
Last Updated: April 7, 2026
Introduction to Cholesterol
Cholesterol is an extremely important biological molecule that has roles in membrane structure as well as being a precursor for the synthesis of the steroid hormones, and the bile acids. The pathway of cholesterol synthesis is also important for the synthesis of 7-dehydrocholesterol, the precursor for vitamin D synthesis.
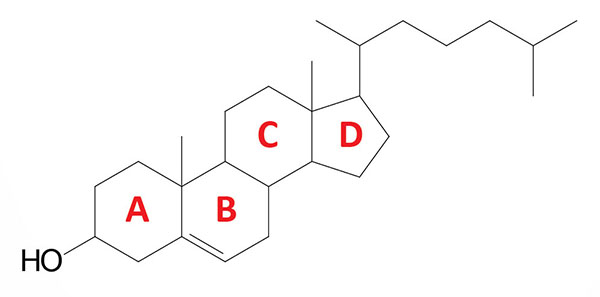
Both dietary cholesterol, and that synthesized de novo, are transported through the circulation in lipoprotein particles. The same is true of cholesteryl esters, the form in which cholesterol is stored in cells. Due to its important role in membrane function, all cells express the enzymes of cholesterol biosynthesis.
The biosynthesis, utilization, and metabolism of cholesterol must be tightly regulated in order to prevent over-accumulation and abnormal deposition within the body. Cholesterol metabolism does not involve the breakdown of the molecule but involves its conversion to the bile acids and the steroid hormones.
Of particular clinical importance is the abnormal deposition of cholesterol and cholesterol-rich lipoproteins in the coronary arteries. Such deposition, eventually leading to atherosclerosis, is the leading contributory factor in diseases of the coronary arteries.
Biosynthesis of Cholesterol
Slightly less than half of the cholesterol in the body derives from biosynthesis de novo. Biosynthesis in the liver accounts for approximately 10%, and in the intestines approximately 15%, of the amount produced each day. The cholesterol biosynthesis pathway involves enzymes that are in the cytoplasm, microsomes (ER), and peroxisomes.
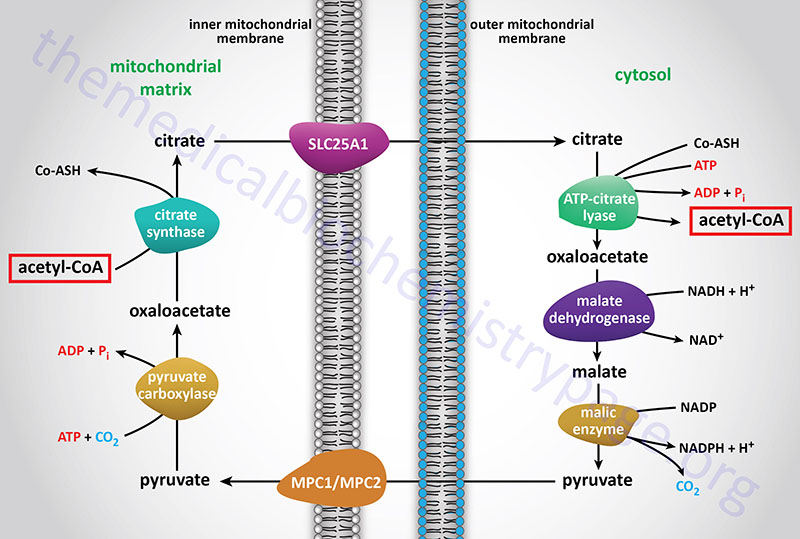
The acetyl-CoA utilized for cholesterol biosynthesis is derived from an oxidation reaction (fatty acids or pyruvate or amino acids) in the mitochondria and is transported to the cytoplasm by the same process as that described for fatty acid synthesis (see the Figure below). Acetyl-CoA can also be synthesized from cytosolic acetate derived from cytoplasmic oxidation of ethanol which is initiated by cytoplasmic alcohol dehydrogenase (ADH). All the reduction reactions of cholesterol biosynthesis use NADPH as a cofactor.
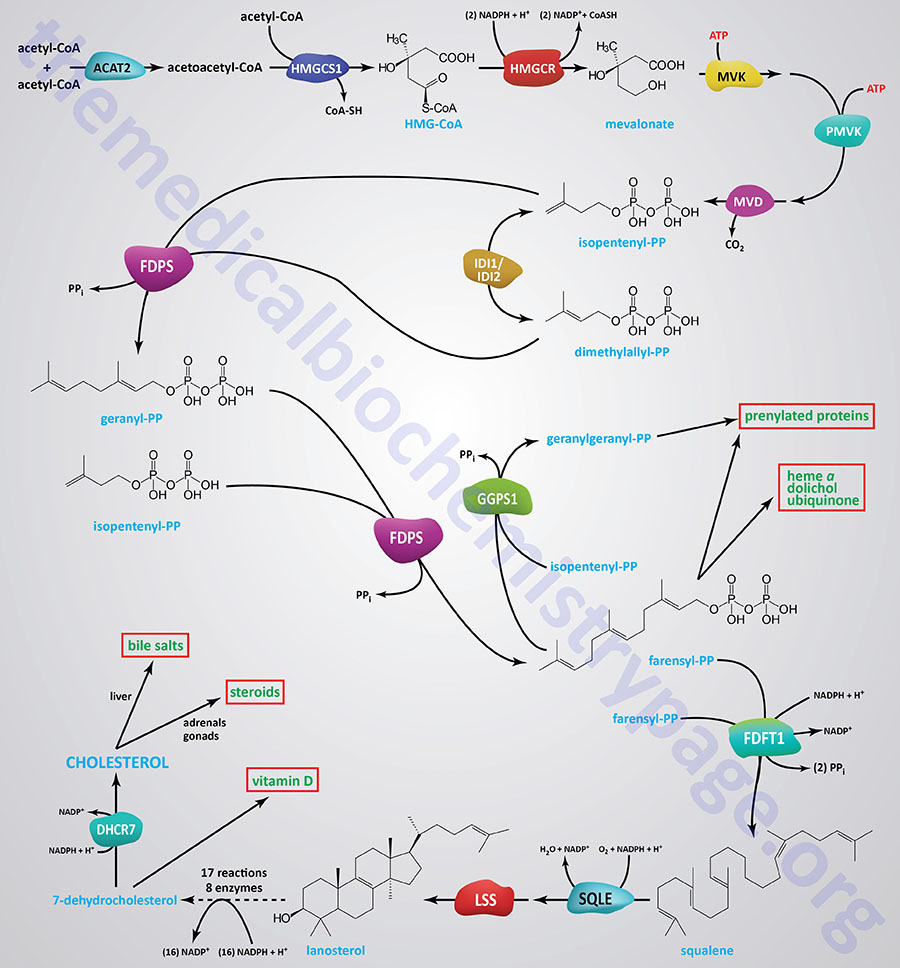
Synthesis of cholesterol, like that of most biological lipids, begins from the two-carbon acetate group of acetyl-CoA and requires a total of 18 different enzymes. Most sources indicate a total of 17 enzymes where 3-hydroxy-3-methylglutaryl-CoA synthase (HMGCS1) is considered the first enzyme in the pathway. The Figure below of the pathway of cholesterol biosynthesis begins with the enzyme acetyl-CoA acetyltransferase 2 (ACAT2) which converts two acetyl-CoA molecules into acetoacetyl-CoA, the substrate for HMGCS1.
The initial steps in the pathway of cholesterol biosynthesis are collectively called the mevalonate pathway which itself culminates with the synthesis of the isoprenoid molecule, isopentenyl pyrophosphate (IPP).
Several of the enzymes of cholesterol synthesis require NADPH as a cofactor. A total of 21 moles of NADPH are required for every mole of cholesterol synthesized. Some sources suggest the total is only 16 moles of NADPH but rigorous examination of the literature clearly shows that 21 moles of NADPH are consumed in the synthesis of cholesterol from acetyl-CoA. The differences may relate to the fact that one of the enzymes, NAD(P) dependent steroid dehydrogenase-like (encoded by the NSDHL gene) can utilize NADH or NADPH and this enzyme catalyzes two of the reactions of the Kandutsch-Russell pathway of lanosterol conversion to cholesterol.
The isoprenoid intermediates of cholesterol biosynthesis can be diverted to other synthesis reactions, such as those for dolichol (used in the synthesis of N-linked glycoproteins), coenzyme Q (of the oxidative phosphorylation pathway), or the side chain of heme-a. Additionally, these intermediates are used in the lipid modification of some proteins.
The process of cholesterol synthesis can be considered to be composed of five major steps where the reactions that culminate in the synthesis of isopentenyl pyrophosphate, and its isomeric form, dimethylallyl pyrophosphate, are commonly referred to as the mevalonate pathway:
- Acetyl-CoAs are converted to 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA)
- HMG-CoA is converted to mevalonate
- Mevalonate is converted to the isoprene based molecule, isopentenyl pyrophosphate (IPP)
- IPP molecules are converted to squalene
- Squalene is converted to cholesterol
HMG-CoA Synthesis
Acetyl-CoA units are converted to mevalonate by a series of reactions that begins with the formation of HMG-CoA. Unlike the HMG-CoA formed during ketone body synthesis in the mitochondria, this form is synthesized in the cytoplasm. However, the pathway and the necessary enzymes are similar to those in the mitochondria.
Two moles of acetyl-CoA are condensed in a reversal of the thiolase reaction, forming acetoacetyl-CoA. The cytoplasmic thiolase enzyme involved in cholesterol biosynthesis is acetyl-CoA acetyltransferase 2 (also called acetoacetyl-CoA thiolase) encoded by the ACAT2 gene. Although the bulk of acetoacetyl-CoA is derived via this process, it is possible for some acetoacetate, generated during ketogenesis, to diffuse out of the mitochondria and be converted to acetoacetyl-CoA in the cytosol via the action of acetoacetyl-CoA synthetase (AACS).
Acetoacetyl-CoA and a third mole of acetyl-CoA are converted to HMG-CoA by the action of the cytosolic version of HMG-CoA synthase encoded by the HMGCS1 gene. The HMGCS1 gene is located on chromosome 5p12 and is composed of 12 exons that generate nine alternatively spliced mRNAs that collectively encode proteins of 520 amino acids (isoform 1) and 478 amino acids (isoform 2).
Mevalonate Synthesis
HMG-CoA is then converted to mevalonate by HMG-CoA reductase, HMGR (this enzyme is bound in the endoplasmic reticulum, ER). HMGR absolutely requires NADPH as a cofactor and two moles of NADPH are consumed during the conversion of HMG-CoA to mevalonate. The reaction catalyzed by HMGR is the rate limiting step of cholesterol biosynthesis, and this enzyme is subject to complex regulatory controls as discussed below.
HMGR is encoded by the HMGCR gene. The HMGCR gene is located on chromosome 5q13.3 and is composed of 23 exons that generate three alternatively spliced mRNAs that collectively encode HMGR isoform 1 (888 amino acids) and HMGR isoform 2 (835 amino acids).
Mevalonate is then activated by two successive phosphorylations (catalyzed by mevalonate kinase, and phosphomevalonate kinase) yielding, sequentially, mevalonate 5-phosphate and then mevalonate 5-diphosphate. The latter compound is also called 5-pyrophosphomevalonate or mevalonate 5-pyrophosphate.
In humans, mevalonate kinase is a peroxisome localized enzyme encoded by the MVK gene. The MVK gene is located on chromosome 12q24.11 and is composed of 12 exons that generate three alternatively spliced mRNAs that collectively encode two distinct protein isoforms.
Phosphomevalonate kinase is also a peroxisomal enzyme and it is derived from the PMVK gene. The PMVK gene is located on chromosome 1q21.3 and is composed of 6 exons that generate four alternatively spliced mRNAs, each of which encode a distinct protein isoform.
Isopentenylpyrophosphate (IPP) Synthesis
Following the formation of mevalonate 5-diphosphate, an ATP-dependent decarboxylation yields isopentenyl pyrophosphate (IPP) which is an activated isoprenoid molecule. The synthesis of IPP is catalyzed by diphosphomevalonate decarboxylase (also called mevalonate-5-pyrophosphate decarboxylase) encoded by the MVD gene.
The MVD gene is located on chromosome 16q24.2 and is composed of 13 exons that encode a 400 amino acid protein.
Isopentenyl pyrophosphate is in equilibrium with its isomer, dimethylallyl pyrophosphate (DMAPP) via the action of isopentenyl-diphosphate delta isomerase (also called isopentenylpyrophosphate isomerase). Humans express two isopentenyl-diphosphate delta isomerase genes, IDI1 and IDI2. The IDI2 gene most likely arose via a gene duplication event.
The IDI1 gene is located on chromosome 10p15.3 and is composed of 8 exons that generate four alternatively spliced mRNAs that collectively encode three protein isoforms that are localized to the peroxisomes.
The IDI2 gene is located on the same chromosomal region as the IDI1 gene but is composed of only 5 exons and encodes a 227 amino acid protein.
Squalene Synthesis
One molecule of IPP condenses with one molecule of DMAPP to generate geranyl pyrophosphate, GPP. GPP further condenses with another IPP molecule to yield farnesyl pyrophosphate, FPP. Synthesis of both GPP and FPP is catalyzed by the enzyme, farnesyl diphosphate synthase that is encoded by the FDPS gene. The FDPS gene is located on chromosome1q22 and is composed of 11 exons that generate seven alternatively spliced mRNAs that collectively encode three different isoforms of the enzyme.
The synthesis of squalene, from FPP, represents the first cholesterol/sterol-specific step in the cholesterol synthesis pathway. This is due to the fact that, as depicted in the pathway Figure above, several intermediates in the pathway can be diverted to the production of other biologically relevant molecules. The synthesis of squalene is catalyzed by the NADPH-requiring enzyme, farnesyl-diphosphate farnesyltransferase 1 (commonly called squalene synthase). Farnesyl-diphosphate farnesyltransferase 1 catalyzes the two-step head-to-head condensation of two molecules of FPP, yielding squalene.
Farnesyl-diphosphate farnesyltransferase 1 is encoded by the FDFT1 gene. The FDFT1 gene is located on chromosome 8p23.1 and is composed of 14 exons that generate 11 alternatively spliced mRNAs. These 11 different FDFT1-encoded mRNAs collectively synthesize five different isoforms of farnesyl-diphosphate farnesyltransferase 1.
Squalene to Lanosterol
Squalene then undergoes a two step cyclization to yield lanosterol. The first reaction in this two-step cyclization is catalyzed by the enzyme, squalene epoxidase (also called squalene monooxygenase). This enzyme uses NADPH as a cofactor to introduce molecular oxygen as an epoxide at the 2,3 position of squalene forming the intermediate, 2,3-oxidosqualene. In the second step, this epoxide intermediate is converted to lanosterol through the action of the enzyme lanosterol synthase (2,3-oxidosqualene-lanosterol cyclase).
Squalene epoxidase is encoded by the SQLE gene. The SQLE gene is located on chromosome 8q24.13 and is composed of 11 exons that encode a protein of 574 amino acids.
Lanosterol synthase is encoded by the LSS gene. The LSS gene is located on chromosome 21q22.3 and is composed of 22 exons that generate four alternatively spliced mRNAs which together generate three distinct isoforms of the enzyme.
Lanosterol to 7-Dehydrocholesterol
The actual fate of lanosterol is determined by the cell in which it is synthesized as well as by the need for various steroids other than cholesterol. There are two pathways that utilize lanosterol for the synthesis of sterols. One pathway is called the Bloch pathway which terminates with the synthesis of desmosterol. Desmosterol can be converted to cholesterol via the action of the NADPH-requiring enzyme, 24-dehydrocholesterol reductase, encoded by the DHCR24 gene. The other pathway is called the Kandutsch-Russell pathway and this represents the major pathway for the conversion of lanosterol to 7-dehydrocholesterol in humans.
Kandutsch-Russell Pathway
In the Kandutsch-Russell pathway, a series of 17 reactions converts lanosterol to 7-dehydrocholesterol. These 17 reaction steps are catalyzed by eight different enzymes that are integral membrane proteins of the ER.
Reaction 1: Lanosterol is first converted to 24,25-dihydrolanosterol by the NADPH-requiring enzyme, 24-dehydrocholesterol reductase, encoded by the DHCR24 gene. The DHCR24 gene is located on chromosome 1p32.3 and is composed of 9 exons that encode a 516 amino acid precursor protein.
Reactions 2, 3, 4: The next step involves demethylation occurring in a series of three reactions that yields 4,4-dimethylcholesta-8,11-dienol. These reactions are catalyzed by the enzyme encoded by the CYP51A1 gene. The CYP51A1 encoded enzyme is commonly called lanosterol 14-alpha-demethylase (14-α-demethylase). Each of the three reactions required for demethylation requires a mole of NADPH. The CYP51A1 gene is located on chromosome 7q21.2 and is composed of 11 exons that generate two alternatively spliced mRNAs, both of which encode distinct protein isoforms.
Reaction 5: The next reaction is catalyzed by the NADPH-requiring enzyme encoded by the TM7SF2 (transmembrane 7 superfamily member 2) gene yielding 4,4-dimethylcholesta-8-enol. The enzymatic activity of the TM7SF2 encoded protein is commonly called delta4-sterol reductase (Δ4-sterol reductase). The TM7SF2 gene is located on chromosome 11q13.1 and is composed of 10 exons that generate two alternatively spliced mRNAs, both of which encode distinct protein isoforms.
Reactions 6, 7, 8: The next three steps in the conversion of lanosterol to 7-dehydrocholesterol each requires NADPH and they are catalyzed by 3beta-hydroxysteroid C4-methyl oxidase (also called methylsterol monooxygenase 1) which is encoded by the MSMO1 gene. The ultimate product of these three reactions is 4-methyl-4-carboxyzymostenone. The MSMO1 gene is located on chromosome 4q32.3 and is composed of 6 exons that generate two alternatively spliced mRNAs, both of which encode distinct protein isoforms.
Reaction 9: The next reaction is catalyzed by the NAD(P)H-requiring enzyme, NAD(P) dependent steroid dehydrogenase-like that is encoded by the NSDHL gene. The product of this reaction is 4-α-methylcholest-8-enone. The NSDHL gene is located on the X chromosome (Xq28) and is composed of 11 exons that generate two alternatively spliced mRNAs, both of which encode the same 373 amino acid protein.
Reaction 10: The next reaction is catalyzed by the NADPH-requiring enzyme, hydroxysteroid 17-beta dehydrogenase 7, encoded by the HSD17B7 gene. The product of this reaction is 4α-methylcholest-8-en-3β-ol. The HSD17B7 gene is located on chromosome 1q23.3 and is composed of 10 exons that generate three alternatively spliced mRNAs, each of which encodes a distinct protein isoform.
Reactions 11, 12, 13: The next three steps, each requiring NADPH, are catalyzed by the MSMO1 encoded enzyme. The ultimate product of these reactions is 4α-carboxy-5α-cholesta-8-en-3β-ol.
Reaction 14: The next reaction is catalyzed by the NSDHL gene and yields zymostenone.
Reaction 15: The next reaction is catalyzed by the HSD17B7 encoded enzyme and yields zymostenol.
Reaction 16: The next reaction is catalyzed by 3-beta-hydroxysteroid-delta(8)-delta(7)-sterol isomerase (also called cholestenol delta-isomerase) which is encoded by the EBP (EBP cholestenol delta-isomerase) gene. The product of this reaction is lathosterol. The EBP gene is located on the X chromosome (Xp11.23) and is composed of 5 exons that encode a 230 amino acid protein.
Reaction 17: The final reaction generating 7-dehydrocholesterol is catalyzed by the NADPH-dependent enzyme, sterol C5-desaturase (also known as lathosterol oxidase), which is encoded by the SC5D gene. The SC5D gene is located on chromosome 11q23.3–q24.1 and is composed of 6 exons that generate two alternatively spliced mRNAs, both of which encode the same 299 amino acid protein.
7-Dehydrocholesterol to Cholesterol
The terminal reaction in cholesterol biosynthesis is catalyzed by the NADPH-dependent enzyme, 7-dehydrocholesterol reductase which is encoded by the DHCR7 gene. Functional DHCR7 protein is a 55.5 kDa NADPH-requiring integral membrane protein localized to the microsomal (ER) membrane.
The DHCR7 gene is located on chromosome 11q13.4 and is composed of 9 exons that generate two alternatively spliced mRNAs, both of which encode the same 475 amino acid protein.
Deficiency in DHCR7 (due to gene mutations) results in the disorder called Smith-Lemli-Opitz syndrome, SLOS. SLOS is characterized by increased levels of 7-dehydrocholesterol and reduced levels (15% to 27% of normal) of cholesterol resulting in multiple developmental malformations and behavioral problems.
Cytochrome P450 Enzymes in Cholesterol Metabolism
Cytochrome P450 enzymes are involved in a diverse array of biological processes that includes lipid, cholesterol, and steroid metabolism as well as the metabolism of xenobiotics. The now common nomenclature used to designate P450 enzymes is CYP. There are at least 57 CYP enzymes in human tissues with eight being involved in cholesterol biosynthesis and metabolism, which includes conversion of cholesterol to bile acids. CYP metabolism of cholesterol yields several oxysterols that function as biologically active molecules such as in the activation of the liver X receptors (LXR) and SREBP (see below).
CYP3A4: CYP3A4 is also known as glucocorticoid-inducible P450 and nifedipine oxidase. Nifedipine is a member of the calcium channel blocker drugs used to treat hypertension. CYP3A4 is a major hepatic P450 enzyme and is responsible for the biotransformation of nearly 60% of all commercially available drugs. With respect to cholesterol metabolism, CYP3A4 catabolizes cholesterol to 4β-hydroxycholesterol. This cholesterol derivative is one of the major circulating oxysterols and is seen at elevated levels in patients treated with anti-seizure medications such as carbamazepine, phenobarbital, and phenytoin. The nuclear receptor, pregnane X receptor (PXR), is known to be an inducer of the CYP3A4 gene.
CYP7A1: CYP7A1 is also known as cholesterol 7α-hydroxylase and is the rate limiting enzyme in the primary pathway of bile acid synthesis referred to as the classic pathway. This reaction of bile acid synthesis plays a major role in hepatic regulation of overall cholesterol balance. Deficiency in CYP7A1 manifests with markedly elevated total cholesterol as well as LDL, premature gallstones, premature coronary and peripheral vascular disease. Treatment of this disorder with members of the statin drug family do not alleviated the elevated serum cholesterol due to the defect in hepatic diversion of cholesterol into bile acids.
CYP7B1: CYP7B1 is also known as oxysterol 7α-hydroxylase and is involved in the synthesis of bile acids via the less active secondary pathway referred to as the acidic pathway. A small percentage (1%) of individuals suffering from autosomal recessive hereditary spastic paraplegia 5A (SPG5A) have been shown to harbor mutations in the CYP7B1 gene.
CYP8B1: CYP8B1 is also known as sterol 12α-hydroxylase and is involved in the conversion of 7-hydroxycholesterol (CYP7A1 product) to cholic acid which is one of two primary bile acids and is derived from the classic pathway of bile acid synthesis. The activity of CYP8B1 controls the ratio of cholic acid over chenodeoxycholic acid in the bile.
CYP27A1: CYP27A1 is also known as sterol 27-hydroxylase and is localized to the mitochondria. CYP27A1 functions with two cofactor proteins called ferredoxin 1 (also called adrenodoxin) and ferredoxin reductase (also called adrenodoxin reductase) to hydroxylate a variety of sterols at the 27 position. CYP27A1 is also involved in the diversion of cholesterol into bile acids via the less active secondary pathway referred to as the acidic pathway. Deficiencies in CYP27A1 result in progressive neurological dysfunction, neonatal cholestasis, bilateral cataracts, and chronic diarrhea.
CYP39A1: CYP39A1 is also known as oxysterol 7α-hydroxylase 2. This P450 enzyme was originally identified in mice in which the CYP7B1 gene had been knocked out. The preferential substrate for CYP39A1 is 24-hydroxycholesterol, which is a major product of CYP46A1, which via CYP39A1 action is diverted into bile acid synthesis.
CYP46A1: CYP46A1 is also known as cholesterol 24-hydroxylase. This enzyme is expressed primarily in neurons of the central nervous system where it plays an important role in metabolism of cholesterol in the brain. The product of CYP46A1 action if 24S-hydroxycholesterol which can readily traverse the blood-brain-barrier to enter the systemic circulation. This pathway of cholesterol metabolism in the brain is a part of the reverse cholesterol transport process and serves as a major route of cholesterol turnover in the brain. 24S-hydroxycholesterol is a known potent activator of LXR and as such serves as an activator of the expression of LXR target genes and thus, can effect regulation of overall cholesterol metabolism not only in the brain but many other tissues as well.
CYP51A1: CYP51A1 is also referred to as lanosterol-14α-demethylase. This CYP enzyme is the only one of the eight that is involved in de novo cholesterol biosynthesis and it catalyzes the removal of the 14α-methyl group from lanosterol resulting in the generation of at least two oxysterols that, in mammalian tissues, are efficiently converted into cholesterol as well as more polar sterols and steryl esters. The oxysterols derived through the action of CYP51A1 inhibit HMGR and are also known to inhibit sterol synthesis. Knock-out of the mouse CYP51A1 homolog results in a phenotype similar to that seen in the human disorder known as Antley-Bixler syndrome (ABS). ABS represents a group of heterogeneous disorders characterized by skeletal, cardiac, and urogenital abnormalities that have frequently been associated with mutations in the fibroblast growth factor receptor 2 (FGFR2) gene.
Important Isoprenoids from Intermediates of Cholesterol Synthesis
Dolichol Phosphate Synthesis
Dolichol phosphate is a polyisoprenoid compound synthesized from the isoprenoid intermediates of the de novo cholesterol biosynthesis pathway. The function of dolichol phosphate is to serve as the foundation for the synthesis of the precursor carbohydrate structure, termed the lipid-linked oligosaccharide, LLO ( also referred to as the en bloc oligosacchariode), required for the attachment of carbohydrate to asparagine residues in N-linked glycoproteins.
As indicated in the Figure showing the pathway of cholesterol biosynthesis, a molecule of geranylpyrophosphate (GPP) and a molecule of isopentenylpyrophosphate (IPP) are condensed into farnesylpyrophosphate (FPP) through the action of the farnesyl diphosphate synthase enzyme which is encoded by the FDPS gene. Through the action of the ER-localized enzyme, dehydrodolichyl diphosphate synthase (encoded by the DHDDS gene), farnesylpyrophosphate is elongated via the sequential head-to-tail addition of multiple isopentenylpyrophosphate groups in a reaction referred to as cis-prenylation. The number of IPP substrates added ultimately determines the overall number of isoprene units in dolichol which in humans ranges from 17 to 21.
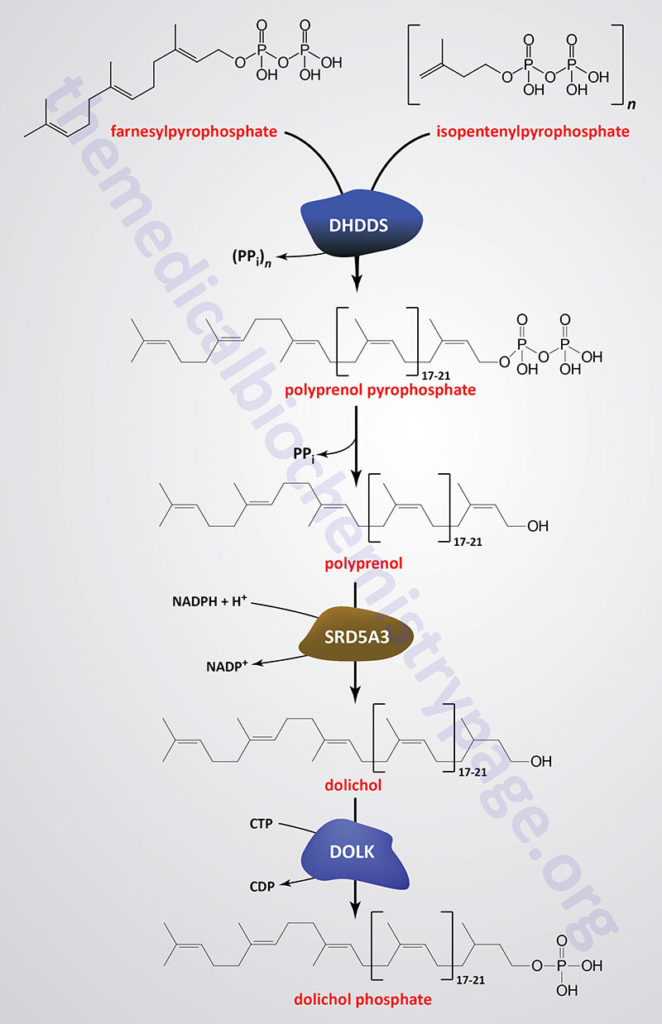
The DHDDS gene is located on chromosome 1p36.11 and is composed of 9 exons that generate five alternatively spliced mRNAs each of which encode a distinct protein isoform.
The product(s) of the DHDDS reaction is referred to as a polyprenolpyrophosphate. The pyrophosphate is removed by an as yet uncharacterized enzyme activity that may be either a polyprenol pyrophosphate phosphatase or a polyprenol phosphatase resulting in the formation of a polyprenol.
The resultant polyprenol(s) is a substrate for steroid 5-α reductase 3 (also called polyprenol reductase) which is encoded by the SRD5A3 gene. Steroid 5-α reductase 3 belongs to the polyprenol reductase subfamily of the steroid 5-α reductase family. The SRD5A3 encoded enzyme reduces the carbon-carbon double bond closest to the hydroxyl end of the polyprenol generating dolichol. In addition to participating in the synthesis of dolichol the SRD5A3 encoded enzyme synthesizes 5-α-dihydrotestosterone from testosterone.
The SRD5A3 gene is located on chromosome 4q12 and is composed of 6 exons that generate two alternatively spliced mRNAs encoding proteins of 318 amino acid protein (isoform 1) and 273 amino acids (isoform 2). Mutations in the SRD5A3 gene are associated with the congenital disorder of glycosylation (CDG) identified as CDG-1q (SRD5A3-CDG).
Dolichol phosphate is then synthesized from dolichol through the action of the ER-localized enzyme dolichol kinase. The phosphate donor for dolichol kinase is CTP and not ATP as is the case for most kinases.
Dolichol kinase is encoded by the DOLK gene. The DOLK gene is an intronless gene located on chromosome 9q34.11 and encodes a 538 amino acid protein. Mutations of the DOLK gene are associated with the CDG identified as CDG-1m (DOLK-CDG).
Coenzyme Q (Ubiquinone) Synthesis
Coenzyme Q (ubiquinone) is a red-ox active molecule that is composed of a benzoquinone ring conjugated to a polyisoprenoid tail that is of variable length in different species and organisms. In humans the polyisoprenoid tail consists of 10 isoprenoid units which impart the common name for the molecule as CoQ10. A minor amount of ubiquinone in humans contains 9 isoprenoid units. In undergoing reduction and oxidation reaction the electrons are accepted and donated from benzoquinone ring. The polyisoprenoid tail of ubiquinone serves to anchor the molecule in the membrane.
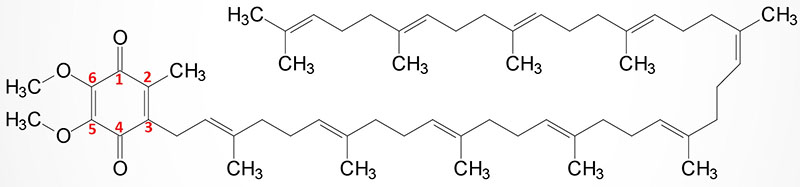
The complete pathway for the synthesis of ubiquinone in eukaryotes has been worked out in yeasts and the round worm, Caenorhabditis elegans. In humans, homologues of all of the yeast genes have been found. The initial steps in the synthesis of ubiquinone involve the formation of the polyisoprenoid tail. In human tissues a molecule of farnesyl pyrophosphate and a molecule of isopentenyl pyrophosphate are condensed to form all trans-decaprenyl diphosphate. This reaction is catalyzed by the heterotetrameric enzyme identified as decaprenyl diphosphate synthase. The two different subunits of the enzyme are encoded by the PDSS1 and PDSS2 genes.
The PDSS1 gene is located on chromosome 10p12.1 and is composed of 14 exons that generate three alternatively spliced mRNAs each of which encode distinct protein isoforms.
The PDSS2 gene is located on chromosome 6q21 and is composed of 12 exons that encode a protein of 399 amino acids. The remainder of the genes involved in human ubiquinone synthesis all have the designation COQ.
Following synthesis of the decaprenyl molecule, the enzyme, 4-hydroxybenzoate polyprenyltransferase (encoded by the COQ2 gene), catalyzes covalent attachment of the decaprenyl diphosphate to the aromatic ring of 4-hydroxybenzoate (para-hydroxybenzoate) forming 3-decaprenyl-4-hydroxybenzoic acid. The COQ2 encoded protein is localized to the mitochondria.
The COQ2 gene is located on chromosome 4q21.23 and is composed of 7 exons that generate two alternatively spliced mRNAs, each of which encode distinct protein isoforms. Mutations in the COQ2 gene are associated with a form of mitochondrial encephalomyopathy as well as a COQ2 nephropathy.
After the attachment of the decaprenyl group the aromatic ring undergoes a series of modifications. The first modification is a hydroxylation reaction at carbon 5 of the benzene ring. This hydroxylation is catalyzed by the FAD-dependent monooxygenase encoded by the COQ6 gene.
The COQ6 gene is located on chromosome 14q24.3 and is composed of 14 exons that generate two alternatively spliced mRNAs each encoding a distinct protein isoform.
In the next reaction the newly attached hydroxyl group undergoes an O-methylation reaction catalyzed by the mitochondrial SAM-dependent O-methyltransferase encoded by the COQ3 gene.
The COQ3 gene is located on chromosome 6q16.2 and is composed of 9 exons that encode a 369 amino acid protein.
The next reaction involves decarboxylation of the carboxylic acid group attached to carbon 1 of the benzene ring leaving a hydroxyl group. The decarboxylation reaction is catalyzed by an as yet uncharacterized enzyme. These three reactions result in the formation of 2-methoxy-6-decaprenylphenol.
In the next reaction, carbon 2 of the benzene ring is methylated. The C-methylation reaction is catalyzed by the mitochondrial SAM-dependent enzyme identified as 2-methoxy-6-polyprenyl-1,4-benzoquinol methylase. This methylase is encoded by the COQ5 gene.
The COQ5 gene is located on chromosome 12q24.31 and is composed of 8 exons that encode a 327 amino acid protein.
The next reaction involves the hydroxylation of carbon 6 of the benzene ring. This hydroxylation is catalyzed by 5-demethoxyubiquinone hydroxylase which is encoded by the COQ7 gene.
The COQ7 gene is located on chromosome 16p12.3 and is composed of 11 exons that generate nine alternatively spliced mRNAs that collectively encode six distinct protein isoforms.
The final reaction in ubiquinone synthesis is a SAM-dependent methylation of the newly added hydroxyl group. This last reaction is catalyzed the COQ3 encoded O-methyltransferase.
Heme a (heme A) Synthesis
Heme a (heme A) is an essential component of the oxidative phosphorylation pathway by serving as the prosthetic group for cytochrome aa3 (also called cytochrome c oxidase) of complex IV. Cytochrome aa3 is so-called due to the presence of two distinct heme a prosthetic groups with heme a being the direct electron donor in the complex IV catalyzed reduction of O2 to H2O. The heme a3 prosthetic group constitutes part of the copper-dependent active site of complex IV.
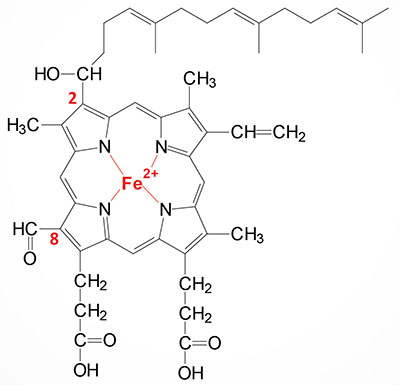
Heme a is synthesized from heme b (iron protoporphyrin IX) through a series of reactions that convert the methyl side group on carbon 8 (C8) of the porphyrin molecule into a formyl group along with conversion of the vinyl group at position C2 to hydroxyethylfarnesyl with the isoprenoid farnesyl pyrophosphate as the substrate. The transfer of the farnesyl group to the C2 vinyl group is catalyzed by the enzyme identified as heme A:farnesyltransferase cytochrome c oxidase assembly factor (also called protoheme IX farnesyltransferase). This enzyme, which is localized to the inner mitochondrial membrane, is encoded by the COX10 gene. The COX10 gene is located on chromosome 17p12 and is composed of 7 exons that encode a 443 amino acid protein. The addition of the farnesyl group to heme a generates the heme identified as heme o (heme O).
Heme o is then converted to heme a through a series of reactions the converts the C8 methyl group into a formyl group. The conversion of heme o to heme a is catalyzed by the enzyme identified as cytochrome c oxidase assembly protein COX15 homolog which is encoded by the COX15 gene. Like the COX10 encoded protein, the COX15 encoded protein is localized to the inner mitochondrial membrane. The COX15 gene is located on chromosome 10q24.2 and is composed of 10 exons that generate five alternatively spliced mRNAs that collectively encode four distinct protein isoforms.
Regulating Cholesterol Synthesis
Normal healthy adults synthesize cholesterol at a rate of approximately 1g/day and consume approximately 0.3g/day. A relatively constant level of cholesterol in the blood (150–200 mg/dL) is maintained primarily by controlling the level of de novo synthesis. The level of cholesterol synthesis is regulated in part by the dietary intake of cholesterol. Cholesterol from both diet and synthesis is utilized in the formation of membranes and in the synthesis of the steroid hormones and bile acids. The greatest proportion of cholesterol is used in bile acid synthesis.
The cellular supply of cholesterol is maintained at a steady level by three distinct mechanisms:
- Regulation of HMGR activity and levels.
- Regulation of excess intracellular free cholesterol through the activity of sterol O-acyltransferases, SOAT1 and SOAT2 with SOAT2 being the predominant activity in liver. The original designation for these enzymes was ACAT for acyl-CoA: cholesterol acyltransferase. However, this conflicts with the official ACAT enzymes, ACAT1 and ACAT2 which are acetyl-CoA acetyltransferases 1 and 2. These latter two enzymes are thiolases discussed in the Lipolysis and the Oxidation of Fatty Acids page.
- Regulation of plasma cholesterol levels via LDL receptor-mediated uptake and HDL-mediated reverse transport.
Regulation of HMGR activity is the primary means for controlling the level of cholesterol biosynthesis. The enzyme is controlled by four distinct mechanisms: feed-back inhibition, control of gene expression, rate of enzyme degradation and phosphorylation-dephosphorylation.
The first three control mechanisms are exerted by cholesterol itself. Cholesterol acts as a feed-back inhibitor of pre-existing HMGR as well as inducing rapid degradation of the enzyme. The latter is the result of cholesterol-induced polyubiquitylation of HMGR and its degradation in the proteasome (see proteolytic degradation below). This ability of cholesterol is a consequence of the sterol sensing domain, SSD of HMGR. In addition, when cholesterol is in excess the amount of mRNA for HMGR is reduced as a result of decreased expression of the gene. The mechanism by which cholesterol (and other sterols) affect the transcription of the HMGR gene is described below under regulation of sterol content.
Regulation of HMGR through covalent modification occurs as a result of phosphorylation and dephosphorylation. The enzyme is most active in its unmodified form. Phosphorylation of the enzyme decreases its activity. HMGR is phosphorylated by AMP-activated protein kinase, AMPK. AMPK itself is activated via phosphorylation. Phosphorylation of AMPK is catalyzed by at least two enzymes. The primary kinase responsible activation of AMPK is LKB1 (liver kinase B1). LKB1 was first identified as a gene in humans carrying an autosomal dominant mutation in Peutz-Jeghers syndrome, PJS. LKB1 is also found mutated in lung adenocarcinomas. The second AMPK phosphorylating enzyme is calmodulin-dependent protein kinase kinase-beta (CaMKKβ). CaMKKβ induces phosphorylation of AMPK in response to increases in intracellular Ca2+ as a result of muscle contraction.
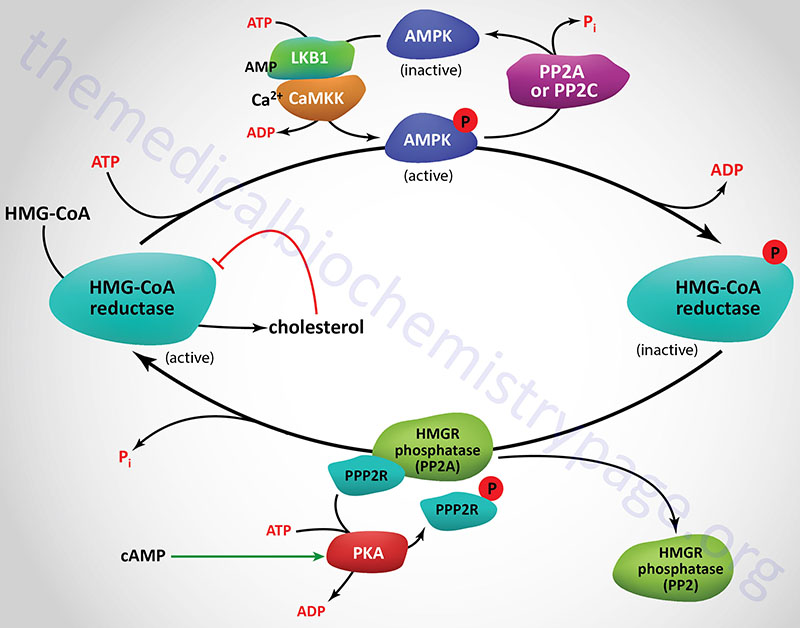
The activity of HMGR is additionally controlled by the cAMP signaling pathway. Increases in cAMP lead to activation of cAMP-dependent protein kinase, PKA. In the context of HMGR regulation, PKA phosphorylates a regulatory subunit of PP2A (PPP2R in Figure) leading to an increase in release of PP2A from HMGR. This prevents PP2A from removing phosphates from HMGR preventing its reactivation. The large family of protein phosphatase regulatory subunits regulate and/or inhibit the activity of numerous phosphatases including members of the PP1, PP2A, and PP2C families.
In addition to PP2A phosphatases that remove phosphates from AMPK and HMGR, phosphatases of the protein phosphatase 2C (PP2C) family also remove phosphates from AMPK. When these regulatory subunits are phosphorylated by PKA the activity of the associated phosphatases is reduced which results in AMPK remaining in the phosphorylated and active state, and HMGR in the phosphorylated and inactive state.
As the stimulus leading to increased cAMP production is removed, the level of phosphorylations decreases and that of dephosphorylations increases. The net result is a return to a higher level of HMGR activity. On the other hand, insulin leads to a decrease in cAMP, which in turn activates cholesterol synthesis.
The ability of insulin to stimulate, and glucagon to inhibit, HMGR activity is consistent with the effects of these hormones on other metabolic pathways. The basic function of these two hormones is to control the availability and delivery of energy to all cells of the body.
Long-term control of HMGR activity is exerted primarily through control over the synthesis and degradation of the enzyme. When levels of cholesterol are high, the level of expression of the HMGR gene is reduced. Conversely, reduced levels of cholesterol activate expression of the gene. Insulin also brings about long-term regulation of cholesterol metabolism by increasing the level of HMGR synthesis.
Role of GPR146 in Regulation of Cholesterol Synthesis
Dietary cholesterol is absorbed from the small intestines via the concerted actions of carboxyl ester lipase (CEL) and the transport protein identified as Niemann-Pick C1-like-1 (NPC1L1). The activity of CEL is to hydrolyze the fatty acylation of cholesterol esters releasing free cholesterol. Free cholesterol is transported into intestinal enterocytes through the action of NPC1L1.
The NPC1L1-mediated cholesterol uptake, into intestinal enterocytes, stimulates the production of the cholesterol-induced gut hormone, cholesin. Cholesin, and its function as a regulator of hepatic cholesterol synthesis, was only recently (2024) identified.
Cholesin binds to the GPR146 receptor on hepatocytes. The GPR146 receptor is coupled to the activation of a Gi-type G-protein. The activation of GPR146 leads to the inhibition of the activation of PKA which in turn leads to reduced activation of ERK1/2 signaling in hepatocytes. ERK1/2 signaling activates the SREBP2 transcription factor which is normally involved in the activation of several genes encoding enzymes involved in cholesterol synthesis, in particular the HMGCR gene encoding HMG-CoA reductase.
The gene encoding GPR146 is found within the gene encoding cholesin and it transcribed in the opposite direction to the transcription of cholesin. Both genes are located on chromosome 7p22.3. The GPR146 gene is composed of 5 exons that generate three alternatively spliced mRNAs, each of which encode the same 333 amino acid protein. The cholesin gene (originally identified as C7orf50) is composed of 18 exons that generate four alternatively spliced mRNAs, each of encode the same 194 amino acid preproprotein.
Polymorphisms in the chromosome 7p22 region had previously been shown to be correlated with hypercholesterolemia and the development of atherosclerosis. Two single nucleotide polymorphisms (SNP), the rs1997243-G allele and the rs2362529-C allele, exert opposing effects on the level of GPR146 expression. The rs1997243-G allele is associated with increased GPR146 expression and correlates with reduced levels of serum cholesterol. The rs2362529-C allele is associated with reduced expression of GPR146 and correlates with increased serum cholesterol and an increased risk for atherosclerosis.
Proteolytic Regulation of HMG-CoA Reductase
The stability of HMGR is regulated as the rate of flux through the mevalonate synthesis pathway changes. When the flux is high the rate of HMGR degradation is also high. When the flux is low, degradation of HMGR decreases. This phenomenon can easily be observed in the presence of the statin drugs as discussed below.
HMGR is localized to the ER and like SREBP (see below) contains a sterol-sensing domain, SSD. When sterol levels increase, in particular the cholesterol pathway intermediate lanosterol, they will interact with the Insig proteins. The interaction of lanosterol with Insig induces its binding to the SSD of HMGR and the recruitment of a ubiquitin ligase. Two ubiquitin ligases have been shown to interact with Insig proteins, RNF139 (ring finger protein 139; also known as TRC8) and AMFR (autocrine motility factor receptor; also known as Gp78). The RNF139 protein also possesses a SSD.
Following Insig interaction with HMGR and its ubiquitylation, the enzyme is degraded via the proteasome. This sterol-mediated ubiquitylation and proteasomal degradation of HMGR is a form of endoplasmic reticulum (ER)-associated degradation (ERAD).
The Utilization of Cholesterol
Cholesterol is transported in the plasma predominantly as cholesteryl esters associated with lipoproteins. Dietary cholesterol is transported from the small intestine to the liver within chylomicrons. Cholesterol synthesized by the liver, as well as any dietary cholesterol in the liver that exceeds hepatic needs, is transported in the serum within very-low density lipoprotein (VLDL) particles. The liver synthesizes VLDL and these are converted to intermediate density lipoprotein (IDL) particles and then to low density lipoprotein (LDL) particles through the action of endothelial cell-associated lipoprotein lipase.
Cholesterol found in plasma membranes can be extracted by high density lipoprotein (HDL) and esterified by the HDL-associated enzyme lecithin-cholesterol acyltransferase, LCAT. The cholesterol acquired from peripheral tissues by HDL can then be transferred to VLDL, IDL, and LDL via the action of cholesteryl ester transfer protein (CETP) which is associated with HDL, a process often termed reverse cholesterol transport (RCT). Reverse cholesterol transport allows peripheral cholesterol to be returned to the liver in LDL.
The functions of de novo synthesized cholesterol or cholesterol derived from the diet include serving as the precursor for adrenal cortical cell steroid hormone biosynthesis, synthesis of the gonadal steroid hormones, synthesis of the bile acids, and as a component of various membranes. Ultimately, cholesterol is excreted in the bile as free cholesterol or as bile salts following conversion to bile acids in the liver.
Regulation of Cellular Sterol Content: Role of SREBP
The continual alteration of the intracellular sterol content occurs through the regulation of key sterol synthetic enzymes as well as by altering the levels of cell-surface LDL receptors. As cells need more sterol they will induce their synthesis and uptake, conversely when the need declines synthesis and uptake are decreased. Regulation of these events is brought about primarily by sterol-regulated transcription of key rate limiting enzymes and by the regulated degradation of HMGR. Activation of transcriptional control occurs through the regulated cleavage of the membrane-bound transcription factor sterol regulated element binding protein, SREBP. As discussed above, degradation of HMGR is controlled by the ubiquitin-mediated pathway for proteolysis.
Sterol control of transcription affects more than 30 genes whose encoded proteins are involved in the biosynthesis of cholesterol, triglycerides, phospholipids and fatty acids. Transcriptional control requires the presence of an octamer sequence in the gene termed the sterol regulatory element, SRE-1. It has been shown that SREBP is the transcription factor that binds to SRE-1 elements.
Humans express two distinct SREBP genes. These genes are identified as sterol regulatory element binding transcription factor 1 (SREBF1) and sterol regulatory element binding transcription factor 2 (SREBF2). In addition, mammalian SREBF1 encodes two major proteins identified as SREBP-1a and SREBP-1c/ADD1 (ADD1 is adipocyte differentiation-1) as a consequence of alternative transcriptional start sites resulting in the utilization of different first exons that are spliced to a common exon 2.
The SREBF1 gene is located on chromosome 17p11.2 and is composed of 22 exons that generate 13 alternatively spliced mRNAs. The human SREBP-1a protein (1147 amino acids) predominates in the spleen and intestines while the SREBP-1c protein (1123 amino acids) predominates in liver, adipose tissue, and muscle.
The SREBF2 gene is located on chromosome 22q13.2 and is composed 24 exons that encode a 1141 amino acid protein.
SREBP-1a regulates all SREBP-responsive genes in both the cholesterol and fatty acid biosynthetic pathways. SREBP-1c controls the expression of genes involved in fatty acid synthesis and is involved in the differentiation of adipocytes. SREBP-1c is also an essential transcription factor downstream of the actions of insulin at the level of carbohydrate and lipid metabolism. SREBP-2 is the predominant form of this transcription factor in the liver and it exhibits preference at controlling the expression of genes involved in cholesterol homeostasis, including all of the genes encoding the sterol biosynthetic enzymes. In addition SREBP-2 controls expression of the LDL receptor (LDLR) gene.
Regulated expression of the SREBP is complex in that the effects of sterols are different on the SREBF1 gene versus the SREBF2 gene. High sterols activate expression of the SREBF1 gene but do not exert this effect on the SREBF2 gene. The sterol-mediated activation of the SREBF1 gene occurs via the action of the liver X receptors (LXR).
The LXR are members of the steroid/thyroid hormone superfamily of nuclear receptors that regulate gene expression by binding to specific target DNA sequences. There are two forms of the LXRs: LXRα and LXRβ. The LXR form heterodimers with the retinoid X receptors (RXR) and as such can regulate gene expression either upon binding oxysterols (e.g. 22R-hydroxycholesterol) or 9-cis-retinoic acid.
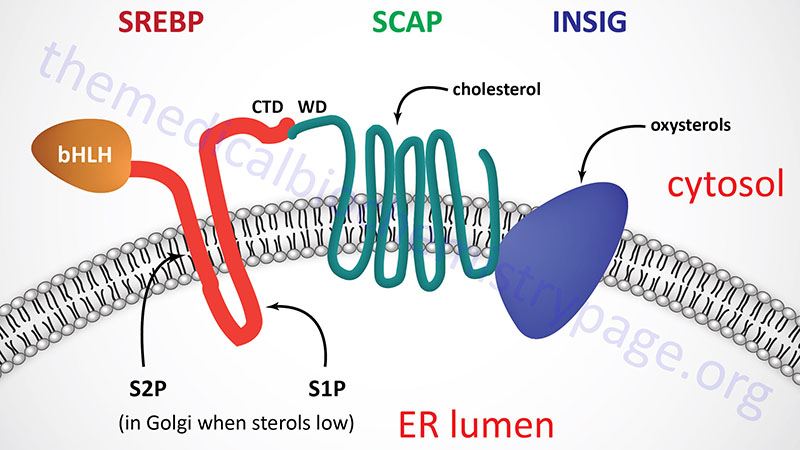
All three SREBP are proteolytically activated and the proteolysis is controlled by the level of sterols in the cell. Full-length SREBP have several domains and are embedded in the membrane of the endoplasmic reticulum (ER). The N-terminal domain contains a transcription factor motif of the basic helix-loop-helix (bHLH) type that is exposed to the cytoplasmic side of the ER. There are two transmembrane spanning domains followed by a large C-terminal domain also exposed to the cytosolic side.
Role of SCAP and INSIG in the Activation of SREBP
The C-terminal domain (CTD) interacts with a protein called SREBP cleavage-activating protein (SCAP). SCAP is a large protein also found in the ER membrane and contains eight transmembrane spanning domains. The C-terminal portion of SCAP, which extends into the cytosol, has been shown to interact with the C-terminal domain of SREBP. This C-terminal region of SCAP contains four motifs called WD40 repeats. The WD40 repeats are required for interaction of SCAP with SREBP.
The SCAP protein is encoded by the SCAP gene. The SCAP gene is located on chromosome 3p21.31 and is composed of 29 exons that generate two alternatively spliced mRNAs encoding proteins of 1279 amino acids (isoform 1) and 1023 amino acids (isoform 2).
The regulation of SREBP activity is further controlled within the ER by the interaction of SCAP with insulin-induced protein-1 and -2 (Insig-1 and Insig-2). When cells have sufficient sterol content, SREBP and SCAP are retained in the ER via the SCAP-Insig interaction. There are two Insig encoding genes identified as INSIG1 and INSIG2.
The INSIG1 gene is located on chromosome 7q36.3 and is composed of 7 exons that generate eight alternatively spliced mRNAs that collectively encode five isoforms of Insig-1. The Insig-1 protein was originally isolated in experiments examining regenerating liver and was subsequently shown to be dramatically induced in fat tissue in experimental animals at the onset of diet-induced obesity. INSIG1 gene expression is highest in human liver.
The INSIG2 gene is located on chromosome 2q14.1-q14.2 and is composed of 7 exons that generate six alternatively spliced mRNAs that collectively encode three distinct protein isoforms. Expression of INSIG2 is ubiquitous.
The Insig proteins bind to oxysterols which in turn affects their interactions with SCAP. The major form of human Insig-1 is a 277 amino acids protein and the major form of Insig-2 is a 225 amino acid protein. These two proteins share 59% amino acid identity with the greatest differences being found in the N- and C-terminal regions. Insig-2 also lacks the 50 amino acids that are found in the N-terminus of Insig-1.
The N-terminus of SCAP, including membrane spans 2–6, resembles the membrane attachment domain of HMGR which itself is subject to cholesterol/sterol-stimulated degradation (see above). This shared motif is called the sterol sensing domain (SSD) and as a consequence of this domain SCAP functions as the cholesterol sensor in the protein complex. When cells have sufficient levels of sterols, SCAP will bind cholesterol which promotes the interaction with Insig and the entire complex will be maintained in the ER.
Both Insig proteins can cause ER retention of the SREBP/SCAP complex. The Insig proteins span the ER membrane six times. It has been shown that a critical aspartate (D) residue in Insig-1 and Insig-2, found in the cytosolic loop between membrane spans 4 and 5, is critical for interaction with SCAP as mutation of this amino acid causes loss of SCAP binding. The third and fourth transmembrane spans in both Insig proteins are required for interaction with oxysterols.
The Insig-1 gene has been shown to be transcriptionally regulated by SREBP with the SRE in the Insig-1 gene residing approximately 380bp upstream of the transcriptional start site. Expression of Insig-1 has also been shown to be regulated by several members of the nuclear receptor family including PPARδ, PXR and CAR. The Insig-2 promoter is activated in response to signals downstream of insulin receptor activation. Nuclear receptors also regulate the expression of the Insig-2 gene which has been shown to contain two FXR response elements.
In addition to their role in regulating sterol-dependent gene regulation, both Insig proteins activate sterol-dependent degradation of HMGR. In the presence of the cholesterol-derived oxysterol, 24,25-dihydrolanosterol, Insig binds to the transmembrane domain of HMGR. The oxysterol-induced interaction between Insig and HMGR within the ER membrane allows Insig to recruit the ubiquitin ligase, gp78, to HMGR resulting in ubiquitination of HMGR and its resultant proteasomal degradation as described above.
When sterols are scarce, SCAP does not interact with Insig. The release of Insig leads to a conformational change in the SCAP protein resulting in the exposure of a hexapeptide motif termed the MELADL motif referencing the amino acids that constitute the motif. The MELADL motif interacts with coat protein complex II (COPII) that facilitates the migration of the SREBP-SCAP complex to the Golgi. Within the Golgi the SREBP is subjected to proteolysis.
Role of SPRING in the Activation of SREBP
Recent studies have identified an addition protein that functions in the control of the activation of SREBP transcriptional activity. This protein has been given the name SPRING for SREBF Pathway Regulator In Golgi. SPRING possesses a single membrane spanning domain in its N-terminal region that anchors the protein into the ER membrane.
SPRING interacts with SCAP and with one of the two proteases (S1P) responsible for the activation and release of SREBP from the membrane. The activity of S1P requires autocatalytic proteolysis and N-glycosylation. The interaction of SPRING with S1P enhances the autocatalytic activation of S1P. The loss of SPRING results in limited ability to activate SREBP due to the reduced level of S1P autocatalytic activity.
Proteolytic Activation of SREBP
The cleavage of SREBP is carried out by two distinct enzymes. The regulated cleavage occurs in the lumen of the Golgi in the loop between the two transmembrane domains. This cleavage is catalyzed by site-1 protease, S1P. S1P is a member of the proprotein convertase subtilisin/kexin type (PCSK) family of proteases and as such is also known as PCSK8. S1P has also been referred to as subtilisin/kexin-isozyme 1 (SKI-1). S1P is officially called membrane-bound transcription factor peptidase, site 1 which is encoded by the MBTPS1 gene.
The MBTPS1 gene is located on chromosome 16q23.3–q24.1 and is composed of 24 exons that encode a 1052 amino acid preproprotein.
MBTPS1 is a member of the subtilisin-like proprotein convertase 2 family of serine proteases. This family of proteases are responsible for the processing of proteins that are in the regulated or constitutive branches of the secretory pathway. The subtilisin-like proprotein convertase 2 family of enzymes are encoded by nine different genes in humans, one of which is the proprotein convertase subtilisin/kexin type 9 (PCSK9) gene whose encoded enzyme is a recent target in the treatment of hypercholesterolemia. The function of SCAP is to positively stimulate S1P-mediated cleavage of SREBP.
The second cleavage, catalyzed by site-2 protease, S2P, occurs in the first transmembrane span, leading to release of active SREBP. The official name for S2P is membrane-bound transcription factor peptidase, site 2 which is encoded by the MBTPS2 gene.
The MBTPS2 gene is located on the X chromosome (Xp22.12) and is composed of 11 exons that encode a 519 amino acid protein.
S2P is an intramembrane zinc metalloprotease. In order for S2P to act on SREBP, site-1 must already have been cleaved. The result of the S2P cleavage is the release of the N-terminal bHLH motif into the cytosol. The bHLH domain then migrates to the nucleus where it will dimerize and form complexes with transcriptional coactivators leading to the activation of genes containing the SRE motif. To control the level of SREBP-mediated transcription, the soluble bHLH domain is itself subject to rapid proteolysis. In addition to the cleavage-activation of SREBP transcriptional activity, S2P is involved in pathways that regulate cellular responses to endoplasmic reticulum stress, primarily the unfolded protein response, UPR.
Several proteins whose functions involve sterols also contain the SSD. The terminal enzyme of cholesterol biosynthesis, 7-dehydrocholesterol reductase, also contains a SSD.
Two proteins involved in cholesterol trafficking also contain the SSD, NPC1 (Niemann-Pick C1) and NPC1L1 (Niemann-Pick C1-like-1). NPC1 is involved in the transport of LDL-derived cholesterol out of lysosomes for delivery to the plasma membrane and ER. NPC1L1 mediates the absorption of cholesterol by the small intestine and is the target for the drug, ezetimibe. The protein patched, an important development regulating receptor whose ligand, hedgehog, is modified by attachment of cholesterol, also contains an SSD.
Regulation of Intracellular Cholesterol Distribution
Cholesterol levels are kept at a relatively constant level in cells with one major mechanism being transcriptional control over cholesterol synthesizing enzymes, such as HMG-CoA reductase, as outlined in the section above. With respect to overall intracellular distribution, cholesterol is localized predominantly to the plasma membrane where it is clustered with sphingolipids in specialized domains referred to as lipid rafts. Compared to the plasma membrane, the amount of cholesterol in membranes of the endoplasmic reticulum (ER) and mitochondria is quite low.
The distribution of cholesterol to various subcellular locations is the function of proteins that belong to one of two families of lipid binding proteins. In addition to cholesterol these proteins are responsible for the transfer of phospholipids from their site of synthesis to the appropriate organelle membrane. The two major lipid transport families are the oxysterol-binding protein (OSBP) family that includes the OSBP-related proteins (ORP) and the STeroidogenic Acute Regulatory protein-related lipid Transfer (START) domain-containing protein family. The START domain is an α/β helix-grip structure, composed of ~210 amino acids, that form a hydrophobic pocket for lipid binding. The START domain is found in proteins that bind sterols, phospholipids, bile acids, and ceramides.
The human OSBP family consists of 12 members with oxysterol binding protein being the founding member. Oxysterol binding protein is referred to as a lipid transfer protein given that it functions to exchange cholesterol for phosphatidylinositol 4-phosphate (PtdIns-4-P) between two apposed membranes. The exchange process is irreversible because of the subsequent hydrolysis of PtdIns-4-P which contributes to the establishment of a cholesterol gradient along organelles of the secretory pathway.
The human START domain-containing protein family consists of 15 members with most being encoded by genes with the STARD nomenclature. Two members of the START domain protein family are acyl-CoA thioesterases (ACOT11, and ACOT12). Based upon sequence similarities, ligand-binding, and overall structure the human START domain proteins are divided into six subfamilies. Three of the subfamilies consist of START domain proteins that also contain other functional domains and so are referred to as multi-domain proteins. The other domains in these proteins are involved in processes such as signal transduction, protein trafficking, or enzymatic function.
The STARD1 family is composed of StAR (also identified as STARD1) and STARD3. The STARD1 family proteins are cholesterol-specific binding proteins that are membrane localized. Details on the function of StAR (steroidogenic acute regulatory protein) are covered in the Steroid Hormones and Their Receptors page. The STARD2 family is composed of PCTP (phosphatidylcholine transfer protein; also identified as STARD2), STARD7, STARD10, and CERT1 (also identified as STARD11). CERT1 is ceramide transporter 1. The STARD2 family proteins bind ceramides and phospholipids. The STARD4 family is composed of STARD4, STARD5, and STARD6 and these proteins are soluble sterol-binding proteins.
The remaining three subfamilies are multi-domain proteins where the ligand that binds the START domain has yet to be determined. The multi-domain protein family that is composed of STARD8, DCL (DLC1 Rho GTPase activating protein; also identified as STARD12), and STARD13 each of which contains a RhoGAP domain. The STARD9 protein represents a single protein multi-domain subfamily and this protein contains a kinesin motor domain. The final multi-domain protein family is composed of two enzymes that are acyl-CoA thioesterases with START domains. The two proteins are ACOT11 (also identified as STARD14) and ACOT12 (also identified as STARD15).
The proteins encoded by the STAR, STARD3, STARD4, STARD5, and STARD6 genes have all been shown to play important roles in cholesterol homeostasis and transport. The primary role of the StAR protein in the regulation of steroid hormone synthesis is described in detail in the Steroid Hormones and Their Receptors page. STARD3 possesses multiple domains including a FFAT [diphenylalanine (FF)–acidic track (AT)] motif that is found in proteins that attach to the endoplasmic reticulum (ER) through protein-protein interactions. Another domain in STARD3 localizes the protein to the late endosomes. These domains allow STARD3 to participate in the movement of cholesterol from the ER to the endosome. STARD4 shuttles cholesterol between the plasma membrane and the ER, between the plasma membrane and the endosome recycling compartment, and possibly to the mitochondria. STARD5 is involved in plasma membrane cholesterol distribution under conditions of ER stress and the expression of the STARD5 gene is induced by conditions of ER stress. Although STARD6 has been shown to facilitate cholesterol delivery to mitochondria, the protein has a higher affinity for cholesterol metabolites such as pregnenolone and testosterone.
Treatment of Hypercholesterolemia
Reductions in circulating cholesterol levels can have profound positive impacts on cardiovascular disease, particularly on atherosclerosis, as well as other metabolic disruptions of the vasculature. Control of dietary intake is one of the easiest and least cost intensive means to achieve reductions in cholesterol. Recent studies in laboratory rats has demonstrated an additional benefit of reductions in dietary cholesterol intake. In these animals it was observed that reductions in dietary cholesterol not only resulted in decreased serum VLDL and LDL, and increased HDL but DNA synthesis was also shown to be increased in the thymus and spleen. Upon histological examination of the spleen, thymus and lymph nodes it was found that there was an increased number of immature cells and enhanced mitotic activity indicative of enhanced proliferation. These results suggest that a marked reduction in serum LDL, induced by reduced cholesterol intake, stimulates enhanced DNA synthesis and cell proliferation.
Drug treatment to lower plasma lipoproteins and/or cholesterol is primarily aimed at reducing the risk of atherosclerosis and subsequent coronary artery disease that exists in patients with elevated circulating lipids. Drug therapy usually is considered as an option only if non-pharmacologic interventions (altered diet and exercise) have failed to lower plasma lipids.
PCSK9 Inhibitors: Alirocumab (Praluent®), Evolcumab (Repatha®)
These drugs are the newest type of anti-hypercholesterolemia drugs recently approved by the FDA for use in the US. The PCSK9 inhibitors are injectable antibodies that block the function of proprotein convertase subtilisin/kexin type 9, PCSK9. A major function of PCSK9 is the endosomal degradation of the LDL receptor (LDLR), thereby reducing the recycling of the LDLR to the plasma membrane. This effect of PCSK9 leads to a reduced ability of the liver to remove IDL and LDL from the blood contributing to the potential for hypercholesterolemia.
The potential for the pharmaceutical benefits of the interference in the activity PCSK9 was recognized by a confluence of several studies. Patients with a specific form of familial hypercholesterolemia not due to mutations in the LDLR gene were shown to have severe hypercholesterolemia due to mutations in the PCSK9 gene resulting in hyperactivity of the enzyme. In addition, it was found that in certain individuals with low serum LDL levels there was an association with the inheritance of nonsense mutations in the PCSK9 gene which result in loss of PCSK9 activity. Hypercholesterolemic patients taking another cholesterol-lowering drug while simultaneously utilizing either of these new PCSK9 inhibitors saw further reductions in serum LDL levels of between 55% and 77%.
A newer class of PCSK9 inhibitor that received US FDA approval in 2021, for the treatment of patients with heterozygous familial hypercholesterolemia, is based upon the RNA interference (RNAi) pathway. The drug, inclisiran (trade name Leqvio) is a small interfering RNA (siRNA) targeting the mRNA encoding the PCSK9 protein. By inducing the degradation of the PCSK9 mRNA the result is a reduction in the level of functional PCSK9 enzyme associated with the LDL receptor.
Statin Drugs: Atorvastatin (Lipitor®), Simvastatin (Zocor®), Lovastatin (Mevacor®)
These drugs are fungal HMG-CoA reductase (HMGR) inhibitors and are members of the family of drugs referred to as the statins. The net result of treatment is an increased cellular uptake of LDL, since the intracellular synthesis of cholesterol is inhibited and cells are therefore dependent on extracellular sources of cholesterol. However, since mevalonate (the product of the HMG-CoA reductase reaction) is required for the synthesis of other important isoprenoid compounds besides cholesterol, long-term treatments carry some risk of toxicity. A component of the natural cholesterol lowering supplement, red yeast rice, is in fact a statin-like compound.
The statins have become recognized as a class of drugs capable of more pharmacologic benefits than just lowering blood cholesterol levels via their actions on HMGR. Part of the cardiac benefit of the statins relates to their ability to regulate the production of S-nitrosylated COX-2 (PTGS-2). COX-2 is an inducible enzyme involved in the synthesis of the prostaglandins and thromboxanes as well as the lipoxins and resolvins. The latter two classes of compounds are anti-inflammatory lipids discussed in the Bioactive Lipid Mediators of Inflammation page. Evidence has shown that statins activate inducible nitric oxide synthase (iNOS) leading to nitrosylation of COX-2. The S-nitrosylated COX-2 enzyme produces the lipid compound 15R-hydroxyeicosatetraenoic acid (15R-HETE) which is then converted via the action of 5-lipoxygenase (5-LOX) to the epimeric lipoxin, 15-epi-LXA4. This latter compound is the same as the aspirin-triggered lipoxin (ATL) that results from the aspirin-induced acetylation of COX-2. Therefore, part of the beneficial effects of the statins is exerted via the actions of the lipoxin family of anti-inflammatory lipids.
Additional anti-inflammatory actions of the statins result from a reduction in the prenylation of numerous pro-inflammatory modulators. Prenylation refers to the addition of the 15 carbon farnesyl group or the 20 carbon geranylgeranyl group to acceptor proteins. The isoprenoid groups are attached to cysteine residues at the carboxy terminus of proteins in a thioether linkage (C-S-C). A common consensus sequence at the C-terminus of prenylated proteins has been identified and is composed of CAAX, where C is cysteine, A is any aliphatic amino acid (except alanine) and X is the C-terminal amino acid.
In addition to numerous prenylated proteins that contain the CAAX consensus, prenylation is known to occur on proteins of the RAB family of RAS-related G-proteins. There are at least 60 proteins in this family that are prenylated at either a CC or CXC element in their C-termini. The RAB family of proteins are involved in signaling pathways that control intracellular membrane trafficking.
The prenylation of proteins allows them to be anchored to cell membranes. In addition to cell membrane attachment, prenylation is known to be important for protein-protein interactions. Thus, inhibition of this post-translational modification by the statins interferes with the important functions of many signaling proteins which is manifest by inhibition of inflammatory responses.
Some of the effects on immune function that have been attributed to the statins are attenuation of autoimmune disease, inhibition of T-cell proliferation, inhibition of inflammatory co-stimulatory molecule expression, decreases in leukocyte infiltration, and promotion of a shift in cytokine profiles of helper T-cell types from Th1 to Th2. Th1 cells are involved in cell-mediated immunity processes, whereas, Th2 cells are involved in humoral immunity process. The cytokines produced by Th2 cells include IL-4, IL-5, IL-10 and IL-13 and these trigger B cells to switch to IgE production and to activate eosinophils.
Complications of Statin Therapies
Although the use of statin class drugs have proven effective at preventing cardiovascular disease, these drugs are not without potentially severe negative outcomes. The most common negative effects of the statins are rhabdomyolysis and impaired liver function. As the use of statins has become quite broad, nearly 1 in 4 adults over 40 years of age are taking these drugs, additional negative outcomes have become apparent including the onset of diabetes and glucose intolerance.
Recent evidence has found that statin-induced dysregulation of insulin and glucose homeostasis is the result of the inhibition of GLP-1 synthesis and release by intestinal enteroendocrine L-cells. A major mechanism, contributing to reduced intestinal GLP-1 production with statin use, is a reduction in the level of gut bacteria of the Clostridium genus.
The role of Clostridium in intestinal GLP-1 production is related to the conversion of the bile acid, chenodeoxycholic acid (CDCA) to ursodeoxycholic acid (UDCA). The Clostridium enzymes of the 7α,β-hydroxysteroid dehydrogenase (HSDH) family are responsible for CDCA conversion to UDCA. Within the gut UDCA bids to and activates the bile acid receptor, G-protein coupled bile acid receptor 1, GPBAR1 (originally identified as TGR5 and also known as GPR131) to enhance the expression of the proglucagon (GCG) gene in enteroendocrine L-cells resulting in increased production and release of GLP-1.
The significance of gut microbiota production of UDCA to the synthesis of GLP-1 and the consequent positive effects on insulin secretion and glucose homeostasis has been demonstrated with the use of dietary UDCA supplementation. Administration of UDCA results in increased GLP-1 levels, enhanced hepatic expression of the gene (CYP7A1) encoding the rate-limiting enzyme in bile acid synthesis, improved insulin sensitivity, and improved glucose tolerance.
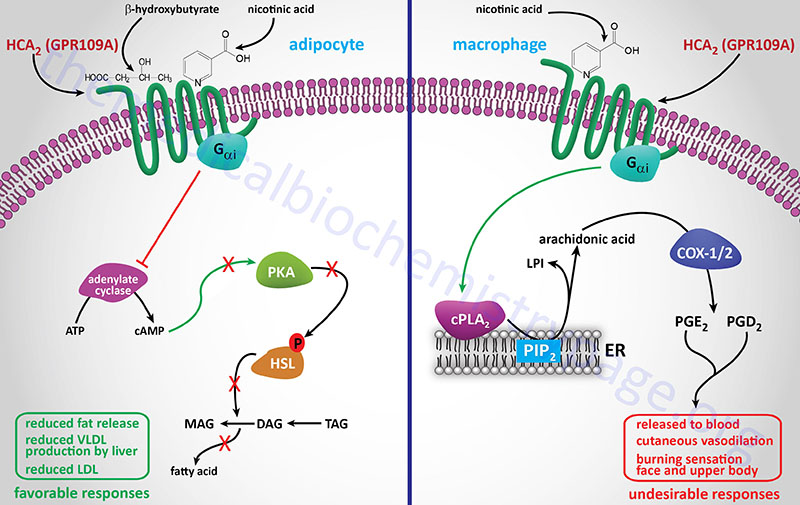
Nicotinic acid (Niacor® and Niaspan®)
Nicotinic acid reduces the plasma levels of both VLDL and LDL by inhibiting hepatic VLDL secretion, as well as suppressing the flux of FFA release from adipose tissue by inhibiting lipolysis. In addition, nicotinic acid administration strongly increases the circulating levels of HDL. Patient compliance with nicotinic acid administration is sometimes compromised because of the unpleasant side-effect of flushing (strong cutaneous vasodilation).
Recent evidence has shown that nicotinic acid binds to and activates the G-protein coupled receptor identified as GPR109A (also called HM74A or PUMA-G). GPR109A is a member of the hydroxycarboxylic acid (HCA) receptor family and as such is now designated as HCA2 (encoded by the HCAR2 gene). For more detailed information on the normal biological function of HCA2 (GPR109A) go to the Bioactive Lipids and Lipid Sensing Receptors page.
The identity of a receptor to which nicotinic acid binds allows for the development of new drug therapies that activate the same receptor but that may lack the negative side-effect of flushing associated with nicotinic acid. Because of its ability to cause large reductions in circulating levels of cholesterol, nicotinic acid is used to treat Type II, III, IV and V hyperlipoproteinemias.
Gemfibrozil (Lopid®), Fenofibrate (TriCor®)
These compounds (called fibrates) are derivatives of fibric acid and although used clinically since the 1930’s were only recently discovered to exert some of their lipid-lowering effects via the activation of peroxisome proliferation. Specifically, the fibrates were found to be activators of the peroxisome proliferator-activated receptor-α (PPARα) class of proteins that are classified as nuclear receptor co-activators. The naturally occurring ligands for PPARα are leukotriene B4 (LTB4, see the Eicosanoid Metabolism: Prostaglandins, Thromboxanes, Leukotrienes, and Lipoxins page), unsaturated fatty acids, and oxidized components of VLDL and LDL. The PPAR interact with another receptor family called the retinoid X receptors (RXR) that bind 9-cis-retinoic acid. Activation of PPAR results in modulation of the expression of genes involved in lipid metabolism.
In addition the PPAR modulate carbohydrate metabolism and adipose tissue differentiation. Fibrates result in the activation of PPARα in liver and muscle. In the liver this leads to increased peroxisomal β-oxidation of fatty acids, thereby decreasing the liver’s secretion of triacylglycerol- and cholesterol-rich VLDL, as well as increased clearance of chylomicron remnants, increased levels of HDL and increased lipoprotein lipase activity which in turn promotes rapid VLDL turnover.
Cholestyramine or colestipol (resins)
These compounds are nonabsorbable resins that bind bile acids which are then not reabsorbed by the liver but excreted. The drop in hepatic reabsorption of bile acids releases a feedback inhibitory mechanism that had been inhibiting bile acid synthesis. As a result, a greater amount of cholesterol is converted to bile acids to maintain a steady level in circulation. Additionally, the synthesis of LDL receptors increases to allow increased cholesterol uptake for bile acid synthesis, and the overall effect is a reduction in plasma cholesterol. This treatment is ineffective in homozygous FH patients, since they are completely deficient in LDL receptors.
Ezetimibe
This drug is sold under the trade names Zetia® or Ezetrol® and is also combined with the statin drug simvastatin and sold as Vytorin® or Inegy®. Ezetimibe functions to reduce intestinal absorption of cholesterol, thus effecting a reduction in circulating cholesterol. The drug functions by inhibiting the intestinal brush border transporter involved in absorption of cholesterol. This transporter is known as Niemann-Pick type C1-like 1 (NPC1L1). NPC1L1 is also highly expressed in human liver. The hepatic function of NPC1L1 is presumed to limit excessive biliary cholesterol loss. NPC1L1-dependent sterol uptake is regulated by cellular cholesterol content. In addition to the cholesterol lowering effects that result from inhibition of NPC1L1, its inhibition has been shown to have beneficial effects on components of the metabolic syndrome, such as obesity, insulin resistance, and fatty liver, in addition to atherosclerosis.
Ezetimibe is usually prescribed for patients who cannot tolerate a statin drug or a high dose statin regimen. There is some controversy as to the efficacy of ezetimibe at lowering serum cholesterol and reducing the production of fatty plaques on arterial walls. The combination drug of ezetimibe and simvastatin has shown efficacy equal to or slightly greater than atorvastatin (Lipitor®) alone at reducing circulating cholesterol levels.
Lomitapide
Certain patient populations, especially individuals that are homozygous for mutations in the LDL receptor are not effectively treated with drugs such as alirocumab and statins. Two recent drugs, that were designed to target liver lipoprotein homeostasis, have been approved for use in humans. One drug, lomitapide (Juxtapid®), is a small molecule inhibitor of the microsomal triglyceride transfer protein (MTTP; also referred to as MTP). MTTP is a heterodimeric complex composed of a large subunit (encoded by the MTTP gene) and a small subunit which is a member of the protein disulfide isomerase (PDI) family of enzymes that are involved in protein folding. The MTTP complex is required for the incorporation of apoB-48 into chylomicrons in the intestines and apoB-100 into VLDL by the liver. Lomitapide has been shown to reduce circulating LDL in homozygous FH patients by up to 50%.
Mipomersen
The drug mipomersen (Kynamro®) is an anti-sense oligonucleotide (ASO) that targets the apoB mRNA in the liver, thereby resulting in reduced synthesis of the apoB-100 protein. Since apoB-100 is required for VLDL assembly in the liver there is reduced VLDL secretion by the liver. In homozygous LDL receptor gene mutation patients with familial hypercholesterolemia (FH) who take mipomersen there was an observed reduction of circulating LDL by approximately 25%.
Bempedoic Acid
Bempedoic acid is a dicarboxylic acid that was demonstrated to inhibit fatty acid and cholesterol synthesis in experimental animals and these effects were correlated to reductions in plasma triglyceride and lipoprotein levels. The ability of bempedoic acid to modulate lipid metabolism, in a manner reflective of the role of fatty acids in these processes, defines the molecule as a decoy fatty acid. Several other decoy fatty acids, such as gemcabene, are being tested for their efficacy at reducing hepatic lipid synthesis and thus improving health outcomes either alone or in combination with statins.
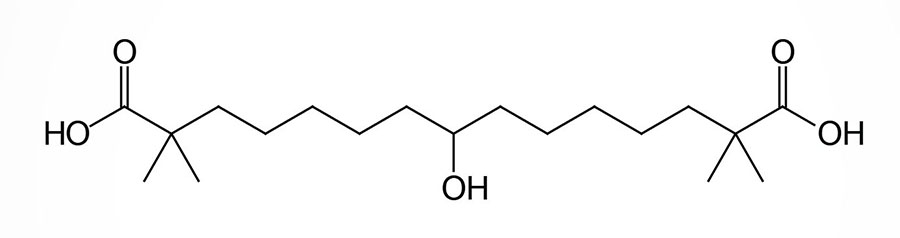
Bempedoic acid is a pro-drug that is converted exclusively in the liver to its active CoA-derivative, bempedoyl-CoA. The CoA addition to bempedoic acid is catalyzed by very long-chain acyl-CoA synthetase-1 (ACSVL1; also known as fatty acid transport protein 2: FATP2) which is encoded by the SLC27A2 gene (see the Lipolysis and the Oxidation of Fatty Acids page). The SLC27A2 gene is highly expressed in the liver but is not expressed in adipose tissue, the intestines, nor in skeletal muscle.
The conversion of bempedoic acid to its CoA derivative is believed to be required for its ability to suppress fatty acid and cholesterol synthesis and to also stimulate mitochondrial fatty acid β-oxidation. One of the targets of bempedoyl-CoA is the enzyme, ATP citrate lyase (ACLY) that hydrolyzes citrate to acetyl-CoA and oxaloacetic acid. This is a major part of the pathway of the conversion of glucose carbons into those of fatty acids and those of cholesterol. Within the liver, bempedoic acid is capable of reducing lipid levels in hepatocytes in the absence of the ACLY gene. These findings indicate that the mode of action of bempedoic acid on the lowering of hepatic lipids, and thus circulating LDL cholesterol levels, occurs through multiple distinct processes.
In addition to ATP-citrate lyase, bempedoic acid also modulates the activity of AMPK. In the case of AMPK, bempedoic acid exerts its effects in a manner similar to that of long-chain fatty acyl-CoAs (LCFA-CoAs). AMPK is a heterotrimeric enzyme composed of α-, β-, and γ-subunits. The effects of LCFA-CoAs is exerted by their activation of AMPK isoforms containing the β1-subunit. The AMPK isoform shown to be activated by bempedoic acid is the α1β1γ1 isoform. Although evidence has demonstrated that bempedoic acid does activate AMPK, this effect is dispensable for the effects of bempedoic acid on lowering plasma LDL cholesterol (LDL-C) levels.
Recent studies (2026) in mice have demonstrated that bempedoic acid can bind to an activate the nuclear receptor, peroxisome proliferator-activated receptor-alpha (PPARα). The ability of bempedoic acid to activate PPARα does not require that it first be activated to the CoA derivative, bempedoyl-CoA by the SLC27A2 encoded synthetase. The consequences of PPARα activation by bempedoic acid is enhanced hepatocyte fatty acid β-oxidation and a consequent reduction in lipid content in the hepatocytes.
One advantage of bempedoic acid over statins in the treatment of hypercholesterolemia is that the lack of SLC27A2 expression in skeletal muscle would prevent any adverse side effects in that tissue. The inhibition of muscle cholesterol synthesis by statins is a cause of the associated myotoxicity of that class of drug. Indeed, during clinical trials of bempedoic acid there was an absence of any muscle related symptoms. The US FDA approved the use of orally administered bempedoic acid alone (Nexletol™) or in combination with ezetimibe (Nexlizet™) in February of 2020.
Bempedoic acid given alone as a single daily dose of 180 mg reduces LDL cholesterol (LDL-C) by a mean of 24.5%. When administered with a statin drug the reduction in LDL-C is an additional 18% compared to a stain drug alone. The level of LDL-C reduction is between 38-40% when bempedoic acid is administered in a fixed-dose in combination with ezetimibe.
Angiopoietin-Like Protein Inhibitors
Humans express two additional LPL inhibitors encoded by the ANGPTL3 and ANGPTL4 genes. These genes encode proteins of the angiopoietin-like family that not only inhibit LPL but also inhibit endothelial lipase (encoded by the LIPG gene). Loss of function (LOF) mutations in the ANGPTL3 gene are associated with reduced levels of circulating triglycerides in a disorder called familial combined hypolipidemia. These genetic observations suggest that blocking the function of ANGPTL3 may be useful in treating hypertriglyceridemias.
However, given that ANGPTL3 activity is positively correlated to increased levels of phospholipid-rich HDL, which is highly active at cholesterol transport, there may be risks to blocking ANGPTL3 activity entirely. Indeed, humans with homozygous loss-of-function mutations in the ANGPTL3 gene have low serum levels of not only triglycerides and apo-B-containing lipoproteins (VLDL/LDL) but also low levels of apoA-I lipoproteins (HDL).
The use of a monoclonal antibody targeting circulating ANGPTL3 (evinacumab: trade name Evkeeza) demonstrated promise at lowering circulating levels of LDL in homozygous familial hypercholesterolemia (designated HoFH) patients. Patients were administered the monoclonal antibody over a period of 24 weeks and showed an average reduction in LDL cholesterol (LDL-c) of 49%. In 2021 the US FDA approved the use of Evkeeza as an adjunct to other lipid-lowering drugs for patients 12 years of age and older who are diagnosed with homozygous familial hypercholesterolemia (HoFH).
The use of RNA-targeting therapies to lower liver specific expression of ANGPTL3 has proved problematic. An antisense oligonucleotide (ASO) to the ANGPTL3 mRNA was developed that was conjugated to three molecules of N-acetylgalactosamine (GalNAc). This drug is called vupanorsen. The advantage of the GalNAc conjugation is that it specifically targets the conjugated molecule to the hepatocyte-specific asialoglycoprotein receptor 1 (encoded by the ASGR1 gene). However, clinical trials with vupanoresen were terminated due to adverse liver effects.
Nonetheless, another RNA-targeting approach has been developed utilizing an RNA interference methodology. A double stranded small-interfering RNA (siRNA) to the ANGPTL3 mRNA, that is conjugated to GalNAc (identified as ARO-ANG3), has shown promise at hepatocyte-specific reduction in ANGPTL3 mRNA levels. The results of early clinical trials with ARO-ANG3 demonstrated reductions in circulating levels of ANGPTL3 of up to 84%, reductions in circulating triglyceride levels of up to 58%, and reductions of non-HDL cholesterol of up to 26%.
Potential Future Therapies for Hyperlipidemia
apoC-III Inhibitors
Several other potential targets have been identified that may prove useful for pharmacological intervention of hyperlipidemias and hypercholesterolemias. The apolipoprotein, apoC-III, (encoded by the APOC3 gene) inhibits the activity of lipoprotein lipase (LPL), endothelial lipoprotein lipase (LPL), as well as hepatic lipase. Individuals harboring loss-of-function mutations in the APOC3 gene have significantly reduced levels of circulating triglycerides suggesting that targeting this protein may be an effective treatment of hypertriglyceridemias.
The drug volanesorsen (Waylivra®), which is a 2’-O-methoxyl-modified single-stranded antisense oligonucleotide (ASO) to the mRNA encoding apoC-III, has been found to reduce triglyceride levels in patients with classic familial chylomicronemia syndrome (FCS) on the order of 50%–80%. ApoC-III is an inhibitor of LPL-dependent and LPL-independent pathways in the clearance of triglyceride-rich lipoproteins by the liver. Thus, its inhibition leads to enhanced hepatic triglyceride clearance. Although approved for use by the European Union, the US FDA has rejected approval of volanesoren due to a high correlation (25%) of thrombocytopenia in trial participants. The EU approval is exclusively for patients with documented FCS.
A next-generation version of an ASO to the apoC-III mRNA has been developed and identified as olezarsen. Olezarsen is an ASO conjugated with three molecules of N-acetylgalactosamine (GalNAc). The GalNAc conjugation allows for highly specific attachment to the hepatocyte specific asialoglycoprotein type 1 receptor which is encoded by the ASGR1 gene. The tissue specificity of this modified ASO allows for a lower dose with no loss of efficacy. The US FDA granted olezarsen an Orphan Drug designation in 2023 for the treatment of familial chylomicronemia syndrome (FCS).
CETP Inhibitors
Numerous epidemiological and clinical studies over the past 15 years have demonstrated a direct correlation between the circulating levels of HDL cholesterol (most often abbreviated HDL-c) and a reduction in the potential for atherosclerosis and coronary heart disease (CHD). Individuals with levels of HDL above 50mg/dL are several time less likely to experience CHD than individuals with levels below 40mg/dL. In addition, clinical studies demonstrated that when apolipoprotein A-I (apoA-I; the predominant protein component of HDL-c), or reconstituted HDL are infused into patients there was an increase in the level of circulating HDL and reduced incidence of CHD. Thus, there is precedence for therapies aimed at raising HDL levels in the treatment and prevention of atherosclerosis and CHD. Unfortunately current therapies only modestly elevate HDL levels.
Both the statins and the fibrates have only been shown to increase HDL levels between 5–20% and niacin is poorly tolerated in many patients. Therefore, alternative strategies aimed at increasing HDL levels are being tested. Cholesterol ester transfer protein (CETP) is secreted primarily from the liver and plays a critical role in HDL metabolism by facilitating the exchange of cholesteryl esters (CE) from HDL for triglycerides (TG) in apoB containing lipoproteins, such as LDL. IDL, and VLDL. The activity of CETP directly lowers the cholesterol levels of HDL and enhances HDL catabolism by providing HDL with the TG substrate of hepatic lipase. Thus, CETP plays a critical role in the regulation of circulating levels of HDL, LDL, and apoA-I.
It has also been shown that in mice naturally lacking CETP most of their cholesterol is found in HDL and these mice are relatively resistant to atherosclerosis. The potential for the therapeutic use of CETP inhibitors in humans was first suggested when it was discovered in 1985 that a small population of Japanese had an inborn error in the CETP gene leading to hyperalphalipoproteinemia and very high HDL levels. To date four CETP inhibitors have been used in clinical trials. These compounds are anacetrapib, torcetrapib, dalcetrapib, and evocetrapib. Although torcetrapib is a potent inhibitor of CETP, its use has been discontinued due to increased negative cardiovascular events and death rates in test subjects. Studies with dalcetrapib and evocetrapib showed no real benefits and the trials were terminated. Treatment with anacetrapib results in a significant increase in both HDL (104%) and LDL (17%). Anacetrapib trials went to completion and it is also being tested in conjunction with statin drugs.