
Last Updated: November 9, 2025
Introduction to Farber Lipogranulomatosis
Farber lipogranulomatosis is a rare autosomal recessive disease that belongs to the family of disorders identified as lysosomal storage disorders. This disorder is characterized by the lysosomal accumulation of ceramides. Farber lipogranulomatosis results from defects in the gene encoding the lysosomal hydrolase: acid ceramidase.
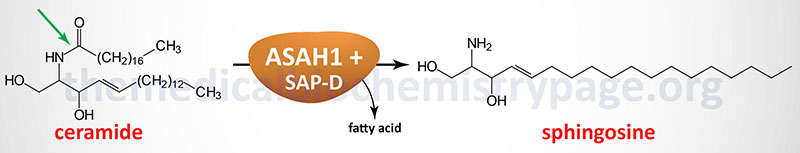
Acid ceramidase catalyzes the hydrolysis of ceramides generating sphingosine and a free fatty acid.
Molecular Biology of Farber Lipogranulomatosis
Acid ceramidase is encoded by the ASAH1 (N-acylsphingosine amidohydrolase 1) gene. The ASAH1 gene is located on chromosome 8p22 and is composed of 17 exons that generate four alternatively spliced mRNAs. These four mRNAs each encode a distinct isoform of acid ceramidase, isoforms a, b, c, and d. Isoform a (395 amino acids) has a different N-terminus compared to isoform b (411 amino acids), which is the longest of the three isoforms. Isoform c (389 amino acids) contains the same N- and C-termini that are included in isoform b, but the overall protein is shorter than the b isoform. Isoform d is a protein of 330 amino acids.
Acid ceramidase is a pseudoheterodimeric protein composed of a nonglucosylated α-peptide and a glucosylated β-peptide. Both peptides are encoded in the acid ceramidase mRNAs and the primary translation products are posttranslationally processed generating the two peptides.
Mutations in the ASAH1 gene resulting in Farber lipogranulomatosis include point mutations, and splice-site mutations. As of 2025 there have been 65 different pathogenic mutations identified in the ASAH1 gene causing Farber lipogranulomatosis.
Clinical Features of Farber Lipogranulomatosis
Symptoms of Farber lipogranulomatosis commonly appear during the first months after birth. The clinical manifestations are characterized by painful and progressively deformed joints, progressive hoarseness due to laryngeal involvement and subcutaneous nodules particularly over the joints. The tissues in afflicted individuals contain granulomatous and lipid-laden macrophages. The liver, spleen, lungs and heart are particularly affected with central nervous system involvement resulting in the progressive degeneration in psychomotor development. Farber lipogranulomatosis is a rapidly progressing disease often leading to death before 2 years of age. There is currently no effective therapy for Farber lipogranulomatosis. There are several clinical phenotypes associated with acid ceramidase deficiencies giving rise to seven subtypes of Farber lipogranulomatosis.
Type 1
Type 1 is the classic Farber lipogranulomatosis. The characteristic clinical presentation of type 1 disease is painful swelling of the joints, especially the ankle, wrist, elbow and knee, and a hoarse cry. These symptoms are evident as early as 2 weeks of age. Evaluation of neurological involvement is difficult as movement causes extreme pain in afflicted infants. In many cases these patients will have an additional symptom characteristic of Tay-Sachs disease which is the “cherry-red spot” on the fundus of the eye.
Type 2
Type 2 is the “intermediate” form of the disease. Type 2 patients have longer survival times than do type 1 infants. In addition the neurologic involvement is much more mild than in type 1.
Type 3
Type 3 is the “mild” form of the disease. Type 3 patients have longer survival times than do type 1 infants. In addition the neurologic involvement is much more mild than in type 1.
Type 4
Type 4 is referred to as the “neonatal-visceral” form of the disease. Infants are extremely ill in the neonatal period. These patients will present with severe hepatosplenomegaly. In the severest cases, type 4 neonates will present as hydrops fetalis (severe fluid accumulation, usually in the brain) and die within days of birth. Unlike type 1 patients, infants with type 4 Farber lipogranulomatosis do not present with the characteristic features of deformed painful joints, thus, requiring biochemical assay for definitive diagnosis.
Type 5
Type 5 is referred to as “neurologic progressive”. As the name implies, the most striking clinical feature of type 5 disease is a progressive neurological deterioration accompanied by seizures. Joint involvement is evident but to a lesser degree than in type 1 disease. Type 5 patients also exhibit the “cherry-red spot” seen in many type 1 patients.
Type 6
Type 6 is characterized by patients exhibiting both Farber lipogranulomatosis as well as Sandoff disease.
Type 7
Type 7 disease results from a deficiency in prosaposin which is a precursor encoding the sphingolipid activator proteins called saposins (saposin A, B, C, and D). See the Gaucher disease page for description of saposins. As of 2025 a total of 47 pathogenic mutations in the PSAP gene, that encodes prosaposin, have been identified, some of which affect the saposin D protein and as such are associated with type 7 Farber lipogranulomatosis.