
Last Updated: October 31, 2025
Introduction to Molecular Biology in Medicine
Modern molecular medicine encompasses the utilization of many molecular biological techniques in the analysis of disease, disease genes and disease gene function. The study of disease genes and their function in an unaffected individual has been possible by the development of recombinant DNA and cloning techniques. The basis of the term recombinant DNA refers to the recombining of different segments of DNA. Cloning refers to the process of preparing multiple copies of an individual type of recombinant DNA molecule. The classical mechanisms for producing recombinant molecules involved the insertion of exogenous fragments of DNA into either bacterially derived plasmid (circular double stranded autonomously replicating DNAs found in bacteria) vectors or bacteriophage (viruses that infect bacteria) based vectors. The term vector refers to the DNA molecule used to carry or transport DNA of interest into cells.
Table of Common Enzymes used in Molecular Biology
| Enzyme(s) | Activity | Comments |
| Restriction endonucleases | recognize specific nucleotide sequences and cleaves the DNA within or near to the recognition sequences | see below |
| Reverse transcriptase (RT) | retrovirally encoded RNA-dependent DNA polymerase | used to convert mRNA into a complimentary DNA (cDNA) copy for the purpose of cloning cDNAs |
| RNase H | recognizes RNA-DNA duplexes and randomly cleaves the phosphodiester backbone of the RNA | used primarily to cleave the mRNA strand that is annealed to the first strand of cDNA generated by reverse transcription |
| DNA polymerase | synthesis of DNA | used during most procedures where DNA synthesis is required, also used in in vitro mutagenesis |
| Klenow DNA polymerase | proteolytic fragment of DNA polymerase that lacks the 5′ → 3′ exonuclease activity | used to incorporate radioactive nucleotides into restriction enzyme generated ends of DNA, also can be used in place of DNA polymerase |
| DNA ligase | covalently attaches a free 5′ phosphate to a 3′ hydroxyl | used in all procedures where to molecules of DNA need to be covalently attached |
| Alkaline phosphatase | removes phosphates from 5′ ends of DNA molecules | used to allow 5′ ends to be subsequently radiolabeled with the γ-phosphate of ATP in the presence of polynucleotide kinase, also used to prevent self-ligation of restriction enzyme digested plasmids and lambda vectors |
| Polynucleotide kinase | introduces γ-phosphate of ATP to 5′ ends of DNA | see above for alkaline phosphatase |
| DNase I | randomly hydrolyzes the phosphodiester bonds of double-stranded DNA | is used in the identification of regions of DNA that are bound by protein and thereby protected from DNase I digestion, also used to identify transcriptionally active regions of chromatin since they are more susceptible to DNase I digestion |
| S1 Nuclease | exonuclease that recognizes single-stranded regions of DNA | used to remove regions of single strandedness in DNA or RNA-DNA duplexes |
| Exonuclease III | exonuclease that removes nucleotides from the 3′ end of DNAs | used to generate deletions in DNA for sequencing, or to map functional domains of DNA duplexes |
| Terminal transferase | DNA polymerase that requires only a 3′-OH, lengthens 3′ ends with any dNTP | used to introduce homopolymeric (same dNTP) tails onto the 3′ ends of DNA duplexes, also used to introduce radiolabeled nucleotides on the 3′ ends of DNA |
| T3, T7, and SP6 RNA polymerases | bacterial virus encoded RNA polymerase, recognize specific nucleotide sequences for initiation of transcription | used to synthesize RNA in vitro |
| Taq and Vent DNA polymerase | thermostable DNA polymerases | used in PCR |
| Taq and Vent DNA ligases | thermostable DNA ligases | used in LCR |
| Recombinases | Cre-lox, Flp-FRT | catalyze site-specific recombination events within DNA to generate chimeric DNA molecules; function both in vitro and in vivo |
| CRISPR-Cas9 | derived from Clustered Regularly Interspaced Short Palindromic Repeats and CRISPR-associated protein 9 | is a method for editing genomes that was adapted from a naturally occurring microbial adaptive immune system that utilizes RNA-guided nucleases |
Restriction Endonucleases
Restriction endonuclease are enzymes that will recognize, bind to and hydrolyze specific nucleic acid sequences in double-stranded DNA. The term restriction endonuclease was given to this class of bacterially derived enzymes since they were identified as being involved in restricting the growth of certain bacteriophages. Bacteria are capable of modifying specific sequences within their genomes by methylation which prevents their own DNA from being recognized by the restriction enzymes encoded by their genomes. This process is termed modification and restriction. Infecting bacteriophage DNA is not modified and, hence, will be digested by the restriction endonucleases present in the bacterium.
The key to the in vitro utilization of restriction endonucleases is their strict nucleotide sequence specificity. The different enzymes are identified by being given a name indicating the bacteria from which they were isolated, e.g. the enzymes EcoRI which recognizes the sequences, 5’–GAATTC–3′, was isolated from Escherichia coli. One unique feature of restriction enzymes is that the nucleotide sequences they recognize are palindromic, i.e. they are the same sequences in the 5′ → 3′ direction of both strands. Some restriction endonucleases make staggered symmetrical cuts away from the center of their recognition site within the DNA duplex, some make symmetrical cuts in the middle of their recognition site while still others cleave the DNA at a distance from the recognition sequence. Enzymes that make staggered cuts leave the resultant DNA with cohesive or sticky ends. Enzymes that cleave the DNA at the center of the recognition sequence leave blunt-ended fragments of DNA.
Any two pieces of DNA containing the same sequences within their sticky ends can anneal together and be covalently ligated together in the presence of DNA ligase. Any two blunt-ended fragments of DNA can be ligated together irrespective of the sequences at the ends of the duplexes. This process is the basic method for the generation of recombinant DNA molecules, particularly with the ligation of a fragment or fragments of DNA into a suitable vector for subsequent cloning (see below).
DNA Sequencing
Sequencing of DNA can be accomplished by either chemical or enzymatic means. The original technique for sequencing, Maxam and Gilbert sequencing, relies on the nucleotide-specific chemical cleavage of DNA and is not routinely used any more. The enzymatic technique, Sanger sequencing, involves the use of dideoxynucleotides (2′,3′-dideoxy) that terminate DNA synthesis and is, therefore, also called dideoxy chain termination sequencing.
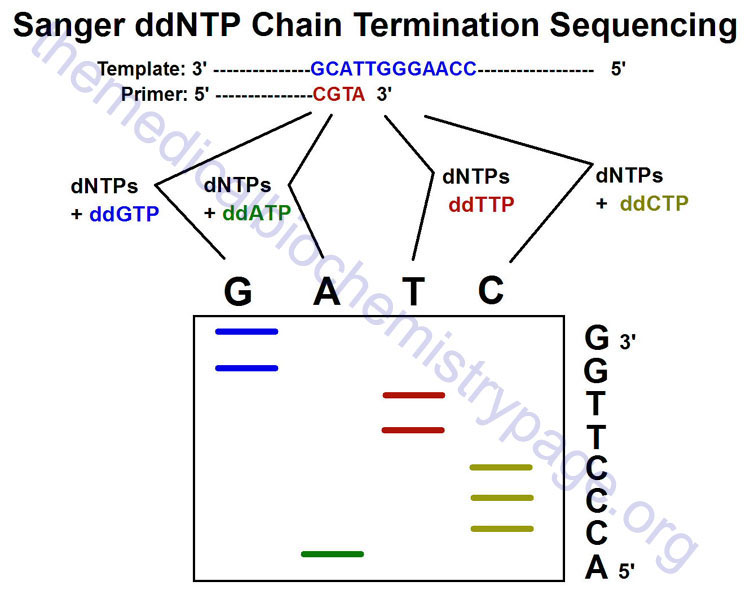
Deep Sequencing DNA
Deep sequencing of DNA simply refers to the process whereby the sequence of nucleotides in the same region of a DNA molecule are independently determined several times. This process can be significant to the accuracy of the sequence of a particular stretch of DNA. This fact is related to the frequency of sequencing errors. Although the accuracy associated with any given nucleotide is generally very high, there are huge numbers of nucleotides in the human genome and, therefore, if the entire sequence is obtained only once, there is the potential for a significant number of sequencing errors. This significance is made most acutely obvious when considering the rare single-nucleotide polymorphisms (SNPs) in the genome of every individual. Therefore, it is necessary to be able to distinguish between sequencing errors and true SNPs and this can only be accomplished through deep (repeated) sequencing to ensure accuracy.
RNA-Seq: Deep Sequencing RNA
Deep sequencing of RNA first involves the conversion of mRNA into cDNA followed by multiple rounds of sequencing of the resulting cDNAs. Deep sequencing of RNA, therefore, allows for information regarding both the sequence and the frequency of the mRNA molecules that are present at any particular time in any specific cell type, tissue, or organ.
Background to Cloning
Any fragment of DNA can be cloned once it is introduced into a suitable vector for transforming a bacterial or eukaryotic host cell. Cloning refers to the production of large quantities of identical DNA molecules and usually involves the use of a bacterial cell as a host for the DNA, although cloning can be done in eukaryotic cells as well. cDNA cloning refers to the production of a library of cloned DNAs that represent all mRNAs present in a particular cell or tissue. Genomic cloning refers to the production of a library of cloned DNAs representing the entire genome of a particular organism. From either of these types of libraries one can isolate (by a variety of screening protocols) a single cDNA or gene clone.
In order to clone either cDNAs or copies of genes, a vector is required to carry the cloned DNA. Vectors used in molecular biology are, principally of two basic types. One class of vectors is derived from bacterial plasmids. Plasmids are circular DNAs found in bacteria that replicate autonomously from the host genome. These DNAs were first identified because they harbored genes that conferred antibiotic resistance to the bacteria. The antibiotic resistance genes found on the original plasmids are used in modern in vitro engineered plasmids to allow selection of bacteria that have taken up the plasmids containing a DNA fragment of interest.
Plasmids are limited in that, in general, fragments of DNA less than 10,000 base pairs (bp) can be cloned. In practice fragments of around 5,000 bp are the limit. Most plasmids used for cloning DNA are engineered to contain an origin of replication, an antibiotic resistance gene, and a multicloning site (MCS). The MCS is composed of a stretch of DNA that contains the recognition sequences for numerous restriction enzymes allowing for the insertion of a wide variety of DNA fragments.
In addition, many plasmids have the MCS inserted into the bacterial gene encoding the enzyme β-galactosidase. This feature allows for the rapid detection of recombinant plasmids by color selection, referred to as the “blue-white” screening protocol. Plasmid transformed bacteria are grown in the presence of both antibiotic and a chromophoric substrate (X-gal) for β-galactosidase. Growth in the presence of the antibiotic indicates the bacteria took up a plasmid and if the resulting colonies are whitish then the bacteria were transformed with a recombinant plasmid that disrupted the β-galactosidase gene and, therefore, could not hydrolyze the X-gal to generate a bluish product.
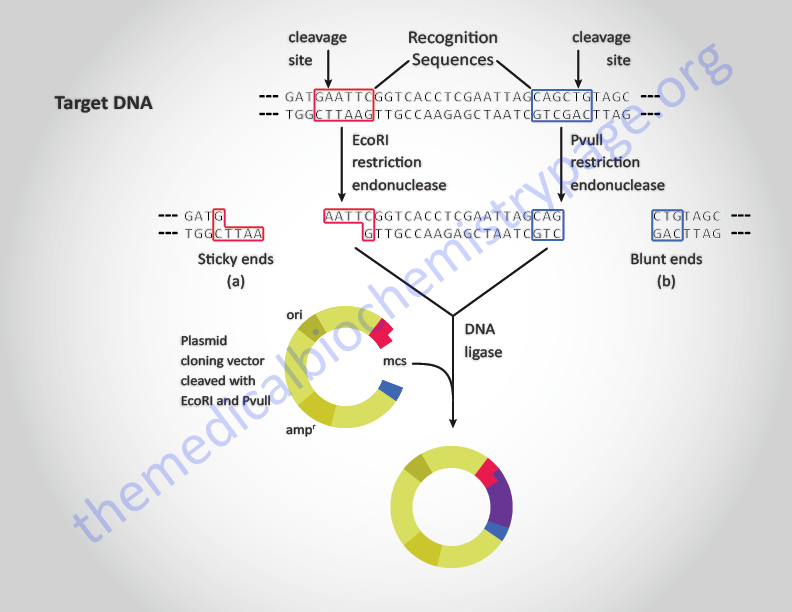
A second class of vectors is derived from the bacteriophage (bacterial virus) lambda. This virus is capable of both lysogeny (integration into the host genome) and lysis (infection followed by lysis of the infected host). The genes required for lysogeny have been removed from the lambda based vectors in order to allow only the lytic life cycle to take place. The advantage to lambda-based vectors is that they can carry fragments of DNA up to 25,000 bp. In the analysis of the human genome even lambda-based vectors are limiting and a yeast artificial chromosome (YAC) vector system has been developed for the cloning of DNA fragments up to 500,000 bp (see below).
cDNA Cloning
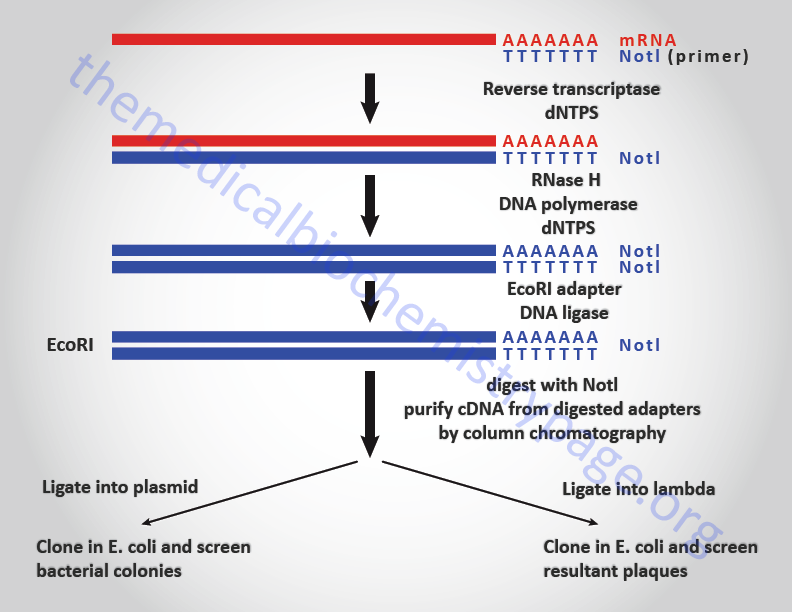
cDNAs are made from the mRNAs of a cell by any number of related techniques. Each technique consists of first reverse transcription of the mRNA followed by synthesis of the second strand of DNA and insertion of the double-stranded cDNA into either a plasmid or lambda vector for cloning. This process creates a library of cloned cDNA representing each mRNA species. Screening of the library for a particular cDNA clone is accomplished using nucleic acid or protein-based (proteins or antibodies) probes. cDNA libraries can also be screened by biological assay of the products produced by the cloned cDNAs. Screening with proteins, antibodies or by biological assay are mechanisms for analysis of the expression of proteins from cloned cDNAs and is given the term expression cloning. Nucleic acids probes can be generated from DNA (including synthetic oligonucleotides, oligos) or RNA. Nucleic acid probes can be radioactively labeled or labeled with modified nucleotides that are recognizable by specific antibodies and detected by colorimetric or chemiluminescent assays.
Genomic Cloning
The majority of genomic cloning utilizes lambda-based vector systems. These vector systems are capable of carrying 15-25,000 bp of DNA. Cloning slightly larger fragments of genomic DNA can be accomplished using a chimeric plasmid-lambda vector system termed a cosmid. Cosmid vectors contain only the cos (cohesive) ends of the lambda genome (required for packaging the DNA into infectious virus particles) along with a plasmid antibiotic resistance gene and origin of DNA replication. Since approximately 30,000 bp of lambda DNA have been removed from cosmid vectors, larger genomic DNA fragments can be cloned. Still larger genomic DNA fragments can be cloned into YAC vectors (see below).
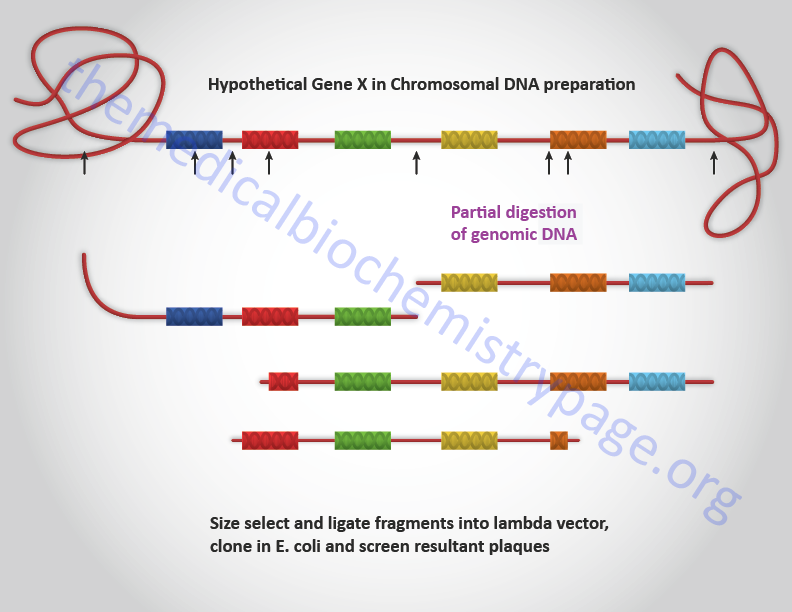
Genomic DNA can be isolated from any cell or tissue for cloning. The genomic DNA is first digested with restriction enzymes to generate fragments in the size range that are optimal for the vector being utilized for cloning. Given that some genes encompass many more base pairs than can be inserted into conventional lambda or cosmid vectors, the clones that are present in a genomic library must be overlapping. In order to generate overlapping clones, the DNA is only partially digested with restriction enzymes. This means that not every restriction site, present in all the copies of a given gene in the preparation of DNA, is cleaved. The partially digested DNA is then size-selected by a variety of techniques (e.g. gel electrophoresis or gradient centrifugation) prior to cloning. Screening of genomic libraries is accomplished primarily with nucleic acid-based probes. However, they can be screened with proteins that are known to bind specific sequences of DNA (e.g. transcription factors). A typical genomic DNA cloning protocol is diagrammed below.
Cloning Genomic DNA in YAC Vectors
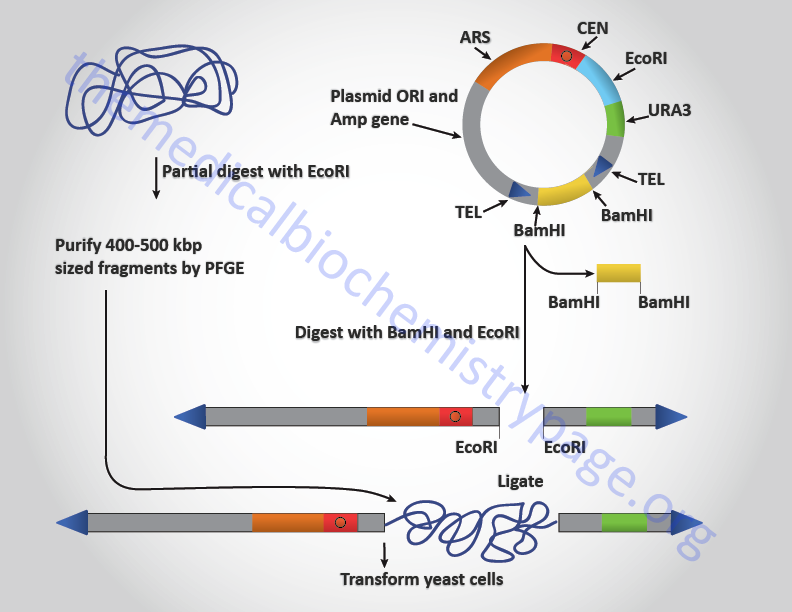
YAC vectors allow the cloning, within yeast cells, fragments of genomic DNA that approach 500,000 bp. These vectors contain several elements of typical yeast chromosomes, hence the term YAC. The YAC vectors contain a yeast centromere (CEN), yeast telomeres (TEL), telomeres are the specific sequences that are present at the ends of chromosomes and are necessary for replication) and a yeast autonomously replicating sequence (ARS). Yeast ARS are essentially origins of replication that function in yeast cells autonomously from the replication of yeast chromosomal replication origins. YAC vectors also contain genes, (e.g. URA3, a gene involved in uracil synthesis) that allow selection of yeast cells that have taken up the vector. In order to propagate the vector in bacterial cells, prior to insertion of genomic DNA, YAC vectors contain a bacterial replication origin and a bacterial selectable marker such as the gene for ampicillin resistance.
In the cloning of genomic DNA in a typical YAC vector, the genomic DNA is partially digested with EcoRI and fragments in the range of 400–500 kilobase pairs (kbp) are purified by pulsed field gel electrophoresis, PFGE. The YAC vector is digested with EcoRI and BamHI which places the telomere sequences at the ends of the linearized vector. The small BamHI fragment is separated from the rest of the YAC vector by standard gel electrophoresis. The genomic DNA is then ligated into the vector and then used to transform yeast cells.
Analysis of Cloned Products
The analysis of cloned cDNAs and genes involves a number of techniques. The initial characterization usually involves mapping of the number and location of different restriction enzyme sites. This information is useful for DNA sequencing since it provides a means to digest the clone into specific fragments for sub-cloning, a process involving the cloning of fragments of a particular cloned DNA. Once the DNA is fully characterized cDNA clones can be used to produce RNA in vitro and the RNA translated in vitro to characterize the protein. Clones of cDNAs also can be used as probes to analyze the structure of a gene by Southern blotting or to analyze the size of the RNA and pattern of its expression by Northern blotting. Northern blotting is also a useful tool in the analysis of the exon-intron organization of gene clones since only fragments of a gene that contain exons will hybridize to the RNA on the blot.
Southern Blotting:
Southern blotting is the analysis of DNA structure following its attachment to a solid support. The DNA to be analyzed is first digested with a given restriction enzyme then the resultant DNA fragments are separated in an agarose gel. The gel is treated with NaOH to denature the DNA, then the NaOH is neutralized. The DNA is transferred from the gel to nitrocellulose or nylon filter paper by either capillary diffusion or under electric current. The DNA is fixed to the filter by baking or ultraviolet light treatment. The filter can then be probed for the presence of a given fragment of DNA by various radioactive or non-radioactive means.
Northern Blotting:
Northern blotting involves the analysis of RNA following its attachment to a solid support. The RNA is sized by gel electrophoresis then transferred to nitrocellulose or nylon filter paper as for Southern blotting. Probing the filter for a particular RNA is done similarly to probing of Southern blots.
Western Blotting:
Western blotting involves the analysis of proteins following attachment to a solid support. The proteins are separated by size SDS-PAGE and electrophoretically transferred to nitrocellulose or nylon filters. The filter is then probed with antibodies raised against a particular protein.
Restriction Fragment Length Polymorphism (RFLP) Analysis
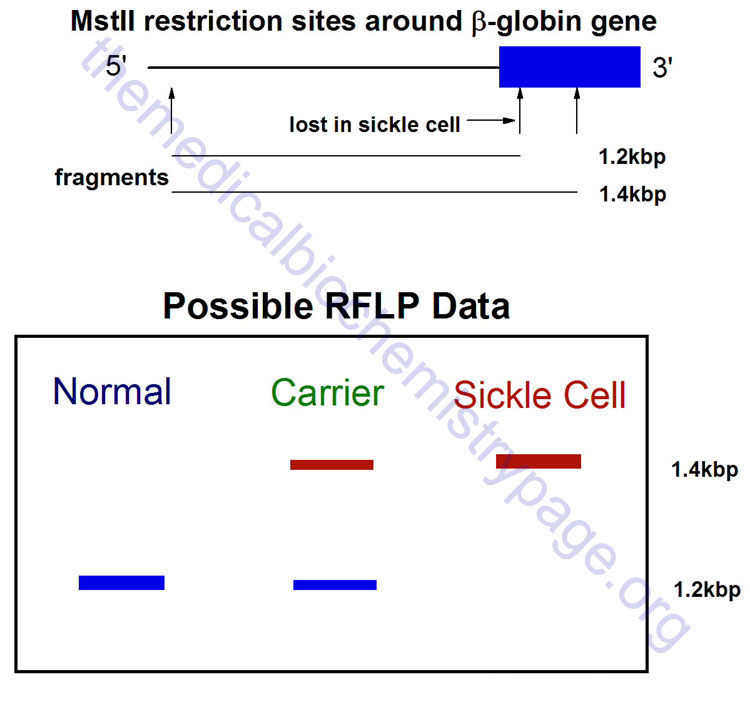
The genetic variability at a particular locus (gene) due to even minor base changes can alter the pattern of restriction enzyme digestion fragments that can be generated. Pathogenic alterations to the genotypic can be due to deletions or insertions within the gene being analyzed or even single nucleotide substitutions that can create or delete a restriction enzyme recognition site. RFLP analysis takes advantage of this and utilizes Southern blotting of restriction enzyme digested genomic DNA to detect familial patterns of the fragments of a given gene, detectable by screening the Southern blot with a probe corresponding to the gene of interest. A classic example of a disease detectable by RFLP is sickle cell anemia.
Sickle cell anemia results (at the level of the gene) from a single nucleotide change (A to T) at codon 6 within the β-globin gene. This alteration leads to a glu (G) to val (V) amino acid substitution, while at the same time abolishing a MstII restriction site. As a result a β-globin gene probe can be used to detect the different MstII restriction fragments. It should be recalled that there are two copies of each gene in all human cells, therefore, RFLP analysis detects both copies: the affected allele and the unaffected allele.
Size variability in detectable fragments within a family pedigree indicate differences in the pattern of restriction sites within and around the gene being analyzed. RFLP patterns are inherited and segregate in Mendelian fashion thus, allowing their use in genotyping such as in cases of paternity dispute or in criminal investigations.
Another form of DNA polymorphism detectable by classical RFLP mapping results from the inherited variations in the number of tandemly repeated DNA sequence elements that are from 2 to 60 bp in length. The number of repeats is also variable from 2 to 40 copies. These elements are termed variable number tandem repeats (VNTR). When restriction enzyme digestion cuts DNA flanking the VNTRs, the lengths of the resultant fragments will be variable depending upon the number of repeats at a given locus. Many different VNTR loci have been identified and are extremely useful for DNA fingerprint analysis such as in forensic and paternity identity cases.
The Polymerase Chain Reaction (PCR)
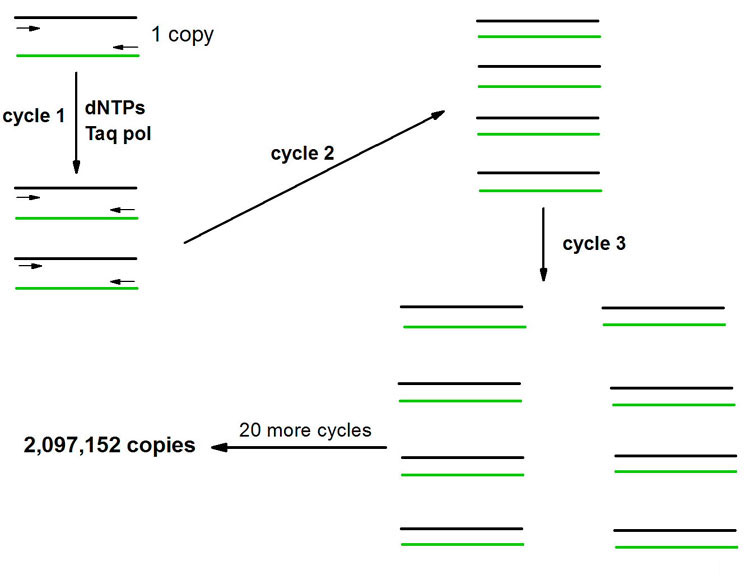
The PCR is a powerful technique used to amplify DNA millions of fold, by repeated replication of a template, in a short period of time. The process utilizes sets of specific in vitro synthesized oligonucleotides to prime DNA synthesis. The design of the primers is dependent upon the sequences of the DNA that is desired to be analyzed. The technique is carried out through many cycles (usually 20–50) of melting the template at high temperature, allowing the primers to anneal to complimentary sequences within the template and then replicating the template with DNA polymerase. The process has been automated with the use of thermostable DNA polymerases isolated from bacteria that grow in thermal vents in the ocean or hot springs. During the first round of replication a single copy of DNA is converted to two copies and so on resulting in an exponential increase in the number of copies of the sequences targeted by the primers. After just 20 cycles a single copy of DNA is amplified over 2,000,000 fold.
The products of PCR reactions are analyzed by separation in agarose gels followed by ethidium bromide staining and visualization with uv transillumination. Alternatively, radioactive dNTPs can be added to the PCR in order to incorporate label into the products. In this case the products of the PCR are visualized by exposure of the gel to x-ray film. The added advantage of radiolabeling PCR products is that the levels of individual amplification products can be quantitated.
PCR can be used in the analysis of disease genes by being able to amplify detectable amounts of specific fragments of DNA. The amplified fragments from disease genes may be larger, due to insertions, or smaller, due to deletions. The dramatic amplification of DNA by PCR allows the analysis of disease genes in extremely small samples of DNA. For example, only a small number of fetal cells need be extracted from amniotic fluid in order to analyze for the presence of specific disease genes. Additionally, single point mutations can be detected by modified PCR techniques such as the ligase chain reaction (LCR) and PCR-single-strand conformational polymorphisms (PCR-SSCP) analysis. The PCR technique also can be used to identify the level of expression of genes in extremely small samples of material, e.g. tissues or cells from the body. This technique is termed reverse transcription-PCR (RT-PCR)
Table of Examples of Inherited Disorders Detectable by PCR
| Disease | Affected Gene |
| Severe-combined immunodeficiency, SCID | adenosine deaminase (ADA) |
| Lesch-Nyhan syndrome | hypoxanthine-guanine phosphoribosyltransferase (HGPRT) |
| α1-Antitrypsin deficiency | α1-Antitrypsin |
| Cystic fibrosis | cystic fibrosis transmembrane conductance (CFTR) protein |
| Fabry disease | α-galactosidase |
| Gaucher disease | acid β-glucosidase (glucocerebrosidase) |
| Sandhoff disease | hexosaminidase A and B |
| Tay-Sachs disease | hexosaminidase A |
| Familial hypercholesterolemia (FH) | LDL receptor |
| Glucose-6-phosphate dehydrogenase deficiency | glucose-6-phosphate dehydrogenase |
| Maple syrup urine disease | branched-chain α-keto acid dehydrogenase |
| Phenylketonuria (PKU) | phenylalanine hydroxylase |
| Ornithine transcarbamylase deficiency | ornithine transcarbamylase |
| Retinoblastoma (Rb) | RB gene product, pRB |
| Sickle-cell anemia | point mutation in β-globin |
| β-Thalassemia | mutations in β-globin gene that result in loss of synthesis of protein |
| Hemophilia A | Factor VIII |
| Hemophilia B | Factor IX |
| von Willebrand disease | von Willebrand factor (vWF) |
Reverse Transcription-PCR (RT-PCR)
RT-PCR is a rapid and quantitative procedure for the analysis of the level of expression of genes. This technique utilizes the ability of reverse transcriptase (RT) to convert RNA into single-stranded cDNA and couples it with the PCR-mediated amplification of specific types of cDNAs present in the RT reaction. The cDNAs that are produced during the RT reaction represent a window into the pattern of genes that are being expressed at the time the RNA was extracted.
Total cellular RNA can be extracted from tissues or cells by any of several techniques and used as a template for RT. In most cases the RNA is primed using random primers. A small aliquot of the RT reaction is then added to a PCR reaction containing primers specific to the sequences one wishes to amplify. The products of the RT-PCR can be then be visualized as described above for standard PCR.
PCR-Single-Strand Conformation Polymorphism (PCR-SSCP)
Many inherited disorders are due to single nucleotide changes within critical regions of the affected gene (eg sickle cell anemia). The PCR-SSCP technique can detect single mutations in genes due to the altered conformation mobility of the single strands of DNA (within an electrophoresis gel) harboring the mutation relative to the wild-type strands that do not. Specific PCR primers are made that span the sequences of a given disease gene where a mutation is known to exist and the region amplified by PCR. The same region of the wild-type gene is PCR amplified. The two strands of wild-type PCR product will migrate differently than the two strands of mutant PCR product. Even single point mutations lead to the strands of amplified DNA existing in different conformations which alter their mobility when subjected to electrophoresis in non-denaturing gels.
In order to accurately visualize the PCR products following gel electrophoresis either the primers are radioactively labeled or radioactive nucleotides are incorporated into the PCR products. The PCR products are separated in a polyacrylamide gel and visualized by exposure of the gel to x-ray film. Individuals that are homozygous wild-type at the locus being analyzed will exhibit two distinct bands in the gel as will those individuals that are homozygous mutant. However, due to the nucleotide change the mutant PCR products will migrate with different mobilities in the gel. Individuals that are heterozygous will exhibit a pattern consisting of all four bands.
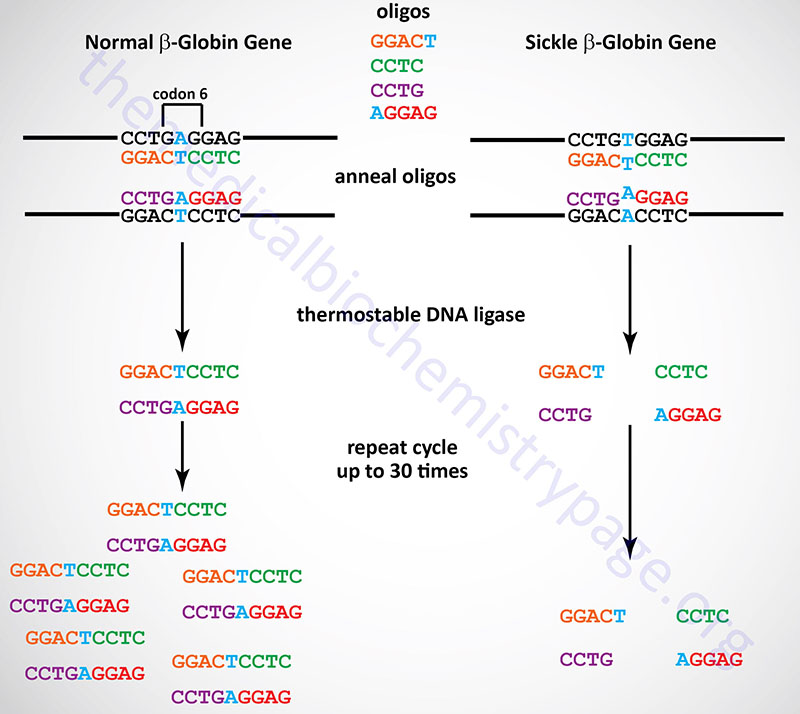
The Ligase Chain Reaction (LCR)
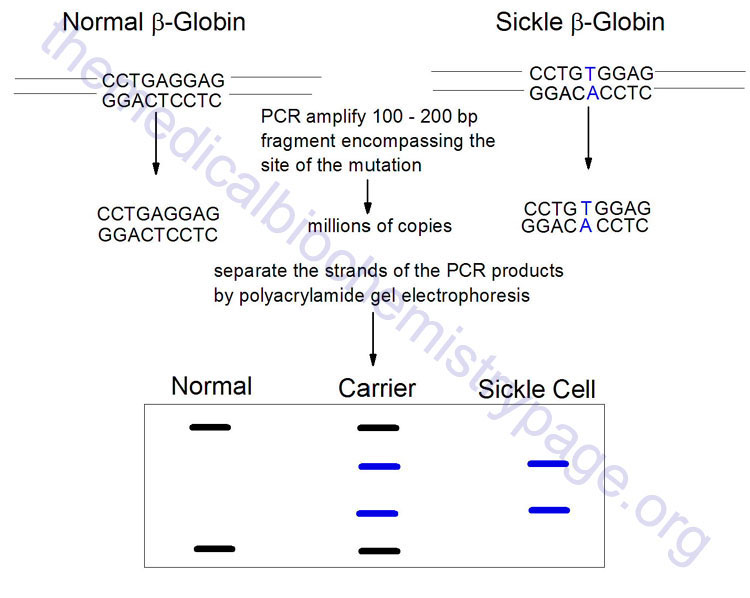
The LCR is another technique that allows detection of single point mutations in disease genes. Although this technique is not generally utilized in a laboratory or diagnostic setting it is briefly described here. The technique utilizes a thermostable DNA ligase to ligate together perfectly adjacent oligos. Two sets of oligos are designed to anneal to one strand of the gene at the site of the mutation, a second set of two oligos anneals to the other strand. The oligos are designed such that they will only completely anneal to the wild-type sequences. In the example shown below for the sickle-cell mutation, the 3′-nucleotide of one oligo in each pair is mismatched. This mismatch prevent the annealing of the oligos directly adjacent to each other. Therefore, DNA ligase will not ligate the two oligos of each pair together. With the wild-type sequence the oligo pairs that are ligated together become targets for annealing the oligos and, therefore, result in an exponential amplification of the wild-type target. Given that prior sequence knowledge is required in order to detect point mutations in disease genes, the LCR technique is utilized for the diagnosis of the presence of a mutant allele in high risk patients.
Microarray Analysis
Microarray analysis involves the use of what are commonly called “gene chips” to determine the expression of a large set of genes at the same time in a single experiment. Gene chips can be purchased from several different companies, eg Affymetrix, or they can be custom prepared in laboratories with the proper equipment. Affymetrix gene chips are created through the covalent attachment of synthetic oligonucleotides (oligos) to a small surface. In general, there are 20 or more different oligos on the chip corresponding to different regions of each gene to be analyzed. In addition, a set of oligos that each contain a nucleotide mismatch are included as negative controls for each gene. The technology of creating gene chips is such that there can be 10’s of thousands of different genes represented on a single chip approximately 2cm square.

Although there are numerous uses for gene chips, the most common experiment involves a comparison of the expression of the genes on the chip between two samples, e.g. cancer cells and normal cells. The assay is carried out by preparing RNA from each sample and converting the RNA to cDNA in the presence of fluorescent nucleotides. For example, one RNA sample is converted to cDNA with a green fluorescent nucleotide and the other RNA sample is converted to cDNA in the presence of a red fluorescent nucleotide. These “tagged” cDNA preparations are called “targets” and equal amounts of each target preparation are mixed together and then hybridized to the gene chip. After washing off the unhybridized targets and image processing of the chip one will see spots that are only green, only red, or a color in between that represents a mix of some red and some green. Thus, some spots will be yellow, some will be orange or degrees of these intermediate combination colors. Spots that are only red indicate that the gene was expressed only in the source of the red labeled targets and vice versa for green spots. Intermediate colors indicate different levels of expression of a gene in both samples. Using a computer to determine hybridization intensity one will get a complete picture of the level of expression of each gene on the chip in each RNA preparation.
Transgenesis
Transgenesis refers to the process of introducing exogenous genes into the germ line of an organism. The first successful transgenesis experiments were carried out in mice. One relatively well known experiment involved the introduction of the rat growth hormone gene into the germ line of mice. These transgenic mice grew to twice their normal size.
To create a transgenic animal the gene of interest must be passed from generation to generation, i.e. it must be inherited in the germ line. To accomplish this with mice or livestock animals, vectors containing the gene of interest with appropriate regulatory elements (e.g. the β-lactoglobulin promoter if expression of the transgene in the milk is desired) are injected into the nucleus of fertilized eggs. The eggs are then transplanted into the uterus of receptive females for development of the potential transgenic offspring. In order to test the resultant animal for germ line transmission of the transgene the chromosomal DNA of their offspring is tested for the presence of the transgene. If the transgene exhibits Mendelian inheritance then it is being transmitted in the germ line.
Currently the process of transgenesis is being utilized in both the plant and livestock industries. The aim of the majority of these experiments is to generate plants and animals that are more resistant to diseases and infections. However, some transgenic farm animal such as sheep and cows are being developed in order to obtain high levels of expression of therapeutically important proteins during milk synthesis. This allows large amounts of the protein of interest to be purified from the milk of the transgenic animals.
Gene Therapy
Transgenesis with humans would allow for the elimination of disease genes in a population of offspring, however, technical as well as ethical issues likely will prevent any transgenic experiments to be carried out with human eggs. Therefore, the ability to replace known disease genes with normal copies in afflicted humans is the ultimate goal of gene therapy. Human gene therapy protocols aim to introduce correcting copies of disease genes into somatic cells of the affected individual. Expression of a correct copy of an affected gene in somatic cells prevents transmission through the germ line, thereby, avoiding many of the ethical issues of transgenesis. This is analogous to treatment of individuals by organ or tissue transplantation.
The most common techniques utilized in gene therapy studies is the introduction of the corrected gene into bone marrow cells, skin fibroblasts or hepatocytes. Early gene therapy techniques utilized vectors derived from retroviruses and these constructs utilized only the transcriptional promoter regions (the long terminal repeats: LTR) of these viruses to drive expression of the gene of interest. The advantage of these retroviral-based vector systems was that expression could be detected in most cell types. Current vector systems used for gene therapy are derived from adenovirus (Ad), adeno-associated virus (AAV), and lentivirus.
A number of human inherited disorders have been corrected in cultured cells and several diseases (e.g. malignant melanoma and severe combined immunodeficiency disease, SCID) are currently being treated by gene therapy techniques indicating that gene therapy is likely to be a powerful therapeutic technique against a host of diseases in coming years. Several clinical trials using Ad vector systems to treat a variety of cancers are currently in various phases. At least 30 different diseases/disorders are currently undergoing clinical trials using AAV vector systems as treatments. At least 15 diseases/disorders are currently undergoing clinical trials using lentivirus vector systems as treatments.
Table of Representative Human Disorders Treated in Cultured Cells by Gene Therapy
| Disorder | Affected Gene |
| SCID | adenosine deaminase (ADA) |
| SCID | purine nucleoside phosphorylase (PNP) |
| Lesch-Nyhan syndrome | hypoxanthine-guanine phosphoribosyltransferase (HGPRT) |
| Gaucher disease | acid β-glucosidase (glucocerebrosidase) |
| Familial hypercholesterolemia (FH) | LDL receptor |
| Phenylketonuria (PKU) | phenylalanine hydroxylase |
| β-Thalassemia | β-Globin |
| Hemophilia B | Factor IX |
CRISPR-Mediated Gene Editing
The CRISPR-Cas9 gene editing system was first described in 2012 by Professor Emmanuelle Charpentier and colleagues. The system was discovered as part of an adaptive bacterial defense system against the invasion of foreign bacteriophages. Since the initial discovery, programmable CRISPR-Cas nucleases have emerged as powerful tools capable of a broad range of activities that includes the ability to specifically edit the genome of cells.
Other forms of gene editing have been developed but do not possess the simplicity nor broad utility of the CRISPR-Cas nuclease system. These other gene editing processes involve the use of the meganucleases, the zinc-finger nucleases (ZFN), and the transcription activator-like effector nucleases (TALENs).
Meganucleases are enzymes that recognize large stretches of DNA sequence ranging from 12 to 45 base pairs and can then cut the DNA at specific sites in that recognition sequence. The limitation to the broad use of meganucleases for gene editing stems from the fact that finding large, specific sequence motifs in a gene of interest, let alone many different genes, is extremely rare.
Zinc-finger nucleases (ZFN) a family of engineered nucleases that, as the name implies, contain zinc-finger motifs. The zinc-finger motif if found in numerous proteins, in particular in many different transcription factors. Each zinc-finger can recognize 3 or 4 base pairs and, when combined, a ZFN can bind to a specific DNA sequence of 24 base pairs or longer in length. Zinc-finger nucleases have been engineered to cut a wide range of DNA with a high degree of specificity. Nonetheless, the use of ZFN is not risk free. Due to specificity issues ZFN can cut DNA unintentionally at undesired, off-target locations leading to undesired results.
Transcription activator-like effector nucleases (TALENs) have increased target specificity due to the fact that the TALE component recognizes a single nucleotide. The TALEN-mediated gene editing systems have been used with a high degree of success in the editing of plant genomes.
Unlike the other gene editing techniques, which require the engineering of the editing nuclease, the CRISPR-Cas system only requires the development of single guide RNA (sgRNA) to target a specific DNA sequence. When the sgRNA is combined with a nuclease protein (such as Cas9) efficient and flexible gene editing is possible. This is because CRISPR-Cas nuclease specificity is not simply a function of its binding affinity. Unlike the ZFN and TALEN systems CRISPR-Cas nucleases do not passively bind to double-stranded DNA, they actively unwind the DNA duplex and recognize their target sequences through a combination of protein:DNA and RNA:DNA interactions.
The utility of the CRISPR-Cas gene editing technique can be appreciated by the fact that the company founded by Emmanuelle Charpentier (CRISPT Therapeutics) has received approval from the US FDA, as well as the related EU and UK entities, for its treatment/cure of sickle cell disease and β-thalassemia called Casgevy (exagamglogene autotemcel).