
Last Updated: October 30, 2025
Introduction to Erythropoietic Protoporphyria 1 (EPP1)
Erythropoietic protoporphyria 1 (EPP1) is disorder that is a member of a family of disorders referred to as the porphyrias. EPP1 can be inherited as an autosomal recessive or dominant disorder and it exhibits a high degree of allelic heterogeneity with incomplete penetrance.
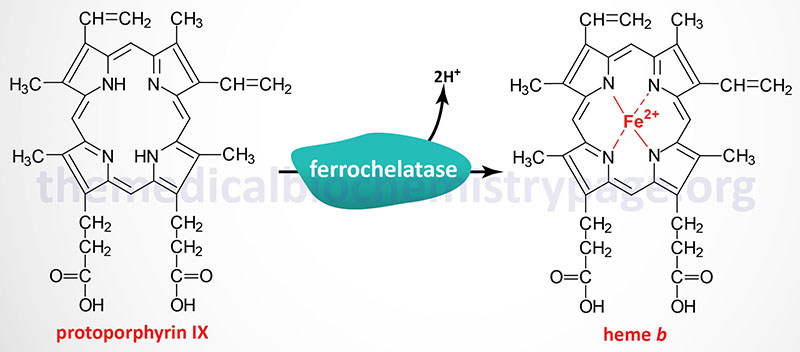
Each disease in the porphyria family results from deficiencies in a specific enzyme involved in the biosynthesis of heme (also called the porphyrin pathway). EPP1 results from defects in ferrochelatase (also called heme synthase). The term porphyria is derived from the Greek term porphura which means “purple pigment” in reference to the coloration of body fluids in patients suffering from the originally described porphyria which is now known as porphyria cutanea tarda (PCT).
The porphyrias are classified on the basis of the tissue that is the predominant site of accumulation of metabolic intermediates. These classifications are “hepatic” or “erythroid”. Each disease is also further characterized as being acute or cutaneous dependent upon the major clinical features of the disease.
As the name would imply, EPP1 is classified as an erythroid porphyria. EPP1 is the most commonly occurring erythroid porphyria and the third most common porphyria. EPP1 has also been referred to simply as protoporphyria since the disorder results in excess accumulation and excretion of protoporphyrin IX. The accumulation of protoporphyrin IX is found primarily in the erythroid cells of the bone marrow, circulating erythrocytes, plasma, and feces.
EPP1 was originally simply referred to as EPP but the identification a child with a related disorder has lead to the identification of this second disorder as EPP2 and the original erythropoietic protoporphyria as EPP1. EPP2 is a disease that results from mutations in the CLPX (caseinolytic mitochondrial matrix peptidase chaperone subunit) gene. EPP2 is inherited as an autosomal dominant disorder.
Molecular Biology of EPP1
The ferrochelatase gene (FECH) is located on chromosome 18q21.31 spanning 45 kb and encompassing 12 exons that generate five alternatively spliced mRNAs, each of which encodes a distinct protein isoform. The most common ferrochelatase is a 429 amino acid protein encoded by the isoform a mRNA.
More than 50 different mutations have been identified in the FECH gene resulting in EPP1. These mutations include missense, nonsense and splicing mutations and both large and small insertions and deletions.
Clinical Features of EPP1
Light-sensitive dermatitis commencing in childhood, usually before 10 years of age, is the presenting finding in erythropoietic protoporphyria. Common symptoms are itching, swelling and painful erythema (skin redness due to capillary congestion) which can develop within minutes on sun-exposed areas of the skin. The cutaneous manifestations of EPP1 are usually non-blistering in contrast to the other cutaneous porphyrias such as CEP (congenital erythropoietic porphyria) and HEP (hepatoerythropoietic porphyria). HEP is the term given to a disease that is the homozygous dominant form of type II porphyria cutanea tarda (PCT). Most EPP1 patients experience only a painful photosensitivity, whereas a small number of them develop liver complications due to the accumulation of excessive amounts of protoporphyrin in the liver. The manifestations in EPP1 remain fairly stable over the span of many years in afflicted individuals. Unlike in the hepatic porphyrias where distinct precipitating factors (e.g. drugs, steroids) lead to acute manifestations of porphyria, there are no known precipitating factors associated with EPP1.
Therapy in Patients with EPP1
Oral administration of β-carotene has been shown to improve sunlight tolerance in EPP1 patients. In EPP1 patients that experience hepatic involvement, treatment with cholestyramine (an intestinal bile acid binding drug used in the treatment of hypercholesterolemia) promotes the excretion of protoporphyrin in the feces reducing the hepatic protoporphyrin levels. The use of cholestyramine also improves the cutaneous symptoms of EPP1 in some individuals.