Last Updated: May 27, 2026
Introduction to DNA Structure, Functions, and Replication
All cells undergo a process of DNA replication and a division cycle during their life span. Some cells are continually dividing (e.g. stem cells), others divide a specific number of times until cell death (apoptosis) occurs, and still others divide a few times before entering a terminally differentiated or quiescent state. Most cells of the body fall into the latter category of cells. During the process of cell division everything within the cell must be duplicated in order to ensure the survival of the two resulting daughter cells. Of particular importance for cell survival is the accurate, efficient and rapid duplication of the cellular genome. This process is termed DNA replication.
Eukaryotic Genomes
The size of eukaryotic genomes is vastly larger than those of prokaryotes. This is partly due to the complexity of eukaryotic organisms compared to prokaryotes. However, the size of a particular eukaryotic genome is not directly correlated to the organisms complexity. This is the result of the presence of a large amount of non-coding DNA. The functions of these non-coding nucleic acid sequences are only partly understood. Some sequences are involved in the control of gene expression while others may simply be present in the genome to act as an evolutionary buffer able to withstand nucleotide mutation without disrupting the integrity of the organism.
One abundant class of non-coding DNA is termed repetitive DNA. There are two primary sub-classes of repetitive DNA, highly repetitive and moderately repetitive. Highly repetitive DNA can be sub-divided into two distinct subclasses termed microsatellite DNA and minisatellite DNA.
Microsatellite DNA consists of short repeat sequences 2–6 bp long reiterated from 100,000–1,000,000 times. These microsatellite sequences of DNA are commonly called short tandem repeats (STR). Since the number of copies of the short repeat sequence is highly variable (i.e. polymorphic) between two different individuals the elements are referred to as short tandem repeat polymorphisms (STRP).
Replication of STR DNA frequently results in mismatching of the DNA strands which is normally corrected by a family of enzymes encoded by mismatch repair (MMR) genes. Defects in MMR genes are highly correlated to an increased likelihood of certain types of cancers, in particular colorectal carcinomas. The involvement of mismatch of the microsatellite DNA in diseases such as colorectal carcinomas is known as microsatellite instability, MSI.
Minisatellite DNA contains repeat sequences that are 10–60 bp in length. The number of copies of these types of repeats is also highly variable (polymorphic) and so the repetitive DNA is most commonly referred to as variable number tandem repeat (VNTR) DNA.
The DNA of the genome consisting of the genes (primarily referring to protein coding sequences) is identified as non-repetitive DNA since most genes occur but once in an organism’s haploid genome. However, it should be pointed out that several genes exist as tandem clusters of multiple copies of the same gene ranging from several copies to hundreds of copies such as is the case for the histone genes. Currently (as of 2020) there are 19,218 protein-coding genes in the human genome. There are also thousands of genes that generate non-coding RNA such as those encoding the rRNAs and tRNAs and those encoding the small non-coding RNAs (sncRNA) and the long non-coding RNAs (lncRNA).
Another characteristic feature that distinguishes eukaryotic from prokaryotic genes is the presence of introns. Introns are stretches of nucleic acid sequences that separate the coding exons of a gene. The existence of introns in prokaryotes is extremely rare or nonexistent. Although essentially all humans genes contain introns there are numerous different mRNA encoding genes in the human genome that contain no introns. Notable intronless genes are the histone genes. In many genes the presence of introns separates exons into coding regions exhibiting distinct functional domains.
Chromatin Structure
Histone Genes and Histone Proteins
Chromatin is a term designating the structure in which DNA exists within cells. The structure of chromatin is determined and stabilized through the interaction of the DNA with DNA-binding proteins. There are two classes of DNA-binding proteins. The histones are the major class of DNA-binding proteins involved in maintaining the compacted structure of chromatin. The other class of DNA-binding proteins is a diverse group of proteins called simply, non-histone proteins. This class of proteins includes the various transcription factors, polymerases, hormone receptors and other nuclear enzymes. In any given cell there are greater than 1000 different types of non-histone proteins bound to the DNA.
There are five primary histone families identified as H1, H2A, H2B, H3, and H4. All of these histones are referred to as the replication-dependent histones. There are approximately 10-20 individual genes encoding each of the core histones of the nucleosome (H2A, H2B, H3, and H4) as well as the linker histone, H1. The majority of the human histone proteins are derived from large clusters of genes located on chromosomes 1 and 6 and are, therefore, defined as cluster histones. Not all of the histone genes express replication-dependent histones. In addition, each of these histone gene families contain several pseudogenes.
Humans express 10 genes of the H1 family, 6 of which constitute the clustered H1 genes on chromosome 1. The H1 family also encompasses two pseudogenes.
Humans express 29 genes of the H2A family, 17 of which constitute the clustered H2A genes on chromosomes 1 and 6. The other 12 non-clustered H2A genes encode what are defined as variant histones. The H2A family also encompasses seven pseudogenes.
Humans express 23 genes of the H2B family, 19 of which constitute the clustered H2B genes on chromosomes 1 and 6. The other four non-clustered H2B genes encoded what are defined ad variant histones. The H2B family also encompasses eight pseudogenes.
Humans express 21 genes of the H3 family, 13 of which constitute the clustered H3 genes on chromosome 6 and one cluster member on chromosome 1.
Humans express 15 genes of the H4 family, all but one of which constitute the clustered H4 genes. Of these 15 genes 12 are on chromosome 6, two are on chromosome 1 and one is on chromosome 12. The H4 family also encompasses one pseudogene.
All of the histone mRNAs derived from the histone gene clusters are synthesized from intronless genes and are produced without a poly(A) tail. The 3′-end of all of these replication-dependent histone mRNAs terminate with a stem-loop structure that is followed by a purine-rich region that is complementary to the 5′-end of the U7 snRNA.
The replication-dependent histone genes encoding the H2A and H2B proteins generate 10-20 different protein isoforms.
The genes encoding the H3 proteins encode two protein isoforms.
The H4 genes all encode identical proteins. An additional histone gene that produces an H4 mRNA (H4-16; formerly HIST4H4), with the stem-loop structure, is located on chromosome 12p12.3.
In addition to the genes encoding the replication-dependent histones there are several genes encoding histones termed replacement variant histones or replication-independent histones. These variant histones include H2A.X (encoded by the H2AX gene; formerly H2AFX), H2A.Z variant 1 and 2 (encoded by the H2AZ1 and H2AZ2 genes), H3.3A (encoded by the H3-3A gene; formerly H3F3A), H3.3B (encoded by the H3-3B gene; formerly H3F3B), and H1.0 (encoded by the H1-0 gene; formerly H1F0), as well as several additional histone variants.
The Nucleosome
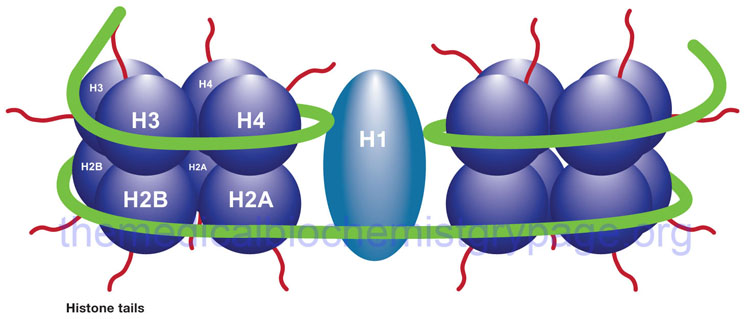
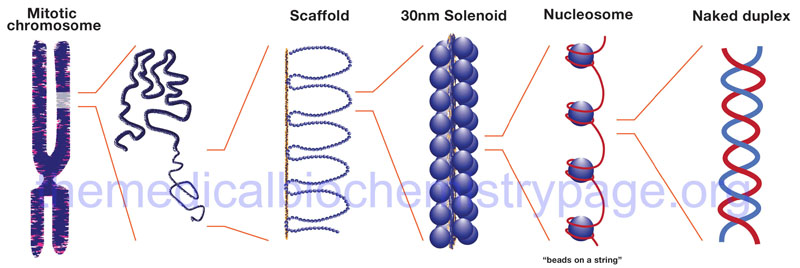
The binding of DNA by the replication-dependent histones generates a structure called the nucleosome. The nucleosome core contains an octameric protein structure consisting of two subunits each of H2A, H2B, H3, and H4. Histone H1 occupies the internucleosomal DNA and is identified as the linker histone. The nucleosome core contains approximately 150 bp of DNA. The linker DNA between each nucleosome can vary from 20 to more than 200 bp. These nucleosomal core structures would appear as “beads-on-a-string” if the DNA were pulled into a linear structure and observed under an electron microscope. The nucleosome cores themselves coil into a solenoid shape which itself coils to further compact the DNA. These final coils are compacted further into the characteristic chromatin seen in a metaphase karyotyping spread.
The protein-DNA structure of chromatin is stabilized by covalent attachment to a non-histone protein scaffold which in turn interacts with the nuclear matrix. The DNA sequences that interact with the scaffold are referred to as scaffold/matrix attachment regions, S/MAR. Thee domains are also known as scaffold attachment regions (SAR) or matrix attachment regions (MAR). The DNA-scaffold interactions organize the chromatin into structural domains. S/MAR allow for the separation of transcriptional units from its neighbors as well as providing platforms for the assembly of transcription factors within a given chromatin domain. Because of their role in the regulation of chromatin structure and thus, transcription, S/MAR are classified as either constitutive, meaning they act as permanent domain boundaries in all cell types, or facultative, meaning they function in a cell type- and activity-related manner. These terms are defined depending on the dynamic properties of a given S/MAR.
In a broad consideration of chromatin structure there are two forms: heterochromatin and euchromatin which were originally designated based on cytological observations of how darkly the two regions were stained. Heterochromatin is more densely packed than euchromatin and is often found near the centromeres of the chromosomes. Heterochromatin is generally transcriptionally silent. Euchromatin on the other hand is more loosely packed and is where active gene transcription will be found to be taking place.
There are several mechanisms operating in a dynamic manner to alter the overall structure of chromatin. The primary mechanisms are methylation of cytidine residues in the DNA that are found in the dinucleotide, –CG– (most often written as a CpG dinucleotide) and histone protein modification. Because both DNA methylation and histone modifications can alter chromatin structure and, thereby, alter the transcriptional activity of genes, both of these types of modification are termed epigenetic processes. The term epigenetics means that changes in phenotype can come about, not by changes in the actual DNA sequences but by changes that occur on the genes. Methylation of cytidine residues, as a post-replication modification of DNA, is discussed below and general processes involved in DNA methylation are covered in the following section.
Nucleotide Modifications Regulating Chromatin Structure
DNA Methylation: Formation of m5C (5mC)
When determining which C residues in DNA are targets for methylation it was discovered that essentially 100% of methyl-C is found in the dinucleotide, CpG. The cytidine is methylated at the 5 position of the pyrimidine ring generating 5-methylcytidine (designated m5C or 5mC). This is not to say that all CpG dinucleotides contain a methylated C residue. When examining the structure of eukaryotic genes and identifying regions of CpG dinucleotides it is the case that the promoter regions of genes contain 10–20 times as many CpGs when compared to the rest of the genome. In an examination of the global methylation status in the human genome it has been shown that 60%–80% of all the CpG dinucleotides present in the genome contain 5mC.
In a general sense what is known about DNA methylation and transcriptional status is that when regions of a gene that can be methylated are methylated, the associated gene(s) is(are) transcriptionally silent and when the region is under-methylated the gene(s) is(are) transcriptionally active or can be activated. When cells undergo differentiation it has been observed that genes that become transcriptionally activated exhibit a reduction in methylation status relative to the level prior to activation and that this under-methylation remains even after transcription ceases. The correlation between DNA methylation and chromatin structure, as it relates to transcriptional activity, is discussed in greater detail in the Regulation of Gene Expression page.
The methylation of DNA is catalyzed by several different DNA methyltransferases (abbreviated DNMT). The methyl donor for the methylation reaction is S-adenosylmethionine (SAM; also abbreviated AdoMet). Humans express three DNMT genes identified as DNMT1, DNMT3a, and DNMT3b.
The DNMT1 gene is located on chromosome 19p13.2 and is composed of 41 exons that generate four alternatively spliced mRNAs that encode four distinct protein isoforms. The DNMT1 isoform a is the largest isoform and is a 1632 amino acid protein.
The DNMT3a gene is located on chromosome 2p23 and is composed of 34 exons that generate six alternatively spliced mRNAs encoding four distinct proteins.
The DNMT3b gene is located on chromosome 20q11.2 and is composed of 24 exons that generate six alternatively spliced mRNAs encoding six distinct protein isoforms.
Of the three DNA methyltransferases, DNMT1 is the most abundant in all cells. Another gene, identified as DNMT3L (for DNMT3-like) has some similarities to the DNA methyltransferases but does not have the methyltransferase catalytic amino acids. The activity of the DNMT3L protein stimulates the DNA methyltransferase activity of DNMT3a. DNMT3L can also affect transcriptional activity through its association with histone deacetylase 1 (HDAC1). Another gene, that was originally designated DNMT2, and thought to be involved in DNA methylation, in fact encodes an enzyme that methylates a specific aspartic acid tRNA. The designation for this gene is now TRDMT1.
When cells divide the DNA contains one strand of parental DNA and one strand of the newly replicated DNA (the daughter strand). If the DNA contains methylated cytidines in CpG dinucleotides the daughter strand must undergo methylation in order to maintain the parental pattern of methylation. This “maintenance” methylation is catalyzed by DNMT1 and thus, this enzyme is called the maintenance methylase. In addition to DNMT1, maintenance methylation requires another protein as an obligate partner for DNMT1. This additional protein is called ubiquitin-like plant homeodomain and RING finger domain 1 and is encoded by the UHRF1 gene. The RING finger domain is a zinc-finger-like domain which gets its name from the term Really Interesting New Gene. The UHRF1 protein is required for the recognition of the hemimethylated sites in the DNA following replication. As might be expected from its characterized primary function, DNMT1 has an up to 100-fold higher level of activity towards hemimethylated DNA compared to unmethylated DNA. The activities of DNMT3a and DNMT3b are relatively equivalent towards unmethylated and hemimethylated DNA. The critical role of DNA methylation in controlling developmental fates was demonstrated in mice by inactivating either DNMT3a or DNMT3b. Loss of either gene resulted in death shortly after birth.
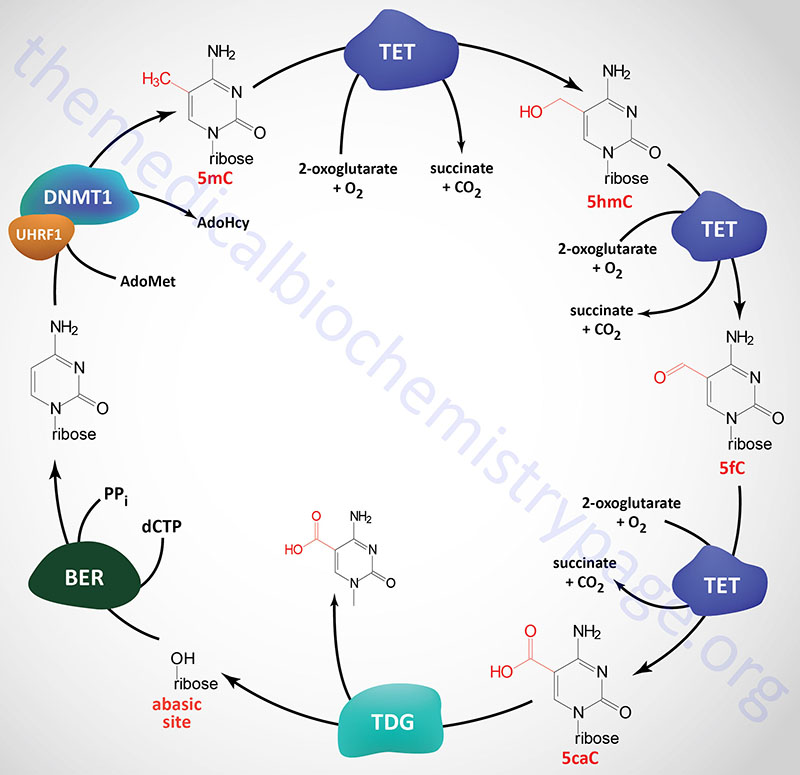
DNA Demethylation: Removal of m5C (5mC)
Given that chromatin structure, and consequently transcriptional activity, can be modified by the addition of a methyl group to cytidine residues, it is not surprising that there are activities in the cell that are responsible for the removal of the methyl group. However, the discovery of enzymes that can remove methyl groups from m5C residues came about rather serendipitously via two independent areas of study. In a search for mammalian homologs of Trypanosoma brucei genes that oxidize thymidine residues in DNA to 5-hydroxymethyluracil (5hmU), a family of three genes was identified. The second approach that resulted in the identification of the same family of genes involved studies aimed at identification of the pathways that led to the introduction of 5-hydroxymethylcytosine (5hmC) in mammalian DNA. The genes that were identified are all related to a gene that was originally identified in rare cases of acute myeloid and lymphocytic leukemia (MLL). This form of leukemia results from a translocation between chromosome 10 and chromosome 11. The translocation results in the fusion of the mixed-lineage leukemia 1 (MLL1) gene on chromosome 10 and a gene on chromosome 11 that was subsequently given the name TET (ten eleven translocation). The MLL1 protein is a lysine methyltransferase (KMT) family enzyme encoded by the KMT2A gene. For details on KMT genes go to the Regulation of Gene Expression page.
Three TET gene are expressed in humans identified as TET1, TET2, and TET3. The official name for these genes is tet methylcytosine dioxygenase 1, 2, and 3. The TET gene encoded enzymes are distantly related to the human ALKB homologs (identified as ALKBH genes) which remove aberrant methylation from damaged DNA bases by an oxidative mechanism.
The TET1 gene is located on chromosome 10q21 and is composed of 20 exons that encode a 2136 amino acid protein.
The TET2 gene is located on chromosome 4q24 and is composed of 11 exons that generate two alternatively spliced mRNAs encoding two distinct protein isoforms.
The TET3 gene is located on chromosome 2p13.1 and is composed of 16 exons that encode a 1795 amino acid protein.
All three TET genes encode proteins that are zinc-finger domain containing 2-oxoglutarate (α-ketoglutarate)- and ferrous iron (Fe2+)-dependent dioxygenases (also identified as 2OG-oxygenases). Each of the three TET enzymes successively oxidize 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) within DNA. All three forms of oxidized methylcytosine have been shown to be present in numerous mammalian tissues.
The primary mechanism for the removal of 5fC and 5caC from DNA involves the action of thymine DNA glycosylase (encoded by the TDG gene). A cytosine is then incorporated, which recovers the original CG base pair, by the process of base excision repair. There are several other proposed mechanisms that may be functional in the removal of 5caC, however, none have been definitively demonstrated experimentally. These mechanisms include direct decarboxylation of 5caC to C by an as yet unknown decarboxylase. Another proposed mechanism involves deamination of 5hmC via the action of activation-induced cytidine deaminase (encoded by the AICDA gene) or via the action of one of the APOBEC (apolipoprotein B mRNA editing enzyme, catalytic polypeptide) family of cytidine deaminases. The resultant products of these two distinct activities are thymine and 5hmU which can then be removed and replaced with cytidine via the actions of single-strand-selective monofunctional uracil DNA glycosylase (encoded by the SMUG1 gene) or TDG.
As indicated, the TET proteins are 2-oxoglutarate and Fe2+-dependent dioxygenases. Many other important enzymes, such as the lysine demethylases that demethylate histone proteins (discussed below and in greater detail in the Regulation of Gene Expression page), are also members of the large family of 2-oxoglutarate- and Fe2+-dependent dioxygenases. Therefore, it has been speculated, and much work directly correlates, that aberrations in the pathways generating 2-oxoglutarate may be important in the development of certain types of tumors.
One of the most significant metabolic pathways in which 2-oxoglutarate is a critical intermediate is the TCA cycle. Several genes, encoding TCA cycle enzymes, have been shown to be mutated in several types of human cancers. Two of these genes are the isocitrate dehydrogenase 1 (IDH1) and IDH2 genes. Although the proteins encoded by IDH1 and IDH2 do not contribute to TCA cycle activity, they are structurally and functionally related to the TCA cycle isocitrate dehydrogenase encoded by the IDH3 gene. Mutations in IDH1 and IDH2 have been found in a large number of different types of cancers. The mutations in these genes are associated with a change in catalytic activity such that instead of oxidizing isocitrate to 2-oxoglutarate, the enzymes oxidize 2-oxoglutarate to 2-hydroxyglutarate (2-HG). This metabolic change can significantly reduce the cellular levels of the 2-oxoglutarate, thereby decreasing its role as a cofactor for dependent dioxygenases such as the TET enzymes and the histone demethylases.
Histone Modifications Regulating Chromatin Structure
Histone proteins are subject to a number of modifications and these modifications are known to affect the structure of chromatin. Greater detail relating to the types and transcriptional consequences of the modification of histones is presented in the Regulation of Gene Expression page.
Histone acetylation is known to result in a more open chromatin structure and these modified histones are found in regions of the chromatin that are transcriptionally active. Conversely, underacetylation of histones is associated with closed chromatin and transcriptional inactivity. A direct correlation between histone acetylation and transcriptional activity was demonstrated when it was discovered that protein complexes, previously known to be transcriptional activators, were found to have histone acetyltransferase activity. And as expected, transcriptional repressor complexes were found to contain histone deacetylase activity.
Linkage between DNA methylation and transcriptional silencing was demonstrated by the observation that proteins that bind to methyl CpG dinucleotides can recruit histone deacetylases to the DNA. Proteins are known to interact with acetylated histones that together lead to a more open chromatin structure. Proteins that bind to acetylated lysines in histones contain a domain called a bromodomain. The bromodomain is composed of a bundle of four α-helices and is a domain involved in protein-protein interactions in a number of cellular systems in addition to acetylated histone binding and chromatin structure modification.
Another histone modification known to affect chromatin structure is methylation. However, with histone methylation there is not a direct correlation between the modification and a specific effect on transcription. The methylation of histone H4 on R4 (arginine at position 4) promotes an open chromatin structure and thereby, leads to transcriptional activation. Methylation of histone H3 on K4 and K79 (lysines 4 and 79) has been shown to act similarly to histone H4 R4 methylation. However, methylation of histone H3 on K9 and K27 is known to be associated with transcriptionally inactive genes.
The methylation of histones provides a site for the binding of other proteins which then leads to alteration of chromatin structure to a more compacted state. Proteins that bind to methylated histones contain a domain called chromodomain. The chromodomain consists of a conserved stretch of 40-50 amino acids and is found in many proteins involved in chromatin remodeling complexes. In addition, chromodomain proteins are found in the RNA-induced transcriptional silencing (RITS) complex which involves small interfering RNA (siRNA) and microRNA (miRNA)-medicated downregulation of transcription. For more details on small non-coding RNAs and transcriptional regulation see the Regulation of Gene Expression page.
Histone proteins can also be modified by addition of the small protein ubiquitin. Ubiquitylation has been observed to occur on all the nucleosomal (replication-dependent) histones but is most often found associated with histones H2A and H2B. When ubiquitylated, H2A is associated with repression of transcription. The exact opposite effect is observed when histone H2B is ubiquitylated, leading to a stimulation of gene activity. The reason that ubiquitylated histone H2B is associated with transcriptional activity is that this modification promotes the methylation of histone H3 at K4 and K79 which, as indicated above, is associated with open chromatin structure.
Phosphorylation of histones occurs primarily in response to outside signals such as growth factor stimulation or stress inducers such as heat shock. Phosphorylated histone are localized to genes that become transcriptionally active as a consequence of these outside signals. The importance of histone phosphorylation in control of gene expression can be demonstrated in patients with Coffin-Lowry syndrome. This disease results from defects in the RPS6KA3 (ribosomal protein S6 kinase A3; also known as ribosomal S6 kinase 2: RSK2) gene. Coffin-Lowry syndrome is a rare form of X-linked intellectual impairment characterized by skeletal malformations, growth impairment, hearing deficit, paroxysmal movement disorders, and cognitive impairment in affected males.
DNA Replication
Replication of DNA occurs during the process of normal cell division cycles. Because the genetic complement of the resultant daughter cells must be the same as the parental cell, DNA replication must possess a very high degree of fidelity. The entire process of DNA replication is complex and involves multiple enzymatic activities.
The mechanics of DNA replication was originally characterized in the bacterium, E. coli which contains 3 distinct enzymes capable of catalyzing the replication of DNA. These have been identified as DNA polymerase (pol) I, II, and III. Pol I is the most abundant replicating activity in E. coli but has as its primary role to ensure the fidelity of replication through the repair of damaged and mismatched DNA. Replication of the E. coli genome is the job of pol III. This enzyme is much less abundant than pol I, however, its activity is nearly 100 times that of pol I.
Up until a few years ago the use of Greek lettering to designate the six known eukaryotic DNA polymerases was sufficient. However, recent evidence indicates that several more members of the eukaryotic DNA polymerase family are present and function in distinct types of DNA replication. These DNA polymerases are divided into four large families designated A, B, X, and Y. In addition, there is a reverse transcriptase activity associated with telomerase as discussed below and the mitochondrial polymerase-primase enzyme. The original six distinct eukaryotic DNA polymerases are identified as α, β, γ, δ, ε, and ζ. The identity of these individual enzymes relates to its subcellular localization, its primary replicative activity and to the order in which it was first described. The known eukaryotic DNA polymerases and descriptions of their activities are indicated in the Table below.
Table of Eukaryotic DNA Polymerases
| Polymerase Family | Common Nomenclature | Enzyme Function, Comments |
| A | γ (gamma) | mitochondrial DNA replication; encoded by the POLG gene on chromosome 15q26.1 |
| A | θ (theta) | DNA repair; encoded by the POLQ gene on chromosome 3q13.33 |
| A | ν (nu) | DNA repair and homologous recombination; encoded by the POLN gene on chromosome 4p16.3 |
| B | α (alpha) | initiation of chromosomal DNA replication, Okazaki fragment priming, also involved in double-strand break repair; functions as a multisubunit complex that includes two primase proteins (encoded by the PRIM1 and PRIM2A genes), an accessory protein (encoded by the POLA2 gene) and the catalytic subunit encoded by the POLA1 gene on chromosome Xp22.1–p21.3 |
| B | δ (delta) | chromosomal DNA replication elongation, nucleotide excision repair, double-strand break repair, mismatch repair; consists of a multisubunit complex that includes the four subunits of the catalytic polymerase encoded by the POLD1, POLD2, POLD3, and POLD4 genes as well as the multisubunit replication factor C protein (RFC1) and proliferating cell nuclear antigen (PCNA) |
| B | ε (epsilon) | chromosomal DNA replication elongation, nucleotide excision repair, double-strand break repair, mismatch repair; consists of a 261kDa catalytic subunit encoded by the POLE gene on chromosome 12q24.33 and a 55kDa accessory protein encoded by the POLE2 gene, additional proteins in the epsilon complex include two histone-fold proteins encoded by the POLE3 and POLE4 genes |
| B | ζ (zeta) | bypass (translesion) DNA synthesis; encoded by the POLZ gene on chromosome 6q21; also known as REV3; enzyme responsible for essentially all DNA damage-induced mutagenesis as well as the majority of spontaneous mutagenesis |
| X | β (beta) | base-excision repair, required for DNA replication and maintenance, recombination, and drug-resistance; encoded by the POLB gene on chromosome 8p11.21 |
| X | λ (lambda) | base-excision repair; encoded by the POLL gene on chromosome 10q24.32 |
| X | μ (mu) | non-homologous end joining (NHEJ); encoded by the POLM gene on chromosome 7p13 |
| X | σ (sigma) | sister chromatid cohesion; encoded by the POLS gene on chromosome 5p15.31; also known as topoisomerase-related function protein 4 (TRF4) and poly(A) polymerase-associated domain-containing protein 7 (PAPD7) |
| Y | η (eta) | bypass (translesion) DNA synthesis; encoded by the POLH gene on chromosome 6p21.1; required for replication through uv-induced cyclobutane pyrimidine dimers (CPD); mutations in POLH result in Xeroderma Pigmentosum variant (XP-V) |
| Y | ι (iota) | bypass (translesion) DNA synthesis; encoded by the POLI gene on chromosome 18q21.2; also known as RAD30B |
| Y | κ (kappa) | bypass (translesion) DNA synthesis; encoded by the POLK gene on chromosome 5q13.3 |
| Y | Rev1L | bypass (translesion) DNA synthesis; encoded by the REV1 gene; interacts with POLK and is essential for POLK function |
| telomerase | telomeric DNA replication; encoded by the TERT gene on chromosome 5p15.33 | |
| PrimPol | primase and DNA directed polymerase; involved in both nuclear and mitochondrial DNA maintenance; functions as a DNA polymerase and is also capable of catalyzing the preferential formation of DNA primers in a zinc finger-dependent manner; encoded by the PRIMPOL gene on chromosome 4q35.1 |
The ability of DNA polymerases to replicate DNA requires a number of additional accessory proteins. The combination of polymerases with several of the accessory proteins yields an activity identified as DNA polymerase holoenzyme. These accessory proteins/complexes include (not ordered with respect to importance):
- Primase complex (DNA polymerase α complex)
- Processivity accessory proteins
- Single strand binding proteins, SSBP
- Helicases
- DNA ligases
- Topoisomerases
The process of DNA replication begins at specific sites in the chromosomes termed origins of replication, requires a primer bearing a free 3’–OH, proceeds specifically in the 5′ → 3′ direction on both strands of DNA concurrently and results in the copying of the template strands in a semiconservative manner. The semiconservative nature of DNA replication means that the newly synthesized daughter strands remain associated with their respective parental template strands.
The large size of eukaryotic chromosomes and the limits of nucleotide incorporation during DNA synthesis, make it necessary for multiple origins of replication to exist in order to complete replication in a reasonable period of time. The precise nature of origins of replication in higher eukaryotic organisms is unclear. However, it is clear that at a replication origin the strands of DNA must dissociate and unwind in order to allow access to all of the accessory proteins and the DNA polymerase complex. Unwinding of the duplex at the origin as well as along the strands as the replication process proceeds is carried out by helicases. Helicases involved in DNA replication are DNA-dependent ATPase with DNA helicase activity. The resultant regions of single-stranded DNA are stabilized by the binding of single-strand binding proteins (SSBPs). The stabilized single-stranded regions are then accessible to the enzymatic activities required for replication to proceed. The site of the unwound template strands is termed the replication fork.
In order for DNA polymerases to synthesize DNA they must encounter a free 3’–OH which is the substrate for attachment of the 5’–phosphate of the incoming nucleotide. During repair of damaged DNA the 3’–OH can arise from the hydrolysis of the backbone of one of the two strands. During replication the 3’–OH is supplied through the use of an RNA primer, synthesized by the activity of the primase complex. The primase complex is composed of four proteins that includes two primase proteins identified as p58 and p49, a p68 accessory subunit of DNA polymerase α and the catalytic subunit of DNA polymerase α. Together these four proteins constitute what is more correctly referred to as the DNA polymerase α complex. The p49 and p58 primase proteins form a heterodimeric complex that interacts with DNA polymerase α and the p68 subunit.
The two primase proteins are encoded by the PRIM1 gene (p49) and the PRIM2A gene (p58). The p68 accessory protein of the complex is encoded by the POLA2 gene and the catalytic DNA polymerase α protein is encoded by the POLA1 gene. The primase complex utilizes the DNA strands as templates and synthesizes a short stretch of RNA generating a primer for DNA polymerase.
The PRIM1 gene is located on chromosome 12q13 and is composed of 13 exons that encode a protein of 420 amino acids.
The PRIM2A gene is located on chromosome 6p12–p11.1 and is composed of 19 exons that generate several alternatively spliced mRNAs.
The POLA1 gene is located on the X chromosome (Xp22.1–p21.3) and is composed of 38 exons that encode a protein of 1462 amino acids.
The POLA2 gene is located on chromosome 11q13.1 and is composed of 21 exons that encode a protein of 598 amino acids.
Synthesis of DNA proceeds in the 5′ → 3′ direction through the attachment of the 5’–phosphate of an incoming dNTP to the existing 3’–OH in the elongating DNA strands with the concomitant release of pyrophosphate. Initiation of synthesis, at origins of replication, occurs simultaneously on both strands of DNA. Synthesis then proceeds bidirectionally, with one strand in each direction being copied continuously and one strand in each direction being copied discontinuously. During the process of DNA polymerases incorporating dNTPs into DNA in the 5′ → 3′ direction they are moving in the 3′ → 5′ direction with respect to the template strand. In order for DNA synthesis to occur simultaneously on both template strands as well as bidirectionally one strand appears to be synthesized in the 3′ → 5′ direction. In actuality one strand of newly synthesized DNA is produced discontinuously.
The strand of DNA synthesized continuously is termed the leading strand and the discontinuous strand is termed the lagging strand. The lagging strand of DNA is composed of short stretches of RNA primer plus newly synthesized DNA approximately 100–200 bases long (the approximate distance between adjacent nucleosomes). The lagging strands of DNA are also called Okazaki fragments. The concept of continuous strand synthesis is somewhat of a misnomer since DNA polymerases do not remain associated with a template strand indefinitely. The ability of a particular polymerase to remain associated with the template strand is termed its’ processivity. The longer it associates the higher the processivity of the enzyme. DNA polymerase processivity is enhanced by additional protein activities of the replisome identified as processivity accessory proteins.
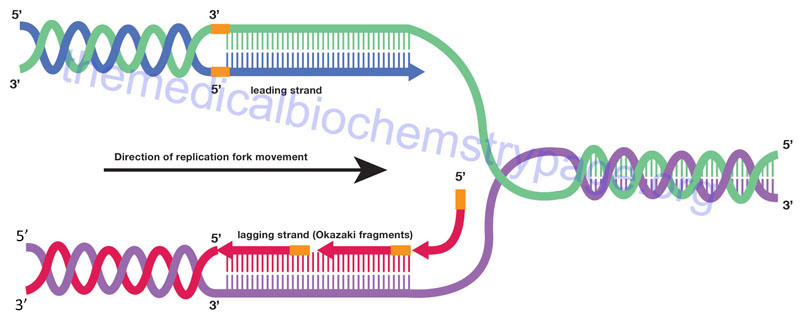
How is it that DNA polymerase can copy both strands of DNA in the 5′ → 3′ direction simultaneously? A model has been proposed where DNA polymerases exist as dimers associated with the other necessary proteins at the replication fork and identified as the replisome. The template for the lagging strand is temporarily looped through the replisome such that the DNA polymerases are moving along both strands in the 3′ → 5′ direction simultaneously for short distances, the distance of an Okazaki fragment. As the replication forks progress along the template strands the newly synthesized daughter strands and parental template strands reform a DNA double helix. The means that only a small stretch of the template duplex is single-stranded at any given time.
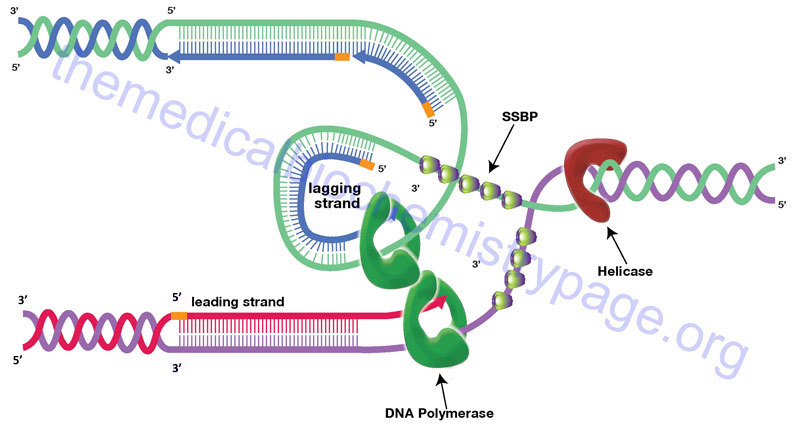
The progression of the replication fork requires that the DNA ahead of the fork be continuously unwound. Due to the fact that eukaryotic chromosomal DNA is attached to a protein scaffold the progressive movement of the replication fork introduces severe torsional stress into the duplex ahead of the fork. This torsional stress is relieved by DNA topoisomerases. Topoisomerases relieve torsional stresses in duplexes of DNA by introducing either double- (type II topoisomerases) or single-stranded (type I topoisomerases) breaks into the backbone of the DNA. These breaks allow unwinding of the duplex and removal of the replication-induced torsional strain. The nicks are then resealed by the topoisomerases. The type I topoisomerases are divided into three subfamilies: type IA, type IB, and type IC. The type II topoisomerases are divided into two subfamilies: type IIA and type IIB.
Humans express a total of six topoisomerase genes, one of which (encoded by the TOP1MT gene) is involved in replication of mitochondrial DNA. Within the type IA family in humans there are the topoisomerases identified as topo IIIα (encoded by the TOP3A gene) and topo IIIβ (encoded by the TOP3B gene). Within the type IB family is human topo I (encoded by the TOP1 gene). Within the type IIA family are the human topoisomerases identified as topo IIα (encoded by the TOP2A gene) and topo IIβ (encoded by the TOP2B gene). Humans do not express type IC or type IIB topoisomerases.
The RNA primers of the leading strands and Okazaki fragments are removed by the repair DNA polymerases simultaneously replacing the ribonucleotides with deoxyribonucleotides. The gaps that exist between the 3’–OH of one leading strand and the 5’–phosphate of another as well as between one Okazaki fragment and another are repaired by DNA ligases thereby, completing the process of replication. Humans express a total of three DNA ligase genes: LIG1 (encoding DNA ligase I), LIG3 (encoding DNA ligase III), and LIG4 (encoding DNA ligase IV). The LIG1 encoded enzyme is involved in both DNA replication and in the processes of base excision repair of damaged DNA.
The LIG3 gene encodes several different enzymes as a result of alternative mRNA splicing and alternative translation start site utilization. The major forms of DNA ligase III are identified as DNA ligase IIIα and IIIβ. DNA ligase IIIα is localized in the nucleus and the mitochondria. In the nucleus DNA ligase IIIα forms a complex with the XRCC1 (X-ray repair cross complementing 1) encoded protein and this complex functions in DNA damage repair processes that include long-patch base excision repair as well as in the repair of single-strand breaks. The mitochondrial form of DNA ligase III is the only DNA ligase activity found in this organelle.
The LIG4 encoded enzyme functions in DNA damage repair in the process of non-homologous end joining (NHEJ) as well as in the process of V(D)J recombination during B and T cell maturation generating the high level of diversity on immunoglobulins (B cells) and the T cell receptor (T cells). The LIG4 encoded protein forms a complex with proteins encoded by the XRCC4 (X-ray repair cross complementing 4), PRKDC (DNA-dependent protein kinase, catalytic subunit: DNA-PKcs), and NHEJ1 (non-homologous end-joining factor 1; also identified as XRCC4-like factor: XLF) genes.
Additional DNA Polymerase Activities
The main enzymatic activity of DNA polymerases is the 5′ → 3′ synthetic activity. However, DNA polymerases possess two additional activities of importance for both replication and repair. These additional activities include a 5′ → 3′ exonuclease function and a 3′ → 5′ exonuclease function. The 5′ → 3′ exonuclease activity allows the removal of ribonucleotides of the RNA primer, utilized to initiate DNA synthesis, along with their simultaneous replacement with deoxyribonucleotides by the 5′ → 3′ polymerase activity. The 5′ → 3′ exonuclease activity is also utilized during the repair of damaged DNA. The 3′ → 5′ exonuclease function is utilized during replication to allow DNA polymerase to remove mismatched bases and is referred to as the proof-reading activity of DNA polymerase. It is possible (but rare) for DNA polymerases to incorporate an incorrect base during replication. These mismatched bases are recognized by the polymerase immediately due to the lack of Watson-Crick base-pairing. The mismatched base is then removed by the 3′ → 5′ exonuclease activity and the correct base inserted prior to progression of replication.
DNA Mismatch Repair: MMR
DNA mismatch repair (MMR) is a highly conserved genome-maintenance system whose primary function is to ensure replication fidelity by removing mis-incorporated bases and insertion-deletion mispairs in newly synthesized DNA. Numerous genes have been identified that encode proteins essential to the processes of MMR. Several of these proteins include homologs of the bacterial MutS family and MutL family of proteins. In humans mismatch recognition is mediated by one of two heterodimeric protein complexes.
One of the complexes is composed of the MutS homologs MSH2 and MSH6 (referred to as MutSα) while the other is composed of MSH2 and MSH3 (referred to as MutSβ). Of the two complexes the MutSα complex is the more abundant. The MutSα complex is involved in the repair of 1 to 2 unpaired nucleotides and single nucleotide mismatches. The MutSβ complex recognizes larger insertion-deletion loops (IDL) as well as selected base-base mismatches. When the MutSα or MutSβ complexes bind to mismatches this action recruits a second heterodimeric complex called MutLα.
The second complex is composed of the MutL homologs MLH1 and PMS2. The human PMS2 (Post Meiotic Segregation increased 2) protein of the MutLα complex possesses endonuclease activity. Additional factors that are critical for the process of MMR include proliferating cell nuclear antigen (PCNA), exonuclease 1 (EXO1), replication protein A (RPA), replication factor C (RFC), DNA polymerase δ, and DNA ligase I.
The endonuclease activity of the MutLα complex introduces random phosphodiester hydrolysis (nicks) in the backbone of the DNA at sites spanning the mismatch. Evidence indicates that these nicks are introduced into the newly synthesized DNA strand. Subsequent loading of EXO1 at the 5’ side of the nick at the mismatch leads to activation of its 5’→ 3’ exonuclease activity resulting in removal of the incorrectly matched DNA fragment. The resulting single-stranded gap is repaired by DNA polymerase δ using the parental strand as the template. This repair synthesis includes the cofactors of DNA polymerase δ, proliferation cell nuclear antigene (PCNA) and replication factor C (RFC). The repair process is completed when the final phosphodiester bond is generated at the remaining 3′ to 5′ gap (nick) via the action of DNA ligase I.
Many chromatin modifying/remodeling factors contain a domain [the Pro-Trp-Trp-Pro (PWWP) domain] that allows then to interact with methylated lysine and arginine residues in histone proteins. The MSH6 subunit of the human MutSα complex possesses a PWWP domain suggesting that it likely interacts with histone. Indeed, studies have demonstrated that MSH6 can “read” the trimethylated lysine of histone H3 (lysine 36 identified as H3K36me3). The presence of H3K36me3 stimulates the recruitment of the MutSα complex to chromatin via, its interactions with the PWWP domain of MSH6, immediately before the onset of DNA replication. Additional studies have demonstrated that cells that are defective in the methyltransferase responsible for the generation of H3K36me3 are also defective in the process of MMR. These results strongly suggest that the H3K36me3 histone mark plays a critical role in MMR. The interaction between histones and the complexes of MMR indicates that the histone code (the nature of modified histones) contributes to the high level of replication fidelity in eukaryotic cells.
The clinical significance of the process of MMR can be seen by the fact that mutations in, or hypermethylation-mediated gene silencing of, key MMR genes causes elevated mutation frequencies and leads to an increased incidence of certain types of cancer. The most commonly associated cancers are colorectal carcinomas that are not associated with prior adenomatous polyp formation and as such are referred to as hereditary non-polyposis colorectal carcinomas, HNPCC. There are at least six defined forms of HNPCC with the first two types also known as Lynch syndromes.
Telomere Replication: Implications for Aging and Disease
Telomeres are the specialized DNA structures at the ends of all chromosomes that consist of repetitive DNA sequences and nucleoproteins, the overall structure of which is referred to as a nucleoprotein cap. The telomere sequence on the lagging strand is composed of the repeat 5’–TTAGGG–3′. The telomeric repeat sequence spans up to several kilobases and is involved in protecting the ends of the chromosomes from exonucleolytic activity. The telomeric ends of the lagging strand of each chromosome requires a unique method of replication which involves the activity of the enzyme complex called telomerase. This is due to the fact that even if the primase activity incorporated a primer sequence for DNA polymerase δ on the extreme 3′-end of the lagging strand, the end of that strand would not be fully replicated and therefore, would be susceptible to degradation.
Telomerase is a complex composed of two copies each of several proteins, two copies of an RNA with sequences complimentary to the telomeric repeats, and two copies of a reverse transcriptase activity that extends the 3′-end of the lagging strands using the telomerase RNA as the template. The reverse transcriptase activity of telomerase is encoded by the TERT gene (telomerase reverse transcriptase) and the RNA component is encoded by the TERC gene (telomerase RNA component). The TERC RNA contains a repeating hexanucleotide sequence, 3’–AAUCCC–5′, that spans between 3 and 20 kilobases. This sequence in the TERC RNA forms a duplex with the lagging DNA strand at the ends of the chromosomes. The 3′-end of the lagging strand then serves as the primer for the reverse transcriptase activity (TERT) which extends the 3′-end of the chromosome using the TERC RNA as a template. The additional proteins involved in the telomerase complex are identified as dyskerin pseudouridine synthase 1 (commonly referred to simply as dyskerin; encoded by the DKC1 gene) and telomerase associated protein 1 (encoded by the TEP1 gene).
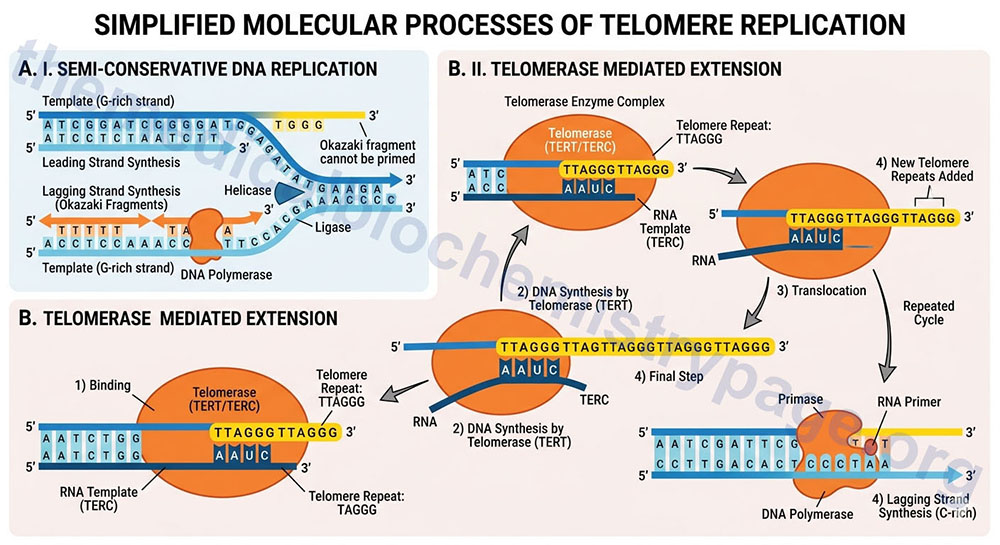
The telomerase process extends the end of the lagging strand that can then be replicated by normal DNA polymerase thereby, preserving the length of the chromosome. Numerous lines of evidence strongly implicate telomere shortening with activation of programmed cell death (apoptosis), loss of tissue stem cells, disease progression, and the overall processes of aging. The importance of telomere length and functional telomerase activity was initially defined in cultures of human fibroblasts as early as the 1960’s. Hayflick and co-workers demonstrated that as fibroblasts went through progressive cell cycles in culture their telomeres became progressively shorter and induced a state of proliferative arrest. The fibroblasts exhibited a finite number of cell divisions leading up to the arrest and this barrier to proliferation was called the Hayflick limit. Forced cell division beyond the limit resulted in further telomere loss culminating in uncontrolled chromosomal instability and the triggering of apoptosis. At the opposite end of the spectrum, forced expression of telomerase, specifically the TERT gene, in cultured fibroblasts results in a preservation of telomere length and the cells gain the ability to divide indefinitely without any malignant properties.
Numerous studies have demonstrated a correlation between telomere shortening and human aging and disease. Decreased telomere length in peripheral blood leukocytes has been shown to correlate with higher mortality rates in older (over 60 years of age) individuals. This is contrasted by studies in centenarians and their offspring that have shown a positive link between telomere length and longevity. These latter studies also demonstrated that individuals with longer telomeres had an overall healthier profile relative to individuals of similar age with telomeres of shorter length. There is also an intriguing correlation between telomere length and psychological stress and the risk for development of psychiatric disease.
Studies in women aged 20–50 years have shown that those individuals with the highest levels of psychological stress had the shortest telomeres. In addition, the level of telomerase activity in peripheral blood leukocytes was lowest in those individuals with the highest levels of stress which also coincided with the highest levels of oxidative stress. This correlation between stress and telomerase activity and telomere length is quite intriguing given that it is known that individuals subject to chronic psychological stress show a shortened lifespan and more rapid onset of diseases that are more typical of an aged population such as cardiovascular disease.
Telomere maintenance correlated to a healthy lifespan is also inferred from studies of various inherited degenerative disorders. As an example, individuals carrying a mutation in either the TERT or TERC genes develop autosomal dominant dyskeratosis congenita, DKS (characterized by a triad of abnormal nails, reticular skin pigmentation, and oral leukoplakia; also called Zinsser-Cole-Engman syndrome). DKS patients have shortened telomeres, a reduced lifespan, and exhibit signs of accelerated ageing. There is another form of DKS that is inherited as an X-linked disease resulting from defects in the DKC1 gene encoding a protein called dyskerin pseudouridine synthase 1 (simply called dyskerin). In addition to being a component of the telomerase complex, dyskerin is also a component of a pseudouridine synthase complex that modifies rRNAs, and another complex that processes small nucleolar RNAs (snoRNAs) into snoRNA-containing ribonucleoprotein complexes (snoRNPs).
Other disorders that manifest with signs of premature aging, such as Werner syndrome and ataxia telangiectasia also correlate with shortened telomeres. Werner syndrome is a rare autosomal recessive disorder resulting from a deficiency of the WRN protein, which is a DNA helicase involved in DNA repair, DNA recombination and telomere maintenance. These patients develop normally until puberty. At this time they begin to manifest signs of multiple progressive premature ageing pathologies, including senile cataracts, osteoporosis, skin atrophy, myocardial infarction and cancer. Werner syndrome fibroblasts show accelerated telomere loss and undergo premature senescence that can be reversed by enforced TERT expression.
In addition to inherited disorders, telomere shortening is also correlated with acquired degenerative conditions associated with chronically elevated tissue turnover. For example, cirrhosis of the liver is associated with a progressive decline in telomere length.
Post-Replicative Modification of DNA: Cytidine Methylation
One of the major post-replicative reactions that modifies the DNA is methylation. DNA methylation can alter chromatin structure, as pointed out above, and as discussed in the Regulation of Gene Expression page can therefore, also alter the transcription of genes. As pointed out earlier, DNA methylation represents a major epigenetic process. The sites of natural methylation (i.e. not chemically induced) of eukaryotic DNA is always on cytosine residues that are present in CpG dinucleotides. However, it should be noted that not all CpG dinucleotides are methylated at the C residue. The cytidine is methylated at the 5 position of the pyrimidine ring generating 5-methylcytidine. Enzymes that incorporate methyl groups into DNA molecules are called DNA methyltransferases, DNMTs. As pointed out earlier, humans express three DNMT genes identified as DNMT1, DNMT3a, and DNMT3b. The enzymes of the DNMT3 family are responsible, principally, for the generation of the initial pattern of CpG dinucleotide methylation. The DNMT1 family enzymes are principally tasked with maintaining the DNMT3-established methylation patterns.
Methylation of DNA in prokaryotic cells also occurs. The function of this methylation is to prevent degradation of host DNA in the presence of enzymatic activities synthesized by bacteria called restriction endonucleases. These enzymes recognize specific nucleotide sequences of DNA. The role of this system in prokaryotic cells (called the restriction-modification system) is to degrade invading viral DNAs. Since the viral DNAs are not modified by methylation they are degraded by the host restriction enzymes. The methylated host genome is resistant to the action of these enzymes.
The role of methylation in eukaryotic DNA serves two clearly defined and overlapping functions. The methylation of cytidine in CpG dinucleotides affects the overall structure of chromatin which in turn broadly alters the availability of the chromatin to the transcriptional machinery. This effect of methylation is one mechanism of epigenetics. Epigenetics, as a means of gene control, is discussed in the Regulation of Gene Expression page.
The effects of methylation on the transcription of specific genes was elegantly demonstrated in experiments that led to the under-methylation of the MyoD gene, a master control gene regulating the differentiation of muscle cells through the control of the expression of muscle-specific genes. Under-methylation of MyoD in fibroblasts results in their conversion to myoblasts. The experiments were carried out by allowing replicating fibroblasts to incorporate 5-azacytidine into their newly synthesized DNA. This analog of cytidine prevents methylation. The net result is that the original pattern of DNA methylation in the fibroblast is lost and numerous genes become under methylated and consequently transcriptionally activated.
Following DNA replication the original pattern of methylation is copied by the maintenance methylase enzyme, DNMT1. The DNMT1 protein recognizes the pattern of methylated C residues in the parental DNA strand following replication and methylates the C residue present in the corresponding CpG dinucleotide of the daughter strand.
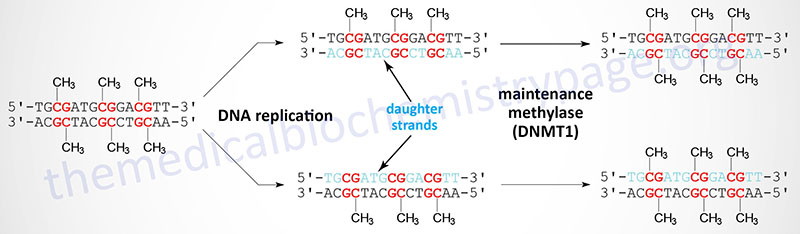
DNA Methylation: Role in Genomic Imprinting
The phenomenon of genomic imprinting refers to the fact that the expression of some genes is exclusive to the maternally or paternally derived alleles. In other words, some genes are only expressed from the mothers chromosomes, whereas some genes are only expressed from the fathers chromosomes. Imprinted genes have been identified to be distributed throughout the genome. The majority of imprinted loci are organized in clusters that are up to one megabase (Mb) in size. The allele-specific expression of imprinted genes is regulated by epigenetic modifications of which DNA methylation is the controlling modification. Thus, these imprinted alleles are “marked” by their state of methylation.
The methylated CpG-rich regions of imprinted loci contain the methylation state on only one of the two parental chromosomes. For this reason these imprinted domains are referred to as differentially methylated regions, DMR. In several of these DMR the differential methylation is also found when comparing sperm and eggs representing the fact that the DMR is gametic in origin. These DMR are, therefore, referred to as germline or gametic DMR. Most of the gametic DMR are found in the maternal germline. To date only four DMR are found in the paternal germline. These four loci are identified as H19, DLK1 (delta-like non-canonical Notch ligand 1), RASGRF1 (RAS protein specific guanine nucleotide releasing factor 1), and ZDBF2 (zinc finger DBF-type containing 2).
The significance of CpG methylation to imprinting can be shown by the fact that mutations in the maintenance DNA methylase, encoded by the DNMT1 gene, result in loss of the parental-origin-specific patterns of expression of imprinted genes. Although CpG methylation is the predominant regulator of the imprinting phenomenon, other epigenetic modifications, such as histone modifications, and other factors such as insulator proteins (e.g. CTCF) and long non-coding RNAs (lncRNA), are also involved in the overall imprinting process.
The CTCF protein, also known as the CCCTC-binding factor, is a zinc-finger domain (11 zinc fingers) containing DNA-binding protein that was first identified as a regulator of MYC gene expression and subsequently shown to be an important regulator of the expression of numerous genes and to be involved in the regulation of the process of X chromosome inactivation, XCI.
Within the human genome there are several thousand CTCF binding sites that are found either around genes (intergenic sites), within genes (intragenic sites), or in the promoter regions. Numerous CTCF binding sites contain CpG dinucleotides and the status of DNA methylation in these sites plays an important role in the occupancy of these sites by CTCF.
The CTCF gene is located on chromosome 16q22.1 and is composed of 13 exons that generate two alternatively spliced mRNAs encoding proteins of 727 amino acids (isoform 1) and 399 amino acids (isoform 2). The CTCF isoform 2 encoding mRNA lacks two internal exons and the encoded protein is initiated at a downstream AUG codon, relative to the isoform 1 start site. CTCF is a major transcriptional regulator with at least 5,000 identified binding sites in the human genome.
To date several hundred imprinted genes have been characterized. One of the very first imprinted genes identified was the insulin-like growth factor-2 gene (symbol: IGF2). The IGF2 gene encoded protein (IGF-2) is required for normal fetal development and growth. Expression of IGF2 occurs exclusively from the paternal copy of the gene. In the case of IGF2 an element in the paternal locus, called an insulator element, is methylated blocking the ability of a transcriptional repressor from binding and inhibiting paternal IGF2 gene transcription.
The function of the un-methylated insulator is to bind a transcriptional repressor protein that when bound blocks activation of IGF2 expression. This protein is called CTCF or, as indicated earlier, is also known as CCCTC-binding factor. The insulator element resides within a differentially methylated region (DMR, also known as an imprinting control region, ICR) in the area of chromosome 11p15.5. This DMR contains seven copies of the sequence CCCTC to which the factor binds when nearby CpG are undermethylated.
When the CpG containing insulator region of the IGF2 gene is methylated, as in the case of the paternal gene, CTCF cannot bind the insulator sequences, thus allowing a distant enhancer element to drive expression of the IGF2 gene. In the maternal genome, the CpG sites in the insulator domain are not methylated, therefore, the CTCF protein binds to it blocking the action of the distant enhancer element. Another important gene in this region is H19 which encodes a long non-coding RNA (lncRNA). The maternal DMR is unmethylated allowing CTCF to bind at the insulator elements within the region of H19 blocking enhancers downstream of H19 from accessing the IGF2 promoters thus, preventing maternal IGF2 expression.
The function of IGF-2 is exerted by its binding to a specific receptor identified as the IGF-2 receptor, IGF2R. Interestingly the IGF2R gene is also imprinted, being expressed exclusively from the maternal gene. Indeed, the IGF2R gene was the very first human gene to be identified as being imprinted. Several of these imprinted loci have been associated with various diseases that result when there is a disruption in the normal pattern of imprinting.
Two of the most clinically interesting disorders are Prader-Willi syndrome (PWS) and Angelman syndrome (AS). These two disorders are phenotypically quite distinct yet arise due to alterations in the same imprinted locus on chromosome 15 that encompasses several genes.
Genomic imprints, that involve CpG methylation, undergo a cycle of establishment, maintenance, and erasure. It is during spermatogenesis and oogenesis when the CpG methylation status is established. In males the CpG methylation imprints are established in pro-spermatogonia while in females the imprints are established only by the fully grown oocyte stage. The patterns of CpG methylation that arise in the germ cells are maintained following fertilization and throughout early development and in the adult. During development of the primordial germ cells (PGC), from which sperm and egg will arise, the pattern of CpG methylation is erased. The erasure of the CpG methylation pattern in the PGC ensures the sex-dependent imprint pattern can be established in later stages of spermatogenesis and oogenesis.
The DNA methyltransferases responsible for the establishment of the germline differential methylation patterns are encoded by the DNMT3A and DNMT3B genes. As pointed out above, the protein encoded by the DNMT3L gene (which is highly expressed in germ cells) functions to enhance the activity of the DNMT3a enzyme. Once established, the maintenance of the state of germ cell CpG methylation is the function the DNMT1 methylase. The erasure of the CpG methylation imprints, that occurs in primordial germ cells, is carried out by the TET cytidine demethylases (TET1, TET2, and TET3) as well as by activation-induced cytidine deaminase (AID) as described above for the general removal of 5mC residues in non-imprinted regions of the DNA.
X Chromosome Inactivation: Contributions from DNA Methylation
In mammals, sex is determined by a pair of sex chromosomes identified as the X and Y chromosomes. Mammalian males are heterogametic and contain one copy of the X chromosome and one copy of the Y chromosome, whereas mammalian females are homogametic and contain two copies of the X chromosome. Whereas the X chromosome has retained a conserved high density of genes (approximately 1000 genes), the Y chromosome has lost the vast majority of its ancestral genes such that modern mammalian Y chromosomes contain few protein coding genes. The fact that females harbor two copies of the X chromosome has led to the potential for differences in X-linked gene dosage between females and males.
These potential gene dosage differences are compensated for through the process of X chromosome inactivation (XCI), a process first characterized in 1961 by Mary Lyon. As a result of her discovery the process of XCI is often referred to as lyonization. The original observation, that led to the discovery of XCI by Mary Lyon, was that of a chromosome in cat nerve cell nuclei that was structurally distinct and characterized by a dense heterochromatic nuclear morphology. This work was published in 1949 by Murray Llewellyn Barr and his student Ewart George Bertram and this highly condensed chromosome is, therefore, referred to as the Barr body. Additional research demonstrated that mice with only one X chromosome were viable and phenotypically normal, indicating that only a single X chromosome was necessary.
The process of XCI takes place very early in development and ensures equivalent X chromosome gene expression levels throughout development in both males and females. Mammalian XCI is a diverse process such that in certain species (e.g. marsupials) a specific X chromosome (the paternal) is inactivated. However, in eutherian mammals, such as humans, the process of XCI is random such that in human females roughly half of the cells in her body have inactivated the paternal X chromosome (Xp) and the other half have inactivated the maternal X chromosome (Xm). This results in all human females being mosaic for all cell populations within the same tissues where either the Xp or the Xm chromosome is inactive. Once a human X chromosome is inactivated it remains inactive in all the progeny cells throughout life.
Although one of the two X chromosomes in females is randomly inactivated not all of the genes on the inactive X chromosome (Xi) are in fact transcriptionally silent. Several of the genes that remain active are found in the pseudo-autosomal region, PAR. The X chromosome PAR is a region of the X chromosome that is homologous to a region in the Y chromosome and is responsible for the X and Y chromosomes pairing up during meiosis. In addition to the genes in the PAR, several other genes on the Xi remain transcriptionally active. These latter genes are referred to as escape genes and approximately 15–20% of human X-linked genes completely escape inactivation while another 10% partially escape inactivation. Because the X chromosome PAR genes are present in the Y chromosome these genes are biallelically expressed in both males and females. X chromosome escape genes, on the other hand, do not have Y chromosome counterparts and are, therefore, differentially expressed between the sexes. The expression of PAR genes, along with the escape genes, most likely accounts for the phenotypes that are observed in Turner syndrome (45,X) females and in triple XXX females.
Mechanisms of X Chromosome Inactivation
The overall process of XCI in eutherian mammals is highly similar although there are known species specific differences. Due to the ease of use of mouse embryonic stem cells in the study of XCI, most of the understanding of the process has been obtained with this model system. XCI requires a family of long non-coding RNAs (lncRNA) with the central regulatory lncRNA being encoded by the XIST gene (X-inactive specific transcript). Like most lncRNA, the primary XIST transcript is spliced and polyadenylated. The XIST gene resides in a region on the X chromosome called the X inactivation center (XIC). The human XIC is located in the q arm at around band 13 (Xq13).
In addition to the XIST gene there are other genes, as well as regulatory DNA sequences, within the XIC that are involved in the X inactivation process. The two major genes (in addition to the XIST gene), that reside within the XIC, that regulate the process of XCI, encode lncRNA. These lncRNA encoding genes are JPX and TSIX. The TSIX gene is transcribed in the antisense direction relative to the XIST gene and is, therefore, referred to as a natural antisense transcript (NAT). There are also protein coding genes involved in the process of XCI including the RNF12 (which itself resides in the XIC), REX1, and CTCF genes. The RNF12 encoded protein is an E3 ubiquitin ligase family member and was the first non-lncRNA factor shown to be required for XCI. The REX1 gene encodes a transcription factor whose activity is regulated by the RNF12 protein and whose function is to regulate the expression of the XIST and TSIX genes. The CTCF gene, as described above, encodes a transcriptional regulator that binds to CCCTC sites in target genes. One of the important DNA elements involved in XCI resides just upstream of the transcriptional start site for the TSIX gene and is identified as XITE (X inactivation intergenic transcription elements).
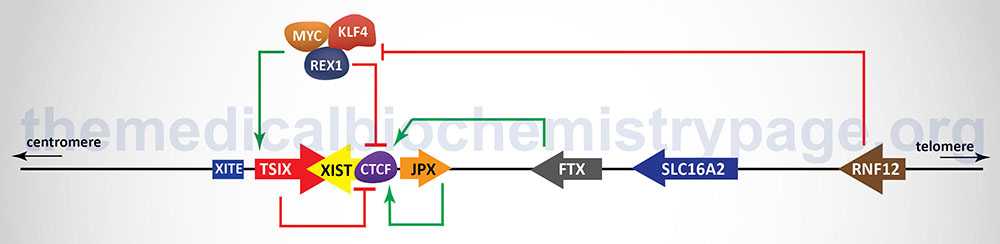
Expression of the XIST gene is exclusive to the future inactive X chromosome (Xi) and the encoded XIST lncRNA coats the X chromosome. The coating of the future Xi with XIST results in the recruitment of chromatin remodeling complexes that lead to the formation of highly condensed inactive heterochromatin, termed the Barr body.
One important remodeling complex involved in the formation and stabilization of the Xi, following XIST coating, is the polycomb repressive complex 2 (PRC2). The catalytic core of PRC2 is composed of six subunits, two of which are enzymes (the EZH1 and EZH2 gene encoded proteins) that trimethylate lysine residues in histones, specifically lysine 27 of histone H3. This modified histone is identified as H3K27me3.
The EZH1 and EZH2 encoded proteins are members of the lysine methyltransferase (KMT) family and as such are also identified as KMT6B and KMT6A, respectively. In addition to the role of PRC2 in the inactivation of one of the X chromosomes, the generation of H3K27me3 by PRC2 is a common feature of transcriptionally repressed chromatin.
Once XIST function has resulted in silencing of one of the X chromosomes, this state is stably inherited such that all progeny cells maintain that specific inactive X chromosome.
The prevention of X inactivation on the other X chromosome in females is accomplished via expression of the TSIX gene, which as indicated earlier, is transcribed in the antisense orientation relative to the XIST gene. The TSIX encoded transcript is also a lncRNA. TSIX expression occurs from the active X chromosome (Xa) prior to and during the process of XCI. The presence of the TSIX lncRNA on the Xa participates in the prevention of XIST gene expression. Like the XIST lncRNA, the TSIX lncRNA recruits chromatin remodeling complexes to the XIST promoter region preventing its expression from the Xa.
As indicated in the Figure above, there are additional regulators of the process of XCI that function either as activators or inhibitors. One significant activator of XIST gene expression is the E3 ubiquitin ligase encoded by the RNF12 gene. The RNF12 gene resides in the XIC towards the telomeric end of the X chromosome about 500 kb upstream of the XIST transcriptional start site. The RNF12 protein was the first regulator of XCI that was not a lncRNA. The activation of XIST expression by RNF12 occurs in a dose-dependent manner. Given that RNF12 expression in males occurs from a single X chromosome the level of expression is not sufficient to induce XCI. However, in female cells the double dose of RNF12 is sufficient to initiate XCI. The major target for ubiquitylation by RNF12 is the zinc-finger protein identified as REX1 (named for Reduced EXpression 1 and encoded by the ZFP42 gene) which is a component of a pluripotency complex that includes the KLF-4 and MYC proteins. This pluripotency complex represses the expression of the XIST gene and activates the TSIX gene. Therefore, when REX1 is degraded following RNF12-mediated ubiquitylation, expression of XIST is activated. Once the inactivation process is started on one X chromosome expression of the RNF12 gene is silenced.
The lncRNA encoded by the JPX and FTX genes also regulate the expression of XIST. The major function of the JPX lncRNA is to displace the CTCF transcriptional repressor protein from the promoter region of the XIST gene. When bound to multiple sites in the XIST gene CTCF interferes with its expression such that its displacement by the JPX lncRNA results in activation of XIST expression. The FTX gene encodes not only a lncRNA but also contains several microRNA (miRNA) genes. The FTX lncRNA has been shown to alter the structure of chromatin around the promoter of the XIST gene by preventing DNA hypermethylation and promoting accumulation of histone H3 lysine 4 dimethylation (H3K4me2). These changes result in direct regulation of XIST expression.
The onset of XCI can be inhibited by autosome encoded proteins that have been shown to be critical in the establishment of pluripotency in mouse embryonic stem cells and in induced pluripotent stem cells (iPS). The key pluripotency proteins include the transcription factors OCT4, REX1, SOX2, KLF-4, NANOG and the reprogramming transcription factor MYC. The binding of different combinations of these pluripotency factors at different locations within the XIC results in either repression of XIST expression or activation of TSIX expression. With respect to the regulation of XIST, evidence has demonstrated that SOX2, OCT4, and NANOG bind to sequences in intron 1 of the XIST gene resulting in direct repression of XIST expression. Within the promoter of the TSIX gene both OCT4 and SOX2 have been shown bind and activate expression of TSIX. Repression of XIST expression falls upon cellular differentiation concomitant with the decline in expression of the pluripotency factor genes. In addition to inhibiting the expression of XIST, OCT4, SOX2, and NANOG have been shown to repress XCI through interference with the function of RNF12.
Once XCI has been initiated and established it must be maintained since the evidence is clear that once an X chromosome is inactivated (Xi) it is maintained in the inactive state throughout the remainder of the life of the cell and within all of progeny of the cell. The maintenance of XCI occurs in part through the coating of the Xi with the XIST lncRNA. The coating results in the recruitment of chromatin remodeling complexes that result in the gradual accumulation of Xi-specific epigenetic marks. In addition, RNA polymerase II (RNA pol II) is excluded from the XIST promoter limiting the overall level of XIST lncRNA. It is believed that the exclusion of RNA pol II from the XIST gene is a contributing factor in the prevention of the spread of XIST RNA to the active X chromosome (Xa) as well as from autosomes. The precise mechanism that prevents XIST lncRNA spreading is not clearly defined but may involve components of the nuclear matrix and heterogeneous ribonucleoprotein complexes, hnRNPs.
DNA Methylation Role in X Chromosome Inactivation
Within the context of the inactivated X chromosome (Xi) and comparison to the active X chromosome (Xa), the state of CpG methylation can be highly correlated to gene expression levels. As an example, the level of CpG methylation in the promoter regions of most of the genes on the X chromosome is higher on the Xi when compared to the same genes on the Xa. However, in the promoter regions of genes in the PAR and of escape genes, the CpG methylation state is lower overall than the level of methylation of XCI-associated silenced genes.
Typical of this pattern of DNA methylation between transcriptional start sites in the Xi versus the Xa is the XIST gene promoter. The XIST gene promoter has several CpG sites that are hypermethylated on the Xi when compared to the Xa and experimental evidence demonstrates that removal of the methylation from the XIST promoter reactivates expression of the gene. However, it is important to note that CpG hypermethylation alone in not sufficient to silence some of the genes on the Xi since examination of methylation state has shown several genes on the Xa that have the same level of CpG methylation when compared to the Xi, yet the Xa genes are active while the Xi genes are not. This clearly demonstrates that, whereas, CpG methylation can be highly correlated to transcriptional silencing associated with XCI, chromatin structure also plays an important role in the transcriptional silencing as well as in the escape of certain genes from XCI. This latter fact is exemplified by the observations that the coating of the Xi by the XIST lncRNA is coupled to the recruitment of chromatin remodeling complexes.
DNA Recombination
DNA recombination refers to the phenomenon whereby two parental strands of DNA are spliced together resulting in an exchange of portions of their respective strands. This process leads to new molecules of DNA that contain a mix of genetic information from each parental strand. There are 3 main forms of genetic recombination. These are homologous recombination, site-specific recombination, and transposition.
Homologous recombination is the process of genetic exchange that occurs between any two molecules of DNA that share a region (or regions) of homologous DNA sequences. This form of recombination occurs frequently while sister chromatids are paired during meiosis. Indeed, it is the process of homologous recombination between the maternal and paternal chromosomes that imparts genetic diversity to an organism. Homologous recombination generally involves exchange of large regions of the chromosomes.
Site-specific recombination involves exchange between much smaller regions of DNA sequence (approximately 20–200 base pairs) and requires the recognition of specific sequences by the proteins involved in the recombination process. Site-specific recombination events occur primarily as a mechanism to alter the program of genes expressed at specific stages of development. The most significant site-specific recombinational events in humans are the somatic cell gene rearrangements that take place in the immunoglobulin genes during B-cell differentiation in response to antigen presentation. These gene rearrangements in the immunoglobulin genes result in an extremely diverse potential for antibody production. A typical antibody molecule is composed of both heavy and light chains. The genes for both these peptide chains undergo somatic cell rearrangement yielding the potential for approximately 3,000 different light chain combinations and approximately 5,000 heavy chain combinations. Then because any given heavy chain can combine with any given light chain the potential diversity exceeds 10,000,000 possible different antibody molecules.
DNA Transposition
Transposition is a unique form of recombination where mobile genetic elements (transposable elements, TEs) can virtually move from one region to another within one chromosome or to another chromosome entirely. There is no requirement for sequence homology for a transpositional event to occur. As a result of the mobilization of transposons, these DNA elements can reshuffle sequences, promote ectopic DNA rearrangements, and create novel genes.
Examples of genes derived through the process of transposition include the phosphoglycerate kinase 2 (PGK2) gene, the paired box 6 (PAX6) gene, and the gene encoding the reverse transcriptase activity (TERT) of the telomerase complex. PAX6 is a DNA-binding protein that regulates transcription and this gene was demonstrated to be derived from a transposase of a class II TE, whereas telomerase was shown to be derived from retrotransposons. In rare cases there has been documentation between transposable element insertions causing mutations which ultimately lead to disease manifestation. To date at least 65 diseases, including cancers and immune system disorders, have been identified as being the result of TE insertions.
Transposable elements (TE) represent up to 50% of the DNA in the human genome. In humans there are two broad classifications for transposable elements. Class I TEs have an intermediate state that is RNA which is then reverse transcribed into DNA prior to insertion into chromatin, a process referred to as “copy and paste”. Because of the reverse transcription process involved in the mobilization of class I TEs these elements are referred to as retrotransposons. Class II TEs are exclusively DNA transposons and mobilize by a “cut and paste” process that requires the enzyme identified as transposase. Class II TEs remain relatively inactive within the human genome. On the other hand, class I TEs remain actively mobile and as a result continuously serve as sources of genetic diversity in the process of generating new transposon insertions.
The movement of TEs are classified as autonomous (move by themselves) or non-autonomous (require another TE to move). The non-autonomous TEs lack the required reverse transcriptase activity (class I TEs) or the lack the transposase enzyme (class II TEs). The most abundant autonomous retrotransposons are the Long Interspersed Nucleotide Elements, (LINE) with LINE-1 (L1) being the common designation for this class of TE. The L1 element is approximately 6,000 bp long and contains two open reading frames (ORF). One of the two ORFs in L1 encodes an RNA binding protein while the other ORF encodes a multi-subunit complex composed of the reverse transcriptase and an endonuclease. The non-autonomous TEs are the SINEs (Short Interspersed Nucleotide Elements), Alu repeats, and the SVAs. The SVAs represent a hybrid TE containing DNA sequences of SINEs, variable number tandem repeats (VNTRs), and Alu repeats.
The Alu repeats represent the most abundant class of TEs in the human genome. The Alu element is approximately 300 bp in length and, therefore, it is a subclass of the SINE class of TE. The name Alu repeat stems from the fact that the element contains sequences recognized by the restriction endonuclease, AluI. Alu elements were derived from the small cytoplasmic RNA (7SL RNA) that is a component of the signal recognition particle (SRP) that recognizes the signal peptide of newly translated proteins and then targets those proteins to the endoplasmic reticulum, ER.
Repair of Damaged DNA
Damage to DNA is a constant ongoing event and, therefore, mechanisms are in place to constantly surveil the structure of DNA and initiate the appropriate response pathways to effect repair. Cancer, in most non-viral induced cases, is the severe medically relevant consequence of the inability to repair damaged DNA. It is clear that multiple somatic cell mutations in DNA can lead to the genesis of the transformed phenotype. Therefore, it should be obvious that complete understanding of DNA repair mechanisms would be invaluable in the design of potential therapeutic agents in the treatment of cancer.
DNA damage can occur as the result of exposure to environmental stimuli such as alkylating chemicals or ultraviolet and other forms of ionizing radiation and free radicals (e.g. reactive oxygen species, ROS) generated spontaneously in the oxidizing environment of the cell. These phenomena can, and do, lead to the introduction of mutations in the coding capacity of the DNA. Mutations in DNA can also, but rarely, arise from the spontaneous tautomerization of the bases. The extent of DNA damage that must be repaired is vast and includes alterations to the DNA structure occurring during replication and recombination, damage to the bases in the DNA (most often due to ROS and ionizing radiation), single- and double-strand breaks in the backbone of the DNA, and strand cross-linking. Coding region mutations in DNA can be of two general types. Transition mutations result from the exchange of one purine, or pyrimidine, for another purine, or pyrimidine. Transversion mutations result from the exchange of a purine for a pyrimidine or vice versa.
In order for the DNA damage repair systems to effect repair of DNA damage, the alterations need to be recognized. Two major damage recognition systems function in human cells to effect DNA repair. One is direct damage recognition and repair which refers to the mechanism by which the repair enzyme or repair complex recognizes the damage and effects repair directly. An example of this type of repair is described below in the context of the base excision repair (BER) process by which alkylated guanine residues are recognized and repaired through the action of O6-methylguanine-DNA methyltransferase. The second major mechanism is a multistep process where an enzyme or complex recognizes the DNA damage and then recruits the repair enzyme or complex to the site of damage to effect repair.
Base Excision Repair: BER
Base excision repair (BER) is a critical mechanism that ensures cells have a means to repair the vast majority of endogenous DNA lesions. The types of DNA lesions that are corrected through the process of BER include alkylations, oxidations, deaminations and depurinations, as well as single-strand breaks (SSB). The primary function of BER is to remove these frequently produced lesions as a means to ensure the integrity of the genome. The initial step in BER is the search for the lesions in DNA by a family of DNA glycosylases. Humans express a total of eleven DNA glycosylases of which four are responsible for the removal of mispaired uracil and thymine, six are responsible for the repair of oxidative damage, and one is responsible for the removal of alkylated bases. These eleven DNA glycosylases are divided into two main functional categories, monofunctional and bifunctional. The monofunctional enzymes only possess glycosylase activity, whereas the bifunctional enzymes possess an apurinic/apyrimindinic (AP) lyase activity in addition to the glycosylase activity.
Table of Mammalian Base Excision Repair Enzymes
| Enzyme | Gene Symbol | Enzyme Type and Functions |
| N-methylpurine DNA glycosylase | MPG | monofunctional; substrates are 3-alkyladenine (3meA) and hypoxanthine |
| uracil DNA glycosylase | UNG | monofunctional; substrate is uracil |
| mutY DNA glycosylase | MUTYH | monofunctional; also known as MYH; substrate is an A:8-oxoG mispair |
| single-strand-selective monofunctional uracil-DNA glycosylase 1 | SMUG1 | monofunctional; although SMUG1 prefers single-stranded DNA it will also remove unmodified uracil from double-stranded DNA; in addition to uracil, SMUG1 substrates include 5-hydroxyuracil (hoU), 5-hydroxymethyluracil (hmU), and 5-formyluracil (fU) |
| thymine DNA glycosylase | TDG | monofunctional; substrates are uracil in a U:G mispair as well as a T:G mispair; T:G mispairing results from the spontaneous deamination of 5-methylcytosine (5mC); specific for double-stranded DNA |
| methyl-CpG binding domain 4, DNA glycosylase | MBD4 | monofunctional; substrates are uracil in a U:G mispair as well as a T:G mispair; T:G mispairing results from the spontaneous deamination of 5-methylcytosine (5mC); specific for double-stranded DNA |
| 8-oxoguanine DNA glycosylase | OGG1 | bifunctional; substrates include 8-oxoguanine (8-oxoG) and 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyG) |
| nth like DNA glycosylase 1 | NTHL1 | bifunctional; NTH is the bacterial enzyme also known as endonuclease III; substrates include 5-hydroxyuracil (hoU), 5-hydroxycytidine (hoC), 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyG) |
| nei like DNA glycosylase 1 | NEIL1 | bifunctional; NEI is the bacterial enzyme also known as endonuclease VIII; substrates include 5-hydroxyuracil (hoU), 5-hydroxycytidine (hoC), 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyG and FapyA); associated with the DNA replication complex where it recognizes oxidized bases before replication |
| nei like DNA glycosylase 2 | NEIL2 | bifunctional; NEI is the bacterial enzyme also known as endonuclease VIII; substrates include 5-hydroxyuracil (hoU), 5-hydroxycytidine (hoC), 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyG and FapyA) |
| nei like DNA glycosylase 3 | NEIL3 | bifunctional; NEI is the bacterial enzyme also known as endonuclease VIII; substrates include 5-hydroxyuracil (hoU), 5-hydroxycytidine (hoC), 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyG and FapyA) |
The DNA glycosylases that recognize unmodified uracil, thymine, as well as alkylated bases are monofunctional. These enzymes remove the damaged base by cleaving the N-glycosyl bond between the base and the sugar releasing the nucleobase resulting in an abasic site. The abasic sites are recognized by an apurinic/apyrimidinic (AP) endonuclease which cleaves the abasic site leaving a sugar attached to the 5′ side of the nick. This endonuclease is encoded by the APEX1 gene in humans. The resulting 3′-OH serves as a substrate for the repair DNA polymerase, DNA polymerase β (polβ). Pol β also has a lyase activity that removes the sugar attached to the 5′-phosphate. Through the polymerase activity the gap is filled in and finally a phosphodiester bond is generated by DNA ligase I to seal the remaining nick.
The DNA glycosylases that recognize oxidation-induced lesions are bifunctional. These enzymes not only remove the damaged base but they also cleave the DNA backbone. The result of the action of bifunctional DNA glycosylases is either an α,β unsaturated aldehyde or a phosphate attached to the 3′ side of the resulting nick. The sugar is removed by the phosphodiesterase activity of APEX1, whereas the phosphate group is removed by polynucleotide kinase (PNK). The remainder of the repair steps are the same as those in the pathway initiated by the monofunctional DNA glycosylases.
BER facilitated by either monofunctional or bifunctional DNA glycosylases is referred to as short-patch BER because it involves the removal of only one nucleotide. On occasion the sugar is inefficiently removed and other polymerases, including the replicative polymerase (pol λ), can take over the repair process. In these cases the damage containing strand is displaced and removed by the enzyme, flap structure-specific endonuclease 1 (encoded by the FEN1 gene). This later process is referred to as long-patch BER. Following these activities, the nick is sealed by DNA ligase I alone or by the complex of DNA ligase IIIα and XRCC1.
The post-translational modification of the XRCC1 protein by lactylation has been shown to promote the repair of DNA damage. This is functionally significant in the context of cancer given that there is an increase in lactate production in the tumor microenvironment. When XRCC1 undergoes lactylation, the process of DNA repair is enhanced which results in resistance of cancer cells to the types of chemotherapy that involve the induction of DNA damage.
Cytidine bases in DNA can undergo spontaneous deamination to uridine, usually as a result of thermal or ionizing radiation. In addition, uridine can be mis-incorporated during DNA replication. Removal of the uracil base from these uridine residues is catalyzed by one of several enzymes that possess uracil DNA N-glycosylase activity. The major enzyme in this family is encoded by the UNG gene. The UNG gene is located on chromosome 12q24.1 and is composed of 7 exons. As a result of alternative promoter usage and alternative splicing, two distinct protein isoforms are generated from the UNG gene. The mitochondrial form of the enzyme is referred to as UNG1, while the nuclear isoform in referred to as UNG2. The removal of the uracil leads to activation of the base excision repair (BER) pathway that leads to replacement of the U with the correct C residue.
Modification of the DNA bases by alkylation (most often the incorporation of methyl groups) occurs nearly exclusively on purine residues. Methylation of G residues allows them to base pair with T instead of C. A unique activity called O6-methylguanine-DNA methyltransferase (encoded by the MGMT gene) removes the methyl group from G residues. The protein itself becomes methylated and is no longer active, thus, a single protein molecule can remove only one alkyl group. Another modified guanine that is recognized as abnormal and removed is 8-oxoguanine (8-oxoG). The generation of 8-oxoG occurs due to exposure of the DNA to reactive oxygen species, the consequences of which are that 8-oxoG base pairs with A instead of C. The removal of 8-oxoG is catalyzed by the enzyme 8-oxoguanine DNA glycosylase which is encoded by the OGG1 gene.
Nucleotide Excision Repair
Nucleotide excision repair (NER) involves the removal of a wide array of structurally unrelated DNA lesions including cyclobutane thymine (pyrimidine) dimers (CPD), [6-4] pyrimidine-pyrimidone photoproducts (6-4PP) produced in human skin by shortwave UV, and DNA helix-distorting chemical adducts induced by xenobiotic chemical carcinogens. Removal of these abnormal DNA lesions is catalyzed by a large family of nucleotide excision repair enzymes that are divided into three major subfamilies comprising a total of 29 different genes. There are 18 genes in the basic NER subfamily of enzymes and seven genes in the ERCC (Excision Repair Cross Complementing) excision repair associated subfamily. The third subfamily encompasses the Xeroderma pigmentosum complementation group which consists of eight genes, four of which are also included in the ERCC subfamily of genes.
Two major NER pathways operate in human cells. One pathway is referred to as the global genomic NER (GG-NER) pathway. The GG-NER enzymes function to repair DNA damage in both transcriptionally active and transcriptionally inactive regions of the genome. The major enzymes of the GG-NER pathway recognize distortions in the DNA duplex and interact with these regions of the DNA activating the repair processes. Mutations in several genes in the GG-NER pathway are associated with the Xeroderma pigmentosum family of disorders. The other NER pathway is termed the transcription-coupled repair (TC-NER) pathway. The TC-NER pathway is activated when RNA polymerase stalls at a region of DNA damage. The stalled RNA polymerase is the recognition signal for the TC-NER enzymes and thus, does not involve recognition of DNA helix distortions.
The prominent DNA adducts resulting from UV irradiation of DNA are pyrimidine dimers, of which thymine dimers represent the most frequent adducts formed. In addition to pyrimidine dimers the formation of 6-4PP adducts is also a common result of UV irradiation. Pyrimidine dimers form from two adjacent pyrimidine residues in the same strand of DNA. Repair of thymine dimers is most understood from consideration of the mechanisms used in E. coli. However, several mechanism are common to both prokaryotes and eukaryotes. Pyrimidine dimers, including thymine dimers, in mammalian DNA are removed by several mechanisms. Specific glycohydrolases recognize the dimer as abnormal and cleave the N-glycosidic bond of the bases in the dimer. This results in the base leaving and generates an apyrimidinic site in the DNA. This is repaired by DNA polymerase and DNA ligase. Glycohydrolases are also responsible for the removal of other abnormal bases, not just pyrimidine dimers.
Clinical Significance of Defective NER
Humans defective in DNA repair, (in particular the repair of UV-induced pyrimidine dimers), due to autosomal recessive genetic defects suffer from the disease, Xeroderma pigmentosum (XP). There are at least eight distinct genetic defects associated with this disease leading to the designation of eight forms of XP identified as XPA (XPA gene), XPB (ERCC3 gene), XPC (XPC gene), XPD (ERCC2 gene), XPE (DDB2 gene), XPF (ERCC4 gene), XPG (ERCC5 gene), and XPV (POLH gene). The genes that are mutated in XP subtypes XPA through XPG are all involved in nucleotide excision repair (NER) while the gene mutated in the XPV form encodes a DNA polymerase involved in replication of damaged DNA on the leading strand. The DDB2 gene encodes damage specific DNA binding protein 2. Many of the protein products of these genes are found in heterodimeric complexes with other proteins involved in repair of damaged DNA. There are two major clinical forms of XP, one which leads to progressive degenerative changes in the eyes and skin and the other which also includes progressive neurological degeneration.
Another inherited disorder affecting DNA repair in which patients suffer from sun sensitivity, short stature and progressive neurological degeneration without an increased incidence of skin cancer is Cockayne Syndrome. Mutations in genes of the TC-NER pathway are responsible for Cockayne syndrome.
Ataxia telangiectasia (AT) is an autosomal recessive disorder resulting in neurological disability and suppressed immune function. Telangiectasias are dilated superficial blood vessels as also seen in patients with rosacea or scleroderma. AT patients develop a disabling cerebellar ataxia early in life and have recurrent infections. Patients suffering from AT have an increased sensitivity to x-irradiation resulting from defects in the ATM gene (ataxia telangiectasia mutated) which encodes a kinase that is induced in response to double-strand breaks in DNA. One important substrate for the ATM kinase is the tumor suppressor protein, p53. There are four distinct complementation groups resulting in AT identified as ATA, ATC, ATD, and ATE. All four are the result of mutations in the ATM gene.
Double Strand Break Repair
The terminology of DNA double-strand break (DSB) refers to the types of DNA lesions where the phosphate-sugar backbones of both DNA strands are broken at the same position (or in close proximity to each other) which allows for physical dissociation of the double helix into two separate molecules. Natural physiological sources of endogenous DSB include DNA replication and meiotic recombination and the programmed rearrangements of the immunoglobulin and T cell receptor genes that occurs during lymphoid cell development. Reactive oxygen species (ROS), produced during cellular metabolism, can oxidize bases and trigger both single-strand breaks (SSB) and DSB.
DNA double-strand breaks are also generated by the action of numerous different exogenous agents that includes ionizing radiation (radioactive decay, cosmic radiation or medical x-rays) or chemicals that mimic the action of ionizing radiation. Chemicals that can induce DSB include base alkylating agents such as methylmethane sulfonate (MMS) and crosslinking agents that introduce covalent crosslinks between bases of the same (intrastrand) or complementary strands (interstrand). DNA crosslinking agents include drugs such as mitomycinC, platinum derivatives, psoralens, and nitrogen mustards. DNA topoisomerase inhibitors can also induce the formation of DSB by trapping topoisomerase-DNA intermediates during isomerization reactions including those occurring during DNA replication. As might be expected, drugs that generate DSB are widely used in cancer chemotherapy since tumor cells are often more sensitive to DSB than normal cells.
Because DSB results in separation of the ends of the two strands of DNA faithful restoration of the phosphodiester bonds can be problematic. The formation of DSB can lead to loss of genetic information, DNA fragmentation, or rearrangement of chromosomes. The various cellular pathways that mediate DSB repair must restore not only the physical integrity of the chromosome but also its precise genetic information. The major pathways of DSB are homology-directed (HDR) and non-homologous end-joining, NHEJ.
Non-homologous end-joining (NHEJ) does not require sequence homology and represents the primary pathway of DSB repair in vertebrates. Non-homologous end-joining is active throughout the cell cycle. Initiation of NHEJ occurs when the Ku70/Ku80 heterodimer (KU) binds to blunt or near-blunt DNA ends. The terms Ku70 and Ku80 are derived from the size of the proteins (70 kDa and 80 kDa, respectively) and the fact that the complexes were first characterized in cells from a Japanese patient whose surname was Ku. The Ku70 protein is encoded by the XRCC6 (X-ray repair cross complementing 6) gene and the Ku80 protein is encoded by the XRCC5 gene.
When DSB are bound by the KU complex the catalytic subunit of the DNA-dependent protein kinase (identified as DNA-PKcs) is recruited to the site and activated. The DNA-PKcs protein is encoded by the PRKDC (protein kinase, DNA-activated, catalytic subunit) gene. Activation of DNA-PKcs triggers an extensive signaling transduction cascade that in turn activates the downstream repair processes. Non-homologous end-joining is facilitated by scaffold proteins encoded by the XRCC4 and NHEJ1 genes. The protein encoded by the XRCC4 gene is also known as SSMED (short stature, microcephaly, and endocrine dysfunction) and that encoded by the NHEJ1 gene is also known as XLF (XRCC4-like factor). These two scaffold proteins bind DNA ligase 4 which is responsible for creating the phosphodiester bond that seals the break. During DSB repair it is possible that the DNA ends need to be processed before ligation. This nucleolytic processing is catalyzed by the Artemis endonuclease, a DNA-PKcs-interacting protein. The Artemis protein is encoded by the DCLRE1C (DNA cross-link repair 1C) gene.
Homology-directed repair (HDR) requires the use of homologous sequences to align DSB ends prior to ligation. The process of HDR occurs primarily during the S phase of the cell cycle. The advantage of this stage of the cell cycle to this repair process is that there is a replicated sister chromatid that can be used as a homologous template to copy and restore the DNA sequence that is found to be missing on the damaged chromatid. A protein complex is required to search for a region of sequence homology to template the HDR repair process. This search requires single-strand DNA at the DSB end and this is generated through an endonucleolytic process referred to as DNA end resection.
The end resection process is initiated by a protein complex that directly binds to the ends of the DSB. In humans this complex is referred to as the MRE11/RAD50 complex and it is composed of at least five distinct protein subunits. The MRE11 protein of the complex possesses both endo- and exonuclease activities and generates 3′ single-stranded overhangs. The nimbrin protein of the complex (encoded by the NBN gene: protein also identified as NBS1) recruits the endonuclease encoded by the RBBP8 (RB binding protein 8, endonuclease) gene whose activity is required for the DNA resection catalyzed by MRE11. The RBBP8 protein is also known as CtIP (C-terminal binding protein 1 interacting protein).
Once the resection process is initiated, the nucleases EXO1 and DNA2 catalyze the bulk of end-resection required for HDR. The cell cycle checkpoint kinases ATM and ATR, are also required for proper DSB resection. These kinases modulate the activities of several of the proteins involved in the resection process including MRE11, RAD50, RBBP8, DNA2 and EXO1. Various mediator proteins are also required for the overall process of HDR including RAD52, BRCA2 and PALB2. Since HDR involves the use of an undamaged DNA template to restore chromosome integrity, it has the potential to repair DSB more faithfully than NHEJ.
Clinical Significance of Defective DSB Repair
The clinical significance of faithful DSB repair can be seen by the fact that inherited mutations in DSB repair genes are associated with a variety of human pathologies, including increased cancer susceptibility, neurological defects and/or immunodeficiencies. Several disease syndromes such as ataxia telangiectasia (AT), Nijmegen breakage syndrome, and some forms of severe combined immunodeficiency (SCID) are associated with defects in DSB repair. The genomic instability that arises from compromised DSB repair function is responsible for the increased cancer susceptibility in women who carry germline mutations in the BRCA1 and/or BRCA2 genes. The proteins encoded by both of these genes are required for proper DSB repair.
Chemotherapies Targeting Replication
The class of compounds that have been used the longest as anticancer drugs are the alkylating agents. The major alkylating agents are derived from nitrogenous mustards that were originally developed for use by the military. Commonly used alkylating agents include cyclophosphamide (Cytoxan, Neosar), ifosphamide, decarbazine, chlorambucil (Leukeran) and procarbazine (Matulane, Natulan).
Alkylating agents function by reacting with and disrupting the structure of DNA. Some agents react with alkyl groups in DNA resulting in fragmentation of the DNA as a consequence of the action of DNA repair enzymes. Some agents catalyze the cross-linking of bases in the DNA which prevents the separation of the two strands during DNA replication. Some agents induce mis-pairing of nucleotides resulting in permanent mutations in the DNA. Alkylating agents act upon DNA at all stages of the cell cycle, thus they are potent anticancer drugs. However, because of their potency, prolonged use of alkylating agents can lead to secondary cancers, particularly leukemias.
Several classes of anticancer drugs function through interference with the actions of the topoisomerases. Two of these classes are the anthracyclines and the camptothecins. The anthracyclines were originally isolated from the fungus, Streptomyces. Doxorubicin (Adriamycin, Doxil, Rubex) and daunorubicin (Cerubidine, DaunoXome, Daunomycin, Rubidomycin) have similar modes of action, although doxorubicin is the more potent of the two and is used in the treatment breast cancers, lymphomas and sarcomas. The anthracyclines inhibit the actions of topoisomerase II whose function is to introduce double-strand breaks in DNA during the process of replication as a means to relive torsional stresses. Anthracyclines also function by inducing the formation of oxygen free radicals that cause DNA strand breaks resulting in inhibition of replication. Another plant-derived anticancer compound that functions through inhibition of topoisomerase II is etoposide (VP-16, Vepesid, Etophos, Eposin). Etoposide is isolated from the mandrake plant and is in a class of compounds referred to as epipodophyllotoxins.
The camptothecins were originally found in the bark of the Camptotheca accuminata tree and include irinotecan (Campto, Camptosar) and topotecan (Hycamtin). Camptothecins inhibit the action of topoisomerase I, an enzyme that induces single-strand breaks in DNA during replication.
Anticancer compounds that are extracted from the periwinkle plant, Catharanthus roseus (also known as Vinca rosea), are called the vinca alkaloids. These compounds bind to GTP-bound β-tubulin monomers which prevents polymerization of the microtubules of the mitotic spindle fibers that are necessary for cell division during mitosis. There are four major vinca alkaloids that are currently used as chemotherapeutics, vincristine (Oncovin, Vincrex), vinblastine (Velbe, Velban, Velsar), vinorelbin (VIN, Navelbine), and vindesine (Eldisine).
The taxanes are another class of plant-derived compounds that act via interference with microtubule function. These compounds were originally isolated from the Pacific yew tree, Taxus brevifolia and include paclitaxel (Taxol) and docetaxel (Taxotere). The taxanes function by stabilizing the GDP-bound form of β-tubulin in microtubules preventing their depolymerization which then prevents cytokinesis (cell division). These compounds are used to treat a wide range of cancers including head and neck, lung, ovarian, breast, bladder, and prostate cancers. Taxol has proven highly effective in the treatment of certain forms of breast cancer.
In order for DNA replication to proceed, proliferating cells require a pool of nucleotides. The class of anticancer drugs that has been developed to interfere with aspects of nucleotide metabolism is known as the antimetabolites. There are two major types of antimetabolites used in the treatment of a broad range of cancers: compounds that inhibit thymidylate synthetase and compounds that inhibit dihydrofolate reductase (DHFR). Both of these enzymes are involved in thymidine nucleotide biosynthesis (see Figure below). Drugs that inhibit thymidylate synthetase include 5-fluorouracil (5-FU, Adrucil, Efudex) and 5-fluorodeoxyuridine. Those that inhibit DHFR are analogs of the vitamin folic acid and include methotrexate (Trexall, Rheumatrex), pyrimethamine (Daraprim), and trimethoprim (Proloprim, Trimpex).