme deficiency, 10-60% of normal activity
- Class 4: very mild or no enzyme deficiency, at least 60% of normal activity
- Class 5: increased enzyme activity
Pathogenic variants in the G6PD gene that result in non-spherocytic hemolytic anemia have all been found to cluster near the carboxy terminal end of the protein, whereas, the pathogenic variants that result in clinically mild symptoms are clustered near the amino terminal end of the protein. Nearly all G6PD pathogenic variants are single nucleotide missense variants resulting in the substitution of a single amino acid. In most instances of pathogenic variants in the G6PD gene the resulting disease it is due to protein instability as a result of the amino acid substitution.
Clinical Features of G6PD Deficiency
The vast majority of individuals that harbor a pathogenic variant of the G6PD gene go their entire lives without knowing they carry a pathogenic variant. The acute hemolysis, that is the only common clinical manifestation associated with pathogenic variants in the G6PD gene, can rapidly be compensated for and thus, may remain undetectable.
The most common symptoms of G6PD deficiency are neonatal jaundice and acute hemolytic anemia. The acute hemolytic attacks are the result of reduced antioxidant capacity of red blood cells with the result being membrane lipid peroxidation and consequent cell lysis.
The accumulation of reactive oxygen species in G6PD deficiency also leads to aberrant oxidation of the cysteine sulfhydryl groups in the globin proteins in hemoglobin. This results in significant cross-linking of hemoglobin tetramers which can be visualized microscopically. The cross-linked hemoglobin forms what is referred to histologically as Heinz bodies. The presence of Heinz bodies is diagnostic for G6PD deficiency versus erythrocyte pyruvate kinase deficiency which is not associated with the formation of these inclusion bodies.
Treatment of G6PD Deficiency
In patients with G6PD deficiency the avoidance of drugs that induce hemolytic anemia, as well as fava beans, is to be avoided. In the case of infections, which can trigger acute hemolytic crisis, it is important to provide fluids to prevent hemodynamic shock which can precipitate acute renal failure. In children, more often than in adults, blood transfusion may be required.