
Last Updated: February 17, 2026
Introduction to the PPP
The pentose phosphate pathway (PPP) is primarily catabolic and serves as an alternative glucose oxidizing pathway. Glucose metabolism via the PPP is essential for the generation of NADPH that is required for reductive biosynthetic reactions such as those of cholesterol biosynthesis, bile acid synthesis, steroid hormone biosynthesis, and fatty acid synthesis.
The pentose phosphate pathway can also function as an anabolic pathway that utilizes the six carbons of glucose to generate five carbon sugars, particularly ribose-5-phosphate (R5P) that is required for purine and pyrimidine nucleotide biosynthesis. The pentose phosphate pathway can, under certain conditions, completely oxidize glucose to CO2 and water.
The primary functions of PPP are:
1. To generate reducing equivalents, in the form of NADPH, for reductive biosynthesis reactions within cells.
2. To provide the cell with ribose-5-phosphate (R5P) for the synthesis of the nucleotides and nucleic acids.
3. Although not a significant function of the PPP, it can operate to metabolize dietary pentose sugars derived from the digestion of nucleic acids as well as to rearrange the carbon skeletons of dietary carbohydrates into glycolytic and /or gluconeogenic intermediates.
Enzymes that function primarily in the reductive direction utilize the NADP+/NADPH co-factor pair as their co-factors as opposed to oxidative enzymes that utilize the NAD+/NADH co-factor pair. The reactions of fatty acid biosynthesis and steroid biosynthesis utilize large amounts of NADPH. As a consequence, cells of the liver, adipose tissue, adrenal cortex, testis, and lactating mammary gland have high levels of the PPP enzymes. In fact 30% of the oxidation of glucose in the liver occurs via the PPP. Additionally, erythrocytes utilize the reactions of the PPP to generate large amounts of NADPH used in the reduction of glutathione (see below). The conversion of ribonucleotides to deoxyribonucleotides (through the action of ribonucleotide reductase) requires NADPH as the electron source, therefore, any rapidly proliferating cell needs large quantities of NADPH.
Although the PPP operates in all cells, with high levels of expression in the above indicated tissues, the highest levels of PPP enzymes (in particular glucose 6-phosphate dehydrogenase) are found in neutrophils and macrophages. These leukocytes are the phagocytic cells of the immune system and they utilize NADPH to generate superoxide radicals from molecular oxygen in a reaction catalyzed by one of the NADPH oxidase complexes. Superoxide anion, in turn, serves to generate other reactive oxygen species (ROS) that kill the phagocytosed microorganisms. Following exposure to bacteria and other foreign substances there is a dramatic increase in O2 consumption by phagocytes. This phenomenon is referred to as the oxidative burst or respiratory burst.
Reactions of the Pentose Phosphate Pathway
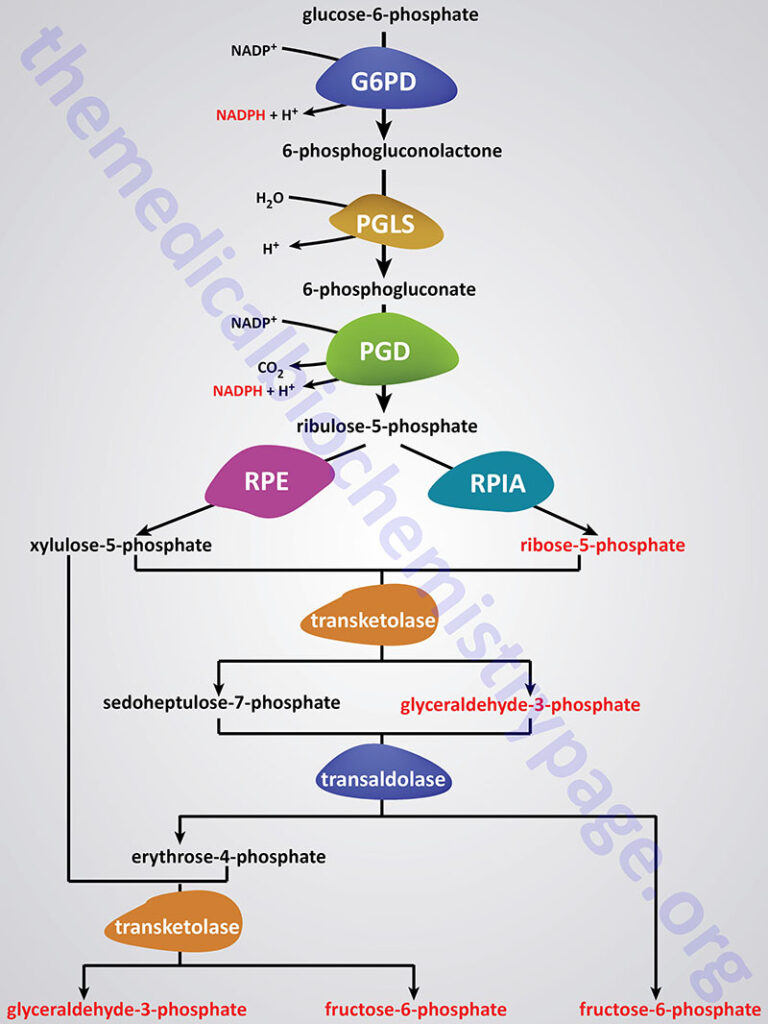
The reactions of the PPP operate exclusively in the cytoplasm. From this perspective it is understandable that fatty acid synthesis (as opposed to oxidation) takes place in the cytoplasm. The pentose phosphate pathway has both an oxidative and a non-oxidative arm. The oxidation steps, utilizing glucose-6-phosphate (G6P) as the substrate, occur at the beginning of the pathway and are the reactions that generate NADPH. The reactions catalyzed by glucose-6-phosphate dehydrogenase (G6PD; also identified ad G6PDH) and 6-phosphogluconate dehydrogenase (PGD) both generate one mole of NADPH for every mole of glucose-6-phosphate that enters the PPP. The conversion of 6-phosphogluconolactone to 6-phosphogluconate is catalyzed by 6-phosphogluconolactonase (PGLS).
Glucose-6-Phosphate Dehydrogenase: G6PD
The glucose-6-phosphate dehydrogenase gene (symbol: G6PD) is located on the X chromosome (Xq28) and is composed of 14 exons that generate three alternatively spliced mRNAs. These three mRNAs collectively encode the G6PD isoform a (545 amino acids) and isoform b (515 amino acids) enzymes.
The G6PD isoform a protein is inactive in the native full-length form but may undergo processing to the smaller 515 amino acid active form of the enzyme. The active G6PD enzyme is also referred to as the G form of glucose-6-phosphate dehydrogenase. The G6PD gene is ubiquitously expressed with high levels of expression in erythrocytes.
Hexose-6-Phosphate Dehydrogenase: H6PDH
Humans express a second glucose-6-phosphate dehydrogenase activity, referred to as the H form. This form of glucose-6-phosphate dehydrogenase activity is identified as hexose-6-phosphate dehydrogenase (encoded by the H6PD gene) and also as glucose-1-dehydrogenase. Whereas the G6PD encoded enzyme resides in the cytosol, the H6PD encoded enzyme resides within the endoplasmic reticulum (ER) and the sarcoplasmic reticulum (SR).
The H6PD gene is located on chromosome 1p36.22 and is composed of 7 exons that generate two alternatively spliced mRNAs encoding precursor proteins of 802 amino acids (isoform 1) and 791 amino acids (isoform 2). The H6PD gene is not expressed in erythrocytes.
Within the ER, hexose-6-phosphate dehydrogenase converts glucose-6-phosphate and NADP+ to 6-phosphogluconate and NADPH in a single step, whereas this process in the cytosol requires two separate enzymes (G6PD and PGLS). In addition to glucose-6-phosphate, H6PD can metabolize other hexose-6-phosphates, as well as glucose-6-sulfate, and glucose.
One of the primary functions of the ER- and SR-localized NADPH is to maintain redox homeostasis within these organelles. Loss of ER redox homeostasis can lead to ER stress and induction of the unfolded protein response (UPR) which, if severe enough will trigger cell death via the apoptotic pathway.
Another principal function of the NADPH produced by ER-localized hexose-6-phosphate dehydrogenase is to provide the reducing energy to ER-localized reductases, specifically those involved in steroid hormone metabolism, with 11β-hydroxysteroid dehydrogenase 1 (11β-HSD1; encoded by the HSD11B1 gene) being particularly important. The primary function of the HSD11B1 encoded enzyme is to reduce the 11-oxo groups in cortisone and 11-dehydrocorticosterone to the active glucocorticoids, cortisol and corticosterone, respectively. However, the enzyme can, under certain conditions, also inactivate cortisol and corticosterone by catalyzing the oxidation reactions converting cortisol to cortisone and corticosterone to 11-dehydrocorticosterone. Of clinical significance to the role of ER-localized NADPH is that mutations in the H6PD gene are associated with glucocorticoid deficiency.
6-Phosphogluconolactonase: PGLS
The product of the glucose-6-phosphate dehydrogenase reaction, 6-phosphogluconolactone, is converted to 6-phosphogluconate by the enzyme, 6-phosphogluconolactonase which is encoded by the PGLS gene. The PGLS gene is located on chromosome 19p13.11 and is composed of 5 exons that encode a 258 amino acid protein.
6-Phosphogluconate Dehydrogenase
The third enzyme of the PPP, 6-phosphogluconate dehydrogenase is encoded by the phosphogluconate dehydrogenase gene (symbol: PGD). The PGD gene is located on chromosome 1p36.33 and is composed of 13 exons that generate three alternatively spliced mRNAs, each of which encode different sized protein isoforms.
Non-Oxidative Reactions of the PPP
The non-oxidative reactions of the PPP are primarily designed to generate ribose-5-phosphate (R5P). Other reactions of the PPP that are of less physiological significance are designed to convert dietary five carbon sugars into both six (fructose-6-phosphate) and three (glyceraldehyde-3-phosphate) carbon sugars which can then be utilized by the pathways of glycolysis.
The enzymes of the non-oxidative reactions of the PPP include transaldolase (encoded by the TALDO1 gene), transketolase (encoded by the TKT gene), ribose-5-phosphate isomerase (encoded by the RPIA gene), and ribulose-5-phosphate-3-epimerase (encoded by the RPE gene).
The primary enzymes involved in the non-oxidative steps of the PPP are transaldolase and transketolase. Transketolase functions to transfer two-carbon groups from substrates of the PPP thus, rearranging the carbon atoms that enter this pathway. Like other enzymes that transfer two-carbon groups, transketolase requires thiamine pyrophosphate (TPP) as a co-factor in the transfer reaction.
Two facts regarding transketolase make it a diagnostically useful enzyme. The enzyme is expressed at high levels in red blood cells, which are easy to isolate and analyze, and the only vitamin-derived cofactor it requires is TPP. Therefore, assay for reduced activity of this enzyme, in red blood cell lysates, is highly diagnostic in cases of suspected thiamine deficiency.
Transaldolase transfers three-carbon groups and thus is also involved in a rearrangement of the carbon skeletons of the substrates of the PPP. The transaldolase reaction involves Schiff base formation between the substrate and a lysine residue in the enzyme.
Transketolase is encoded by the TKT gene. The TKT gene is located on chromosome 3p21.1 and is composed of 15 exons that generate three alternatively spliced mRNAs that collectively encode two distinct protein isoforms.
Transaldolase is encoded by the TALDO1 gene. The TALDO1 gene is located on chromosome 11p15.5 and is composed of 8 exons that encode a protein of 337 amino acids. A molecularly interesting fact about the TALDO1 gene is that exons 2 and 3 were derived as a result of the insertion of a retrotransposon into the gene.
There are two transketolase-related genes in the human genome. One is found on the X chromosome (Xq28) and is identified as the transketolase-like 1 gene (symbol: TKTL1). The other gene (symbol: TKTL2) is located on chromosome 4q32.2.
The TKTL1 encoded protein lacks 38 amino acids, compared to the TKT encoded protein, in the TPP-binding region. All TPP-dependent enzymes contain a highly similar TPP-binding domain and its lack in the TKTL1 encoded protein strongly suggests that it is unlikely that TKTL1 is a TPP-dependent protein capable of catalyzing the transketolase reaction. Indeed, biochemical evidence has indicated that the TKTL1 protein is not capable of catalyzing a transketolase reaction. Intense interest in the TKTL1 gene, and its encoded protein, was stimulated because it was shown that the level of TKTL1 expression correlated with poor patient outcomes and metastasis in many solid tumors. In addition, specific inhibition of TKTL1 mRNA has been shown to inhibit cancer cell proliferation in functional studies.
The net result of the PPP, if not used solely for R5P production, is the oxidation of G6P, a six-carbon sugar, into a five-carbon sugar. In turn, three moles of five-carbon sugar are converted, via the enzymes of the PPP, back into two moles of six-carbon sugars and one mole of three-carbon sugar. The six-carbon sugars can be recycled into the pathway in the form of G6P, generating more NADPH. The three-carbon sugar generated is glyceraldehyde-3-phosphate which can be shunted to glycolysis and oxidized to pyruvate. Alternatively, it can be utilized by the gluconeogenic enzymes to generate more six-carbon sugars, fructose-6-phosphate or glucose-6-phosphate.
Role of Transketolase in the Development of Fatty Liver Disease
Metabolic dysfunction-associated fatty liver disease (MAFLD), which was originally defined as non-alcoholic fatty liver disease (NAFLD), is a major disease occurring in up to 25% of individuals world-wide. Mitochondrial dysregulation within the liver has been found to be a hallmark of MAFLD. MAFLD is also referred to as metabolic dysfunction-associated steatotic liver disease, MASLD.
Expression of the transketolase gene (TKT) has been found to be elevated in hepatocytes from patients manifesting the hallmark symptoms of MAFLD. In laboratory animals, the forced overexpression of the TKT gene promotes the development of MAFLD as well as the advancement of MAFLD to the more severe state referred to as metabolic dysfunction-associated steatohepatitis (MASH). Conversely, when the TKT gene is deleted, specifically in the liver, there is an amelioration of the development of MAFLD in mice fed a high-fat diet.
In hepatocytes from mice in which the TKT gene was knocked out there is an accumulation of ribose-5-phosphate as well as the nucleotides, inosine, adenosine, and deoxyadenosine. The significance of the accumulation of inosine stems from the fact that experiments have previously demonstrated that inosine promotes mitochondrial function in adipose tissue. The effect of inosine in adipocytes was shown to be exerted through the activation of PKA which in turn enhances fat metabolism via increased UCP-1 activity.
Within hepatocytes, inosine also activates PKA which in turn activates CREB signaling. The net effect, of TKT deficiency in hepatocytes, is enhanced mitochondrial DNA content, increased ATP production, and overall mitochondrial activity. The enhanced hepatic mitochondrial activity in the context of TKT deficiency has been shown to be the result of increased phospholipid synthesis, particularly increased synthesis of phosphatidylcholine which represents the major phospholipid in the mitochondrial membrane. The increase in phosphatidylcholine synthesis is the result of inosine activation of PKA activity.
Insulin activity has been shown to result in increased expression of the TKT gene. Indeed, hyperinsulinemia-mediated increases in TKT expression are directly correlated to the development of MAFLD. Even in the context of high-fat diet-induced hepatic insulin resistance it has been found the TKT expression in hepatocytes is still sensitive to insulin. The mechanism of this insulin-mediated effect on TKT expression in the resistant state is as yet undetermined.
Regulation of the Pentose Phosphate Pathway
The major regulator of the PPP is the cellular response to oxidative stress. The response of the PPP, under these conditions, is due to the increased demand for NADPH by cells experiencing increased oxidative stress. Within seconds of the sensing of increased reactive oxygen species (ROS), several enzymes of glycolysis (particularly glyceraldehyde-3-phosphate dehydrogenase and pyruvate kinase) are inhibiting allowing for the diversion of glucose carbons into the PPP. Another major contributor to carbon delivery to the PPP, in periods of oxidative stress, is the glucose from glycogen.
A major regulator of the rate of NADPH production by the PPP in response to oxidative stress is the allosteric activation of G6PDH. When glucose is removed from glycogen by glycogen phosphorylase the product is glucose-1-phosphate (G1P). Glucose-1-phosphate is an allosteric activator of G6PDH resulting in an enhanced level of NADPH production in response to oxidative stress.
In addition to allosteric activation of G6PDH, the G6PD gene is transcriptionally activated in response to oxidative stress. The G6PD gene is also transcriptionally activated by increases in anabolic metabolism which requires increased levels of NADPH as well. The transcriptional activation of genes encoding other enzymes of the PPP also occurs in response to oxidative stress and the demands of increased anabolic metabolism.
Transcriptional activation of G6PD in response to oxidative stress is carried out by the transcription factor, NRF2 (nuclear factor erythroid 2–related factor 2). NRF2 is encoded by the NFE2L2 (NFE2 like bZIP transcription factor 2) gene. The major transcription factors regulating transcription of genes encoding PPP enzymes in response to increased anabolic metabolism, particularly increased lipid synthesis, are members of the sterol regulated element binding protein (SREBP) family.
Metabolic Disorders Associated with the PPP
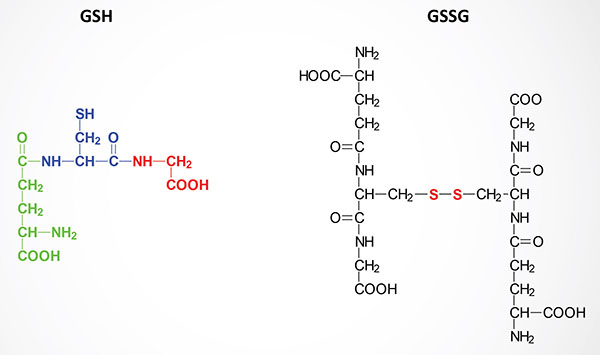
Accumulation of reactive molecules, such as reactive oxygen and reactive nitrogen species (ROS and RNS), lipid hydroperoxides, etc. results in disturbances in the homeostatic redox state of cells. These conditions are controlled primarily by the action of the peptide, glutathione, GSH. In addition to GSH there are various low molecular weight antioxidants such as reduced vitamin C and vitamin E, bilirubin, and urate. There are also several non-catalytic antioxidant proteins such as the glutaredoxins, the thioredoxins (Trx), and the metallothioneins (MT). The catalytic antioxidant proteins include the superoxide dismutases (SOD), catalase, the peroxiredoxins (Prx), and the glutathione peroxidases (GPx).
GSH is a tripeptide composed of γ-glutamate, cysteine and glycine. The sulfhydryl side chains of the cysteine residues of two glutathione molecules form a disulfide bond (GSSG) during the course of being oxidized in reactions with various oxides and peroxides in cells. Reduction of GSSG to two moles of GSH is the function of glutathione reductase, an enzyme that requires coupled oxidation of NADPH.
There are at least three inborn errors in the pentose phosphate pathway that have been identified. The most common being the result of mutations in glucose-6-phosphate dehydrogenase (G6PD; also identified as G6PDH). Extremely rare occurrences of ribose-5-phosphate isomerase and transaldolase deficiency have also been documented.
Transaldolase deficiency is an autosomal recessive disorder resulting from inherited mutations in the TALDO1 gene. The symptoms associated with transaldolase deficiency can be quite variable from patient to patient with most patients manifesting symptoms in the neonatal period. Neonatal presentation includes hepatosplenomegaly, bleeding diathesis, abnormal liver function, cholestatic jaundice, and elevated liver function enzymes. When presenting later patients manifest with hepatic fibrosis or cirrhosis. Most transaldolase deficient patients will be associated with dysmorphic features, congenital heart defects, and hemolytic anemia. Biochemical characteristics of transaldolase deficiency include the accumulation of seven carbon carbohydrates including sedoheptulose, sedoheptulose 7-phosphate, and mannoheptulose as well as open-chain polyols that includes erythritol, arabitol, ribitol, and sedoheptitol.
Ribose-5-phopshate isomerase deficiency is an extremely rare autosomal recessive disorder that has been identified in only a few individuals. Indeed, deficiency in ribose-5-phopshate isomerase is considered to be second rarest disorder world-wide. In these individuals the most common manifesting symptoms are leukoencephalopathy and peripheral neuropathy. Biochemically these individuals manifest with elevated levels of polyols, particularly ribitol, D-arabitol, in body fluids and in the brain.
Although there are other pathways for the generation of NADPH that can be used in the regulation of the red-ox state of cells, the PPP represents the major and most important pathway for NADPH production, especially in erythrocytes. Thus, it should be clear that any disruption in the level of NADPH may have a profound effect upon a cells ability to deal with oxidative stress. No other cell than the erythrocyte is exposed to greater oxidizing conditions. After all it is the oxygen carrier of the body.
Chronic Granulomatous Disease
Because of the need for NADPH in phagocytic cells, by the NADPH oxidase (NOX) system, any defect in enzymes in this process can result in impaired killing of infectious organisms. Humans express several complexes of the NADPH oxidase family. The specific NOX system of phagocytic cells is a multisubunit complex whose function is to generate reactive oxygen species (ROS) as a means to destroy invading pathogens. This particular NADPH oxidase system is referred to as NOX2.
Chronic granulomatous disease (CGD) is a syndrome that results in individuals harboring defects in the NOX2 form of the NADPH oxidase system. The NOX2 system is a multisubunit enzyme complex preferentially expressed in cells of the myeloid lineage. NADPH oxidase complexes are composed of protein subunits that interact to form the functional enzyme complex. The NOX2 system is designed to generate reactive oxygen species (ROS), referred to as the oxidative burst, as a means to eliminate invading microorganisms in macrophages and neutrophils.
All NAPDH oxidase systems are multisubunit enzyme complexes. Two of the proteins of the NOX2 complex are integral membrane proteins and they are identified as p22-PHOX (p22phox; also known as the α-subunit) and gp91-PHOX (gp91phox; also known as the β-subunit). The PHOX designation refers to phagocyte oxidase. These two subunits form a large heterodimeric complex that is referred to as cytochrome b558 (cyt b558). Three of the subunits of NADPH oxidase are regulatory subunits and these remain in the cytosol in the unstimulated state. These regulatory subunits are identified as p40-PHOX, p47-PHOX, and p67-PHOX. When cells are appropriately stimulated, the p47-PHOX subunit is phosphorylated and the entire heterotrimeric complex migrates to the membrane where it associates with the cyt b558 complex.
Two additional proteins are involved in the function of NADPH oxidase systems and both are members of the low molecular weight monomeric G-protein family. These G-proteins are identified as RAC2 (RAC family small GTPase 2) and RAP1A (RAS-related protein 1A). The formation of this membrane associated enzyme complex results in the generation of superoxide anion which in turn is converted to hydrogen peroxide through the action of Cu-Zn superoxide dismutase (SOD1). The generation of these ROS within the phagocytic complexes of macrophages and neutrophils is the means by which these cells degrade phagocytosed microorganisms.
There are several forms of CGD involving defects in various components of the NADPH oxidase 2 (NOX2) system. The majority of patients (67%) with CGD harbor mutations in an X-chromosome gene (CYBB) that encodes the β-subunit (gp91-PHOX) of cytochrome b558. This form of the disorder is referred to as cytochrome b-negative X-linked CGD.
There is an autosomal recessive cytochrome b-negative form of CGD due to defects in the α-subunit (p22-PHOX) of cytochrome b558. The p22-PHOX protein is encoded by the CYBA gene. Mutations in the CYBA gene account for approximately 5% of CGD cases.
There are also two autosomal recessive cytochrome b-positive forms of CGD identified as cytochrome b-positive CGD type I and type II. The type I form represents about 20% of all CGD cases and is caused by mutations in the neutrophil cytosolic factor 1 (NCF1) gene, which encodes the p47-PHOX protein. The type II form represents about 5% of all cases of CGD and is the result of mutations in the NFC2 gene which encodes the p67-PHOX protein. The p40-PHOX protein is encoded by the neutrophil cytosolic factor 4 (NCF4) gene. Mutations in the NFC4 component of the NADPH oxidase system has been reported in a single case of CGD.
In addition to the NOX2 complexes that function in phagocytic cells there are several additional NADPH oxidase systems that function in many other types of cells. These complexes are referred to as NOX1, NOX3, NOX4, NOX5, DUOX1 (dual oxidase 1), and DUOX2. The DUOX1 and DUOX2 members of the NADPH oxidase family are involved in the generation of the the thyroid hormones.
Given the role of NADPH in the process of phagocytic killing it should be clear that individuals with reduced ability to produce NADPH (such as those with G6PD deficiencies) may also manifest with symptoms that have similarities to those of classically inherited forms of CGD.
Microbiological Integration
Individuals with CGD are at increased risk for specific recurrent infections. The most common are pneumonia, abscesses of the skin, tissues, and organs, suppurative arthritis (invasion of the joints by infectious agent leading to generation of pus), and osteomyelitis (infection of the bone). The infections found in CGD patients are primarily associated with a subset of catalase-positive microorganisms. This is due to the fact that in CGD the production of the ROS, hydrogen peroxide, is deficient and so these catalase-positive microorganisms can defeat the limited levels of ROS produced by phagocytic cells. The most common infections in individuals with CGD in North America and Europe are from various Aspergillus species such as A. fumigatus and A. flavus, Burkholderia cepacia, various Norcadia species such a N. asteroides, Salmonella, Serratia marcescens, and Staphylococcus aureus. Infections with Granulibacter bethesdensis (a Gram-negative rod bacterium that causes chronic necrotizing lymphadenitis and sepsis) and Burkholderia gladioli (a Gram-negative rod bacterium which can cause osteomyelitis and sepsis) should prompt evaluation for CGD. Also associated with CGD is a high prevalence of invasive fungal infections affecting 20%–40% of CGD patients.
Erythrocytes and the Pentose Phosphate Pathway
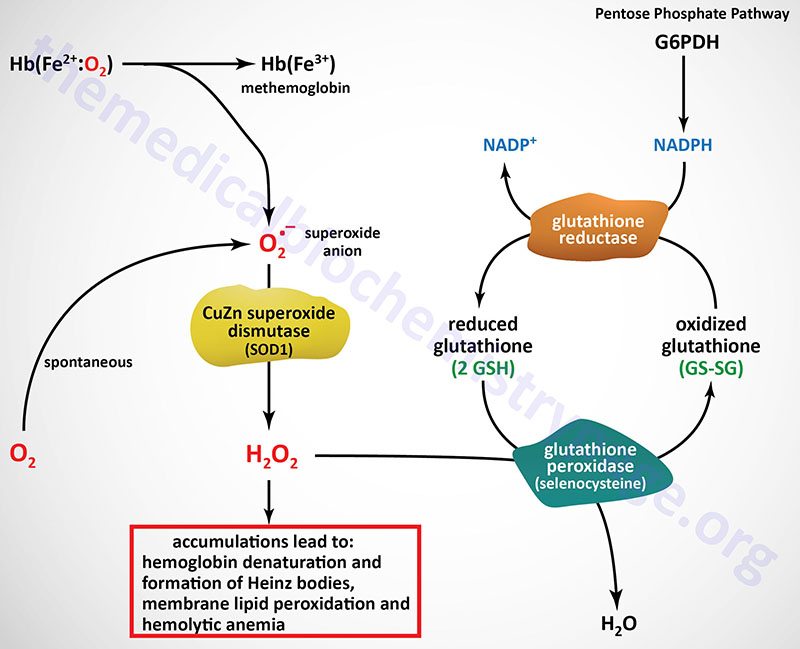
The predominant pathways of carbohydrate metabolism in the red blood cell (RBC) are glycolysis, the PPP and 2,3-bisphosphoglycerate (2,3-BPG) metabolism (refer to discussion of hemoglobin for review of the synthesis and role role of 2,3-BPG). Glycolysis provides ATP for membrane ion pumps and NADH for re-oxidation of methemoglobin. The PPP supplies the RBC with NADPH to maintain the reduced state of glutathione. The inability to maintain reduced glutathione in RBCs leads to increased accumulation of peroxides, predominantly H2O2, that in turn results in a weakening of the cell wall as a result of membrane lipid peroxidation resulting in concomitant hemolysis.
Accumulation of H2O2 also leads to increased rates of cysteine sulfhydryl oxidation in hemoglobin resulting in the formation of cross-linked complexes of denatured hemoglobin. These large complexes of denatured hemoglobin can be visualized by light microscopy and are referred to as Heinz bodies. Glutathione removes peroxides from membrane lipids and serves as a substrate for H2O2 reduction to H2O via the action of glutathione peroxidase. The PPP in erythrocytes is the only pathway for these cells to produce NADPH, therefore, any defect in the production of NADPH, such as due to deficiencies in glucose-6-phosphate dehydrogenase, will have profound effects on erythrocyte survival.
The glutathione reductase gene (GSR; official name is glutathione-disulfide reductase) is located on chromosome 8p12 and is composed of 15 exons that generate four alternatively spliced mRNAs that encode four distinct protein isoforms.
Humans express eight distinct glutathione peroxidase genes identified as GPX1–GPX8.
The GPX1 gene is located on chromosome 3p21.31 and is composed of 2 exons that generate five alternatively spliced mRNAs due to the use of alternative splice acceptor sites in the 3′-exon. These five mRNAs each encode distinct protein isoforms, one of which lacks the critical UGA codon that is utilized for the incorporation of selenocysteine into the protein.
The GPX2 gene is located on chromosome 14q23.3 and is composed of 4 exons that encode a 190 amino acid protein.
The GPX3 gene is located on chromosome 5q33.1 and is composed of 6 exons that generate two alternatively spliced mRNAs that encode precursor proteins of 226 amino acids (isoform 1) and 235 amino acids (isoform 2).
The GPX4 gene is located on chromosome 19p13.3 and is composed of 8 exons that generate four alternatively spliced mRNAs, each of which encode a distinct precursor protein. The GPX4 encoded proteins exhibit distinct subcellular sites of localization including the nucleus, the mitochondria, and the cytosol. In addition, one of the GPX4 encoded proteins plays a role in sperm maturation, a process that is structural not enzymatic.
The GPX5 gene is located on chromosome 6p22.1 and is composed of 6 exons that generate two alternatively spliced mRNAs encoding two protein isoforms. The GPX5 encoded mRNAs do not contain a selenocysteine (UGA) codon and, therefore, they encode selenium-independent enzymes. Expression of the GPX5 gene is restricted to the epididymis in the male gonads and the encoded proteins function to protect the membranes of spermatozoa during their maturation.
The GPX6 gene is located on chromosome 6p22.1 near the GPX5 gene and is composed of 5 exons that encode a 221 amino acid precursor protein. Expression of the GPX6 gene is is near exclusive to the gonads with much higher levels of expression in the testes that the ovaries.
The GPX7 gene is located on chromosome 1p32.3 and is composed of 4 exons that encode a 187 amino acid precursor protein.
The GPX8 gene is located on chromosome 5q11.2 and is composed of 4 exons that generate four alternatively spliced mRNAs. The proteins encoded by the GPX8 gene are currently identified as putative GPx enzymes.
Deficiency in the level of activity of glucose-6-phosphate dehydrogenase (G6PD) is the basis of favism, primaquine (an anti-malarial drug) sensitivity and several other drug-sensitive hemolytic anemias, anemia and jaundice in the newborn, and chronic nonspherocytic hemolytic anemia. In addition, G6PD deficiencies are associated with resistance to the malarial parasite, Plasmodium falciparum, among individuals of Mediterranean and African descent. The basis for this resistance is the weakening of the red cell membrane (the erythrocyte is the host cell for the parasite) such that it cannot sustain the parasitic life cycle long enough for productive growth.