

Last Updated: June 2, 2026
Introduction to Cystinuria
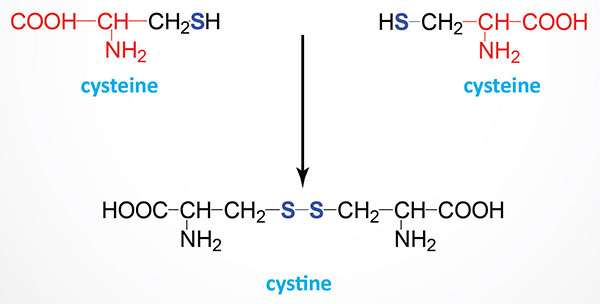
As the name implies, cystinuria is a disorder associated with excess cystine in the urine. Cystine is the oxidized disulfide homodimer of two cysteines. Type I cystinuria is an autosomal recessive disorder that results from a failure of the renal proximal tubules to reabsorb cystine that was filtered by the glomerulus. The accumulation of cystine and its formation of crystals within the distal tubules results in the formation of cystine stones in the kidneys (nephrolithiasis).
Among the various forms and causes of kidney stones, the presence of cystine stones represents 1-2% of all adult urinary nephrolithiasis patients, but in children 6-8% of patients suffer from cystine stones. The etiology of nephrolithiasis can be complex and includes factors such as diet and fluid intake. However, of all causes of nephrolithiasis, cystinuria is the only one caused solely by genetic factors. The overall prevalence of cystinuria is 1:7,000 with population specific variation such that in Americans the rate is 1:25,000 and the highest incidence of 1:2,500 being found in Libyan Jews.
Molecular Biology of Cystinuria
Cystinuria results from defects in either of the two protein subunits one of the cystine transporter which is distinct from the renal cysteine transporter. The same cystine transporter is expressed within the small intestine but a defect in its function in this tissue does not contribute to pathology. In addition to excess cystine in the urine, the disorder is associated with increased urinary excretion of the dibasic amino acids arginine, lysine, and ornithine. However, clinical consequences are only associated with the increased urinary cystine and is due to the poor solubility of this homodimeric compound. Cystine will precipitate in the urine resulting in the formation of renal calculi (stones) within the distal tubules and this can lead to renal failure.
The two subunits of the cystine transporter are both members of the SLC family of transporter proteins and they are encoded by the SLC3A1 and SLC7A9 genes. The SLC3A1 encoded protein is one of the heavy subunits of the heteromeric amino acid transporters. The protein is commonly referred to as the basic amino acid transport protein (rBAT). The functional subunit of the transporter is one of the light chain subunits of the heteromeric amino acid transporters and is encoded by the SLC7A9 gene. The SLC7A9 encoded protein is a Na+-independent amino acid transporter most often referred to as b(0,+)AT. The “b” refers to broad specificity and the 0 and + signify that the transporter transports both neutral (0) and basic (+) amino acids. The “AT” refers to Amino acid Transporter.
The SLC3A1 gene is located on chromosome 2p21 and is composed of 10 exons that encode a 685 amino acid protein. Although several alternatively spliced mRNAs have been detected from the SLC3A1 gene, they do not appear to encode functional protein. Expression of the SLC3A1 gene is highest in the kidney.
The SLC7A9 gene is located on chromosome 19q13.11 and is composed of 13 exons that generate three alternatively spliced mRNAs, all of which encode the same 487 amino acid protein. Expression of the SLC7A9 gene is almost exclusive to the kidney and small intestine.
Mutations in the SLC3A1 gene are associated with the autosomal recessive type I cystinurias while mutations in the SLC7A9 gene are associated with non-type I cystinurias that exhibit broad clinical variability even within the same family.
Clinical Features of Cystinuria
The initial diagnosis of cystinuria is usually precipitated by the finding of kidney stones (renal lithiasis) which show typical cystine crystals. Cystine crystals are long hexagonal translucent crystals that when removed from a patient often appear pink or yellow but turn greenish upon exposure to air. Determination as to whether or not a person is suffering from renal lithiasis, due to cystinuria, is accomplished via the sodium cyanide-nitroprusside test. In this assay urine is exposed to the reagent which turns purple within 2-10 minutes. The sodium cyanide reduces the cystine to cysteine which then binds the nitroprusside causing the purple color.
The clinical classifications of cystinuria was originally divided into three major types. These types are distinguished by the urinary phenotype of the parents (obligate heterozygotes) of afflicted patients. Type I cystinuria is distinguished by heterozygotes that excrete cystine at normal levels. The original designations of type II and type III cystinuria were defined by heterozygotes that had high or moderately elevated cystine excretion. However, since the genetic mutations that predispose an individual to cystinuria could not be correlated to a type II versus a type III individual, these two designations were changed to the classification of non-type I cystinurias. The distinction of non-type I heterozygotes is that they exhibit variable hyperexcretion of cystine and the dibasic amino acids in their urine. Due to these designations of the phenotype, type I cystinuria mainly exhibits an autosomal recessive inheritance trait while non-type I cystinuria can be regarded as an autosomal dominant disorder with incomplete penetrance for cystine lithiasis.
The diagnosis of cystinuria can be further complicated by the fact that some patients exhibit a mixed cystinuria due to their carrying both type I and non-type I alleles. The majority of cystinuria patients will manifest renal calculi formation some time with in the first two decades of life. However, it is important to note that even within the same family there can be variation in disease phenotype and severity. Males with cystinuria are affected more severely and with a greater frequency than females. In addition, the cystine stones that form in males are often larger than those in female patients. Generally, type I homozygous patients have earlier manifestation of stone formation while non-type I homozygous patients as well as mixed type patients will develop stones later in life.
Treatment of Cystinuria
Common treatments for patients with cystinuria are to decrease protein and salt intake as well as to ensure increased hydration as this will dilute the cystine in the urine reducing the potential for crystal formation. In addition, patients are given drugs, such as acetazolamide (a carbonic anhydrase inhibitor principally utilized in the treatment of glaucoma and certain forms of hypertension), which alkalizes the urine thereby reducing the potential for urinary precipitation of cystine.
In addition, thiol drugs can be used to compete for the formation of cystine. These drugs include captopril [an angiotensin converting enzyme (ACE) inhibitor used principally in the treatment of hypertension], D-penicillamine (a chelator drug used in the treatment of Wilson disease, lead poisoning, and rheumatoid arthritis), and alpha-mercaptopropionylglycine, α-MPG (a second-generation chelating drug).
The composition of cystine stones makes their removal by extracoporal lithothripsy difficult. In patients that are diagnosed with large stones their treatment will require percutaneous nephrostomy placement in order to remove the stones. Due to the fact that patients with cystinuria, in particular type I patients, experience episodic stone symptoms it is necessary for repeated stone removal. This episodic character to cystine stone formation and removal can lead to serious renal damage as well as damage to surrounding organs. In rare cases, where proper interventional therapy is not activated, patient deaths have been reported.
Cystinosis
Another disorder related to cystine transport is called cystinosis. Cystinosis is a rare autosomal recessive disorder that is associated with lysosomal accumulation of cystine. The causes of cystinosis are mutations in the lysosomal localized cystine transporter called cystinosin. Cystinosin is responsible for transporting cystine out of the lysosome to the cytosol. The inability to transport cystine out of the lysosomes results in cystine crystal formation in the organelle leading to multi-organ involvement.
There are three classified forms of cystinosis that includes the infantile (nephropathic) form, the juvenile (intermediate and late-onset) form, and the adult (benign, ocular, and non-nephropathic) form. The infantile form is the most common, representing nearly 95% of all identified cases. The infantile form is a severe form of the disease resulting in end-stage renal disease between the ages of 10 to 12. In addition to the kidneys, the infantile form of cystinosis includes pathology in the eyes and endocrine organs which contributes to the overall morbidity of this form of the disorder.
The cystinosin transporter is encoded by the CTNS gene. The CTNS gene is located on chromosome 17p13.2 and is composed of 13 exons that generate seven alternatively spliced mRNA that collectively encode three different precursor proteins. The isoform 3 protein is encoded for by four of the mRNAs and is a 220 amino acid precursor. As of 2025 there have been a total of 109 pathogenic mutations identified in the CTNS gene.