
Last Updated: July 13, 2026
Introduction to Fucosidosis
Fucosidosis belongs to the family of disorders identified as lysosomal storage disorders. Fucosidosis is an autosomal recessive disorder that results from deficiencies in the fucosidase, alpha-L-1 (α-fucosidase-1) gene. This disorder is characterized by the lysosomal accumulation of a variety of glycoproteins, glycolipids, and oligosaccharides that contain fucose moieties. These glycoconjugates accumulate in the lysosomes as a consequence of defects in the lysosomal hydrolase, α-fucosidase.
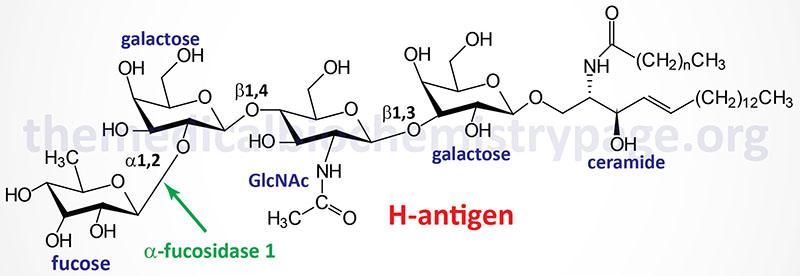
Fucosidosis is the consequence of faulty degradation of both sphingolipids and polysaccharides. Major accumulation of the H-antigen (a member of the ABO blood group antigens), a glycolipid, is seen primarily in the liver of fucosidosis patients.
Molecular Biology of Fucosidosis
The α-fucosidase-1 enzyme is encoded by the FUCA1 gene. The FUCA1 gene is located on chromosome 1p36.11 and is composed of 10 exons that encode a 466 amino acid preprotein. The α-fucosidase-1 activity is associated with a homotetrameric glycoprotein complex.
As of 2025 there have been 55 different pathogenic variants in the FUCA1 gene identified as resulting in fucosidosis. Most of the pathogenic variants in the FUCA1 gene associated with fucosidosis result in unstable or defective mRNAs. The most common pathogenic variant is a nonsense variant where a glutamine (Q) codon is changed to a stop codon at amino acid position 422. This pathogenic variant is designated as: Q422X.
Clinical Features of Fucosidosis
The spectrum of clinical findings in fucosidosis patients has allowed for a division of patients into to classes of disease. Type 1 is the more severe infantile form which manifests between 3 and 18 months of age. Type 2 is a milder form that manifests between 1 and 2 years of age. Both forms of fucosidosis are associated with intellectual impairment and coarse facial features.
Type 1 patients also have severe dystosis multiplex, a constellation of skeletal abnormalities characterized by an enlarged skull, thickened calvarium, premature closure of lamboid and sagittal sutures, shallow orbits, enlarged J-shaped sella turcica (a saddle-shaped skull structure into which sits the bottom of the pituitary gland), and abnormal spacing of the teeth with dentigerous cysts. There is anterior hypoplasia of the lumbar vertebrae, the long bone diaphyses are enlarged and an irregular appearance of the metaphyses. The epiphyseal centers not well developed, the pelvis is poorly formed with small femoral heads and coxa valga. The clavicles are short, thick and irregular and the ribs are oar shaped. Phalanges are shortened and trapezoidal in shape. In addition, type I patients show a marked increase in the content of sodium chloride in their sweat.
The clinical symptoms of type 2 fucosidosis are similar to, but milder than, those of type 1 patients and as indicated onset of symptoms occurs later than in type 1 disease. The major clinical symptom that distinguishes type 2 fucosidosis from type I is the presence of angiokeratoma corporis diffusum (hyperkeratinized deep-red to blue-black skin lesions) that are identical to those seen in Fabry disease. Because the major accumulating glycoconjugate in fucosidosis patients is the blood group H-antigen it is intriguing to speculate, but the evidence is not clear at this time, that blood type may affect the course of the disease.