

Last Updated: November 11, 2025
Introduction to CPS1 Deficiency
Carbamoyl phosphate synthetase 1 deficiency (CPSD) is an autosomal recessive disorder that, as the name implies, is due to mutations in the gene (CPS1) encoding the urea cycle enzyme, carbamoyl phosphate synthetase 1. The functional CPS1 enzyme exists as a homodimer.
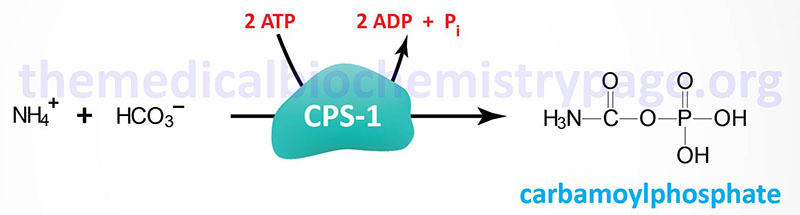
The CPS1 enzyme of the urea cycle is distinct from the CPS2 enzyme activity of pyrimidine nucleotide biosynthesis, although both the CPS1 and CPS2 catalyzed reactions yield carbamoyl phosphate. The CPS2 activity of pyrimidine nucleotide synthesis is one of three enzymatic activities of the tri-functional enzyme encoded by the CAD gene.
Molecular Biology of CPS1 Deficiency
The CPS1 gene is located on chromosome 2q34 composed of 43 exons that generate four alternatively spliced mRNAs that collectively encode two distinct protein isoforms. CPS1 isoform b is a protein of 1500 amino acids. CPS1 isoform d is a protein of 1511 amino acids.
There are at least 25 known mutations in the CPS1 gene that result in carbamoyl phosphate synthetase 1 deficiency (CPSD). Mutations in the CPS1 gene that cause CPSD include missense, nonsense, and exon deletions. The frequency of CPSD is approximately 1 per 62,000 live births. Because CPS1 is responsible for the first reaction of the urea cycle, defects in this gene manifest with the most severe symptoms of all UCDs.
Clinical Features of CPS1 Deficiency
CPSD is, like the other neonatal onset forms of UCD, most severe when presenting in newborn infants. As with each of the four neonatal onset UCD, CPSD is characterized by the accumulation of ammonium ion and glutamine with clinical manifestations appearing in full-term infants with no prior obstetric risk factors. The classic symptoms appear between 24hrs and 48hrs after birth (but not prior to 24hrs) and include convulsions, hyperventilation, ataxia, hypothermia, lethargy, vomiting and poor feeding. If left untreated the hyperammonemia with result in coma and death. The severe effects of hyperammonemia are described in the Nitrogen Metabolism and Urea Cycle page. Even though sepsis is a rare event in a normal term infant with no prior obstetric complications, this disorder is misdiagnosed in almost half of neonatal UCD cases.
Initial laboratory findings will include respiratory alkalosis which is the earliest objective indication of encephalopathy. The encephalopathy will progress to the point where mechanical ventilation is required. Another routine laboratory finding is reduced serum (blood) urea nitrogen (BUN) which may be as low as 1mg/dl (normal for newborns is 3–12mg/dl). If plasma ammonia levels are not measured the infants’ death will be attributed to sepsis, intracranial hemorrhage, or some other disorder that would normally be associated with a pre-term delivery.
Treatment of CPS1 Deficiency
CPSD patients are treated in much the same ways as for other neonatal UCD in that protein intake must me highly regulated and the hyperammonemia must be controlled. Hemodialysis is the only effective means to rapidly lower serum ammonia levels in these patients. Acute episodes of hyperammonemia can be treated with intravenous administration of Ammunol® and with oral Buphenyl® for chronic adjunctive therapy of hyperammonemia. The mechanism of action of these compounds is detailed in the Urea Cycle Disorders: Overview page.
Additionally, CPSD patients are treated with a strict dietary regimen that includes 0.7g/kg/day of protein and 0.7g/kg/day of an essential amino acid mixture. The diet is also supplemented with citrulline to serve as a source of arginine. As development proceeds and the need for protein declines it is necessary to adjust the dietary intake of protein accordingly.
In 2025 the first report (in the New England Journal of Medicine) of the use of gene editing therapy (CRISPR/Cas9 technology) to correct a mutation in the CPS1 gene in an infant was announced. This therapeutic approach was initiated 7 months after the infant was diagnosed with a CPS1 deficiency. The infant was administered two doses of the editing vector, once at 7 months of age and the second at 8 months of age. To prevent immune rejection of the transgenic enzyme the infant was also treated with immunosuppressant drugs. Efficacy of the treatment was evidenced by several criteria including good control of plasma ammonia and glutamine levels and an increase of orotic acid which allowed for a well-tolerated normalization of dietary protein intake and reduction, by half, of the nitrogen scavenger Ammunol.