
Last Updated: October 28, 2025
Introduction to Fabry Disease
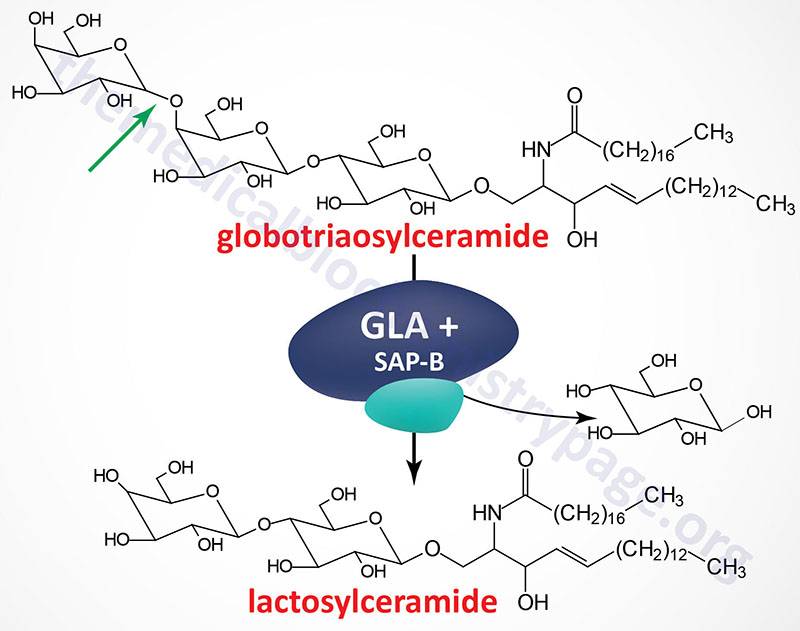
Fabry disease belongs to a family of disorders identified as lysosomal storage diseases. This disorder is characterized by the lysosomal accumulation of glycosphingolipids with terminal α-galactosyl residues. The predominant glycolipid is globotriaosylceramide (abbreviated: Gb3) and to a lesser extent blood group antigen B (of the ABO blood group antigens) derived compounds. Gb3 is also commonly referred to as ceramide trihexoside. These glycolipids accumulate in the lysosomes as a consequence of defects in the lysosomal hydrolase, α-galactosidase A. Fabry disease is inherited as an X-linked recessive disorder.
Molecular Biology of Fabry Disease
The α-galactosidase A enzyme is encoded by the GLA (galactosidase alpha) gene. The GLA is located on the X chromosome (Xq22.1) and is composed of 9 exons that generate four alternatively spliced mRNAs, each of which encode a distinct protein isoform. The primary mRNA encodes a 429 amino acid precursor protein (isoform b) that is processed to a 370 amino acid glycoprotein. α-Galactosidase functions as a homodimer.
Like all enzymes destined for the lysosomes the α-galactosidase A protein is co-translationally modified with mannose-6-phosphate residues. A portion of the phosphorylated enzyme is actually secreted from cells and then taken up by receptor-mediated endocytosis via mannose-6-phosphate receptors on the plasma membrane of cells. This secretion and re-uptake of α-galactosidase A provides the basis for the rationale behind enzyme replacement therapy (ERT, see below).
As of 2025 there have been 349 pathogenic mutations identified in the GLA gene resulting in Fabry disease. The largest number of mutations are missense mutations resulting in single amino acid changes. Insertions, complex rearrangements, and duplications account for approximately 10% of Fabry-causing mutations. Because the gene is X-linked most patients have a blood relative who is an affected male or a carrier female.
Clinical Features of Fabry Disease
The clinical manifestations of Fabry disease begin in childhood or adolescence with the average age of presentation between 6 and 9 years of age. Classic symptoms include pain and paresthesia (burning or prickly sensation) in the extremities, particularly the hands and feet, reduced capacity to sweat, gastrointestinal disturbances, cardiomyopathy, progressive renal impairment, corneal and lenticular opacities and characteristic skin lesions called angiokeratoma (angiokeratoma corporis diffusum). The typical corneal opacity seen via slit lamp examination is termed cornea verticillata, also known as vortex keratopathy, and is characterized by a distinctive swirling pattern of opacities in the lower cornea, primarily affecting the superficial corneal layer. Angiokeratoma is characterized by small dark red or purple raised papules that typically appear on the thighs and around the umbilicus. Angiokeratoma is caused by dilation of vessels near the surface of the skin.
These symptoms are all due to the deposition of Gb3 in the walls of small vessels, kidney tubule and glomerular cells, nerves and dorsal root ganglia. The characteristic skin lesions (angiokeratoma) of Fabry disease are the earliest signs that may lead to diagnosis in childhood. Death usually occurs in early adulthood from renal and cardiac complications of the vascular disease. Carrier females are usually asymptomatic but can, in rare cases, be as severely affected as hemizygous males. The most consistent clinical phenotype found in carrier females is the distinctive corneal opacity, vortex keratopathy.
Treatment of Fabry Disease
Current treatment for Fabry disease involves enzyme replacement therapy (ERT) with intravenous infusions of recombinant human α-galactosidase A. This therapeutic regimen consistently decreases Gb3 levels in plasma and clears lysosomal inclusions from vascular endothelial cells. The effects of ERT on other tissues are not as obvious, suggesting that treatment must be initiated early in the course of the disease to be optimally effective or that some complications of the disease are not responsive to enzymes delivered intravenously. The biotech company Genzyme, a unit of Sanofi SA, currently markets an ERT drug called Fabryzyme ®.