

Last Updated: May 22, 2026
Introduction to Blood Coagulation
The ability of the body to control the flow of blood following vascular injury is paramount to continued survival. The process of blood coagulation (clotting) and then the subsequent dissolution of the clot, following repair of the injured tissue, is termed hemostasis. Hemostasis comprises four major events that occur in a set order following the loss of vascular integrity:
1. The initial phase of the process is vascular constriction. This limits the flow of blood to the area of injury.
2. Next, platelets become activated by thrombin and aggregate at the site of injury, forming a temporary, loose platelet plug. The protein fibrinogen is primarily responsible for stimulating platelet clumping. Platelets clump by binding to collagen that becomes exposed following rupture of the endothelial lining of vessels. Upon activation, platelets release the nucleotide, ADP and the eicosanoid, TXA2 (both of which activate additional platelets), serotonin, phospholipids, lipoproteins, and other proteins important for the coagulation cascade. In addition to induced secretion, activated platelets change their shape to accommodate the formation of the plug.
3. To ensure stability of the initially loose platelet plug, a fibrin mesh (also called the clot) forms and entraps the plug. If the plug contains only platelets it is termed a white thrombus; if red blood cells are present it is called a red thrombus
4. Finally, the clot must be dissolved in order for normal blood flow to resume following tissue repair. The dissolution of the clot occurs through the action of plasmin
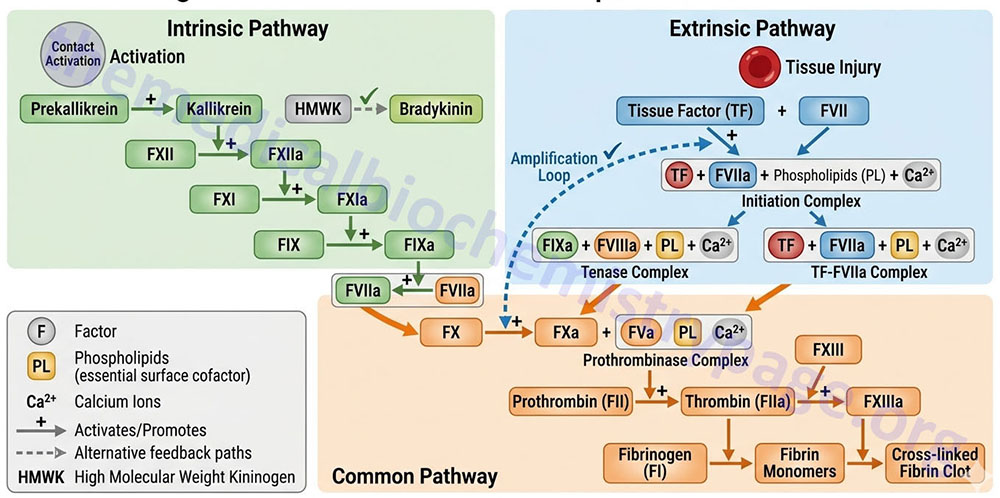
Two pathways lead to the formation of a fibrin clot: the intrinsic and extrinsic pathway. Although they are initiated by distinct mechanisms, the two converge on a common pathway that leads to clot formation. Both pathways are complex and involve numerous different proteins termed clotting factors. Fibrin clot formation in response to tissue injury is the most clinically relevant event of hemostasis under normal physiological conditions. This process is the result of the activation of the extrinsic pathway. The formation of a red thrombus or a clot in response to an abnormal vessel wall in the absence of tissue injury is the result of the intrinsic pathway. The intrinsic pathway has low significance under normal physiological conditions. Most significant clinically is the activation of the intrinsic pathway by contact of the vessel wall with lipoprotein particles, VLDLs and chylomicrons. This process clearly demonstrates the role of hyperlipidemia in the generation of atherosclerosis. The intrinsic pathway can also be activated by vessel wall contact with bacteria.
Platelet Activation and von Willebrand Factor (vWF)
In order for hemostasis to occur, platelets must adhere to exposed collagen, release the contents of their granules, and aggregate. Platelets contain several different types of granules, each of which store and secrete numerous compounds and molecules necessary for the functions of platelets. Indeed, more than 300 distinct molecules have been characterized in the different platelet granules.
Although megakaryocytes (producers of platelets) represent a site of synthesis and secretion of vWF, the major cell types that produce and secrete vWF are the vascular endothelial cells. von Willebrand Factor is synthesized by the VWF gene. The VWF gene is located on chromosome 12p13.31 and is composed of 53 exons that encode a 2813 amino acid preproprotein.
Platelet dense granules contain primarily small molecules such as the nucleotide, ADP and the neurotransmitter/hormone, serotonin.
Platelet α-granules contain primarily proteins such as von Willebrand factor (vWF), fibrinogen, factor V, platelet factor 4 (PF4), and several growth factors (e.g. PDGF, FGF, and VEGF).
Platelet lysosomes also represent a category of secretory vesicle that contain numerous hydrolytic enzymes.
The adhesion of platelets to the collagen exposed on endothelial cell surfaces is mediated by vWF. Inherited deficiencies of vWF are the causes of von Willebrand disease, (vWD) (also see below for more details). The function of vWF is to act as a bridge between a specific glycoprotein complex on the surface of platelets (GPIb-GPIX-GPV) and collagen fibrils. The GPIb part of the complex is composed of two proteins, GPIbα and GPIbβ encoded by separate genes. The importance of this interaction between vWF and the GPIb-GPIX-GPV complex of platelets is demonstrated by the inheritance of bleeding disorders caused by defects in three of the four proteins of the complex, the most common of which is Bernard-Soulier syndrome (also called giant platelet syndrome).
In addition to its role as a bridge between platelets and exposed collagen on endothelial surfaces, vWF binds to and stabilizes coagulation factor VIII. Binding of factor VIII by vWF is required for normal survival of factor VIII in the circulation. von Willebrand factor is a complex multimeric glycoprotein that is produced by and stored in the α-granules of platelets. It is also synthesized by megakaryocytes and found associated with subendothelial connective tissue.
The initial activation of platelets is induced by thrombin binding to specific receptors on the surface of platelets, thereby initiating a signal transduction cascade (see below for Figure). The thrombin receptors are protease-activated receptors (PAR), G-protein coupled receptors (GPCRs) that are activated by the action of thrombin cleaving a peptide from the extracellular domain of the receptor which then binds to the ligand-binding domain activating the receptor-mediated signal transduction process. There are three PARs to which thrombin binds identified as PAR-1 (PAR1), PAR-3 (PAR3), and PAR-4 (PAR4). These thrombin receptors are coupled to either Gq– or G12/13-type G-proteins. The Gq-type G-proteins activate PLCβ that in turn hydrolyzes phosphatidylinositol-4,5-bisphosphate (PIP2; PtdIns-4,5-P2) leading to the formation of inositol trisphosphate (IP3; Ins-1,4,5-P3) and diacylglycerol (DAG). IP3 induces the release of intracellular Ca2+ stores, and DAG activates kinases of the protein kinase C (PKC) family. Several of the PKC isoforms are involved in functions of platelets including PKCα, PKCβ, PKCδ, and PKCθ. The G12/13-type G-proteins in turn activate the small monomeric Rho G-proteins.
The collagen to which platelets adhere, as well as the release of intracellular Ca2+, leads to the activation of phospholipase A2 (PLA2), which then hydrolyzes membrane phospholipids, leading to liberation of arachidonic acid. The arachidonic acid release leads to an increase in the production and subsequent release of thromboxane A2 (TXA2). TXA2 is a potent vasoconstrictor and inducer of platelet aggregation that functions by binding to GPCR termed the TP receptors. These receptors are coupled to Gq and, therefore, binding of TXA2 activates the PLCβ pathway. Another enzyme activated by the released intracellular Ca2+ stores is myosin light chain kinase (MLCK). Activated MLCK phosphorylates the light chain of myosin which then interacts with actin, resulting in altered platelet morphology and motility.
The activation of PKC isoforms leads to numerous effects within platelets with the activation of granule release being of significant importance. Activated PKC isoforms in turn phosphorylate and activate multiple proteins involved in platelet granule secretion. These proteins include members of the phosphatidylinositol-5-phosphate 4-kinase type 2 family, several cytoskeletal-associated proteins [e.g. plekstrin and MARCKS, and several SNARE complex proteins. The MARCKS protein name is derived from [Myristoylated Alanine-Rich C–Kinase (PKC) Substrate]. MARCKS is involved in modulating cytoskeletal actin dynamics and vesicular trafficking which contributes to the activation of various signal transduction pathways.
One important dense granule molecule is ADP. The release of ADP leads to the further stimulation iof platelets, increasing the overall activation cascade. The important role of ADP in platelet activation can be appreciated from the use of the ADP receptor (P2Y12) antagonist, Plavix® (clopidogrel), in the control of thrombosis (see below). ADP also modifies the platelet membranes leading to exposure platelet glycoprotein receptor complex: GPIIb-GPIIIa.
GPIIb-GPIIIa constitutes a receptor for vWF and fibrinogen, resulting in fibrinogen-induced platelet aggregation. The GPIIb-GPIIIa complex is a member of the integrin family of cell-surface receptors that interact with the extracellular matrix. The GPIIb-GPIIIa complex is also called integrin α2bβ3.
The importance of the GPIIb-GPIIIa in platelet activation and coagulation is demonstrated by the fact that bleeding disorders result from inherited defects in this glycoprotein complex. The most commonly inherited platelet dysfunction is Glanzmann thrombasthenia which results from defects in the GPIIb protein of this complex. In addition, the importance of this complex in overall hemostasis is demonstrated by the use of antibodies that block this receptor as anti-coagulants (e.g. ReoPro®, abciximab: see below).
Activation of platelets is required for their consequent aggregation to a platelet plug. However, equally significant is the role of activated platelet surface phospholipids in the activation of the coagulation cascade.
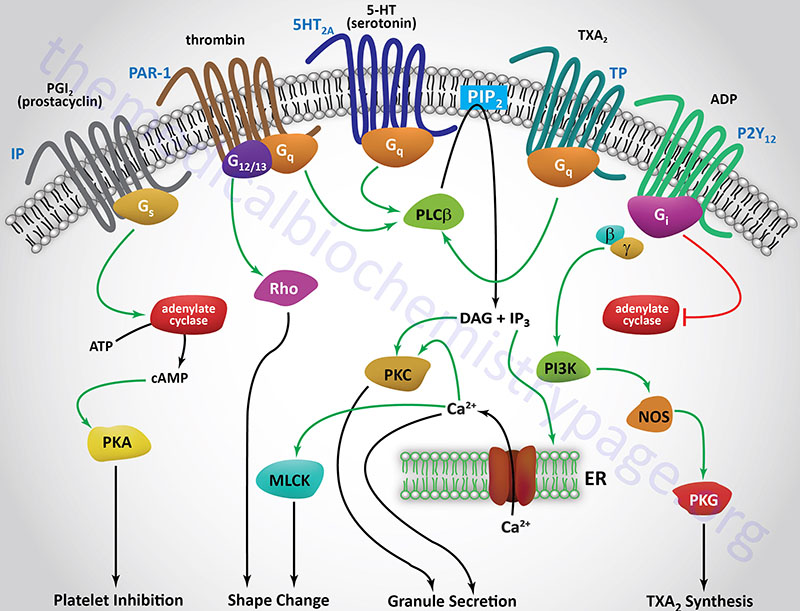
Table of the Primary Coagulation Factors
| Factor | Trivial Name(s) | Pathway | Characteristic |
| Prekallikrein (PK) | Fletcher factor | Intrinsic | Functions with HMWK and factor XII |
| High molecular weight kininogen (HMWK) | contact activation cofactor; Fitzgerald, Flaujeac Williams factor | Intrinsic | Co-factor in kallikrein and factor XII activation, necessary in factor XIIa activation of XI, precursor for bradykinin (a potent vasodilator and inducer of smooth muscle contraction |
| Fibrinogen | Factor I | Both | cleaved by thrombin to form fibrin monomers which are cross-linked by factor XIIIa to form fibrin clot |
| Prothrombin | Factor II | Both | modified by vitamin K to contain gla residues; activated on surface of activated platelets by prothrombinase complex |
| Tissue factor | Factor III | Extrinsic | is a subendothelial cell-surface glycoprotein that acts as a cofactor for factor VII |
| Calcium ions | Factor IV | Both | |
| V | Proaccelerin, labile factor, accelerator (Ac-) globulin | Both | Protein cofactor; activated by thrombin; factor Va is a cofactor in the activation of prothrombin by factor Xa |
| VI (same as Va) | Accelerin | Both | This is Va, redundant to Factor V |
| VII | Proconvertin, serum prothrombin conversion accelerator (SPCA), cothromboplastin | Extrinsic | Endopeptidase; modified by vitamin K to contain gla residues; activated by thrombin in presence of Ca2+ |
| VIII | Antihemophiliac factor A, antihemophilic globulin (AHG) | Intrinsic | Protein cofactor; activated by thrombin; factor VIIIa is a cofactor in the activation of factor X by factor IXa |
| IX | Christmas Factor, antihemophilic factor B, plasma thromboplastin component (PTC) | Intrinsic | Endopeptidase; modified by vitamin K to contain gla residues; activated by factor XIa in presence of Ca2+ |
| X | Stuart-Prower Factor | Both | Endopeptidase; modified by vitamin K to contain gla residues; activated on surface of activated platelets by tenase complex and by factor VIIa in presence of tissue factor and Ca2+ |
| XI | Plasma thromboplastin antecedent (PTA) | Intrinsic | Endopeptidase; activated by factor XIIa |
| XII | Hageman Factor | Intrinsic | Endopeptidase; binds to exposed collagen at site of vessel wall injury, activated by high-MW kininogen and kallikrein |
| XIII | Protransglutaminase, fibrin stabilizing factor (FSF), fibrinoligase | Both | Plasma version is a heterodimeric enzyme composed of two A and two B subunits; A subunit possesses the catalytic activity and is encoded by F13A1 gene; B subunit encoded by F13B gene; the platelet version of factor XIII is composed of the A subunits only; when activated XIII functions as a transglutaminase; activated by thrombin in presence of Ca2+; following activation of plasma XIII the B subunits are dissociated resulting in the same enzyme as in platelets; cross-links fibrin monomers thereby stabilizing fibrin clot |
| von Willebrand factor | Both | associated with subendothelial connective tissue; serves as a bridge between platelet glycoprotein GPIb/IX and collagen | |
| Protein C | Both | activated to protein Ca by thrombin bound to thrombomodulin; then degrades factors VIIIa and Va; modified by vitamin K to contain gla residues | |
| Protein S | Both | acts as a cofactor of protein C; modified by vitamin K to contain gla residues | |
| Thrombomodulin | Both | protein on the surface of endothelial cells; binds thrombin, which then activates protein C | |
| Antithrombin III | most important coagulation inhibitor, controls activities of thrombin, and factors IXa, Xa, XIa and XIIa; is a member of the serpin superfamily of serine protease inhibitors; is encoded by the SERPINC1 (serpin family C member 1) gene | ||
| Protein Z | Both | Required cofactor for the activation of PZI; modified by vitamin K to contain gla residues; deficiency is associated with a procoagulant state caused by excessive Xa and thrombin production, dysfunction is linked with several thrombotic disorders, including arterial vascular and venous thromboembolic diseases | |
| Serpin family A member 10 (SERPINA10) | Protein Z-dependent protease inhibitor (PZI) | Both | Degrades activated factor X (Xa) and activated factor XI (XIa) in complex with protein Z, Ca2+, and phospholipids |
The Clotting Cascades
The Kallikrein-Kinin System in Coagulation
The kallikrein-kinin system comprises a complex of proteins that when activated leads to the release of vasoactive kinins. The kinins are released from both high molecular weight kininogen (HMWK) and low molecular weight kininogen (LMWK) as a result of activation of either plasma kallikrein or tissue kallikrein, respectively. The kallikreins themselves exist in inactive pre-forms. The kinins are involved in many physiological and pathological processes including regulation of blood pressure and flow (via modulation of the renin-angiotensin pathway), blood coagulation, cellular proliferation and growth, angiogenesis, apoptosis, and inflammation. Kinin action on endothelial cells leads to vasodilation, increased vascular permeability, release of tissue plasminogen activator (tPA), production of nitric oxide (NO), and the mobilization of arachidonic acid, primarily resulting in prostacyclin (PGI2) production by endothelial cells. Although the activities of the kallikrein-kinin system are involved in numerous processes, this section will deal only with their function in blood coagulation. With respect to hemostasis the most important kinin is bradykinin which is released from HMWK.
Prekallikrein
The two forms of prekallikrein, plasma and tissue, are derived from distinct genes on different chromosomes.
Plasma kallikrein is encoded by the KLKB1 gene. The KLKB1 gene is on chromosome 4q35.2 and is composed of 17 exons that generate three alternatively spliced mRNAs, each of which encode a distinct protein isoform.
Tissue kallikrein is encoded by the KLK1 gene. The KLK1 gene is located on chromosome 19q13.33 and is composed of 6 exons that encode a 262 amino acid preproprotein.
The two kallikreins are serine proteases whose major substrates are HMWK (plasma kallikrein) and LMWK (tissue kallikrein). When plasma prekallikrein is activated to kallikrein it cleaves HMWK in a two-step process that ultimately releases bradykinin.
HMWK and LMWK
Both HMWK and LMWK are derived from the same gene, KNG1. The KNG1 gene is located on chromosome 3q27.3 and is composed of 11 exons. Exons 1 to 9 encode the heavy chain of both kininogens. Exon 10 encodes bradykinin as well as the light chain of HMWK. Exon 11 encodes the light chain of LMWK. The heavy and light chain nomenclature refers to the disulfide-bonded structure of each kininogen after their activation, which results from kallikrein cleavage. Expression of the KNG1 gene is exclusive to the liver and kidney with the level of expression in the liver being around four times higher than in the kidney.
HMWK is considered an α-globulin and is composed of six functional domains. The protein circulates in the plasma as single-chain polypeptide with a molecular weight of 88–120 kDa dependent upon the level of glycosylation. The heavy chain is 64 kDa and contains domains 1, 2, and 3 whereas the light chain is 45–56 kDa and comprises domains 5 and 6. The heavy and light chains are linked together through domain 4 which also contains the bradykinin sequence. Domain 1 contains a low affinity calcium-binding site. Domains 2 and 3 contain amino acid sequences (QVVAG) that inhibit cysteine proteases. Domain 3 also has platelet and endothelial cell-binding activity. Domain 5 has sequences for heparin binding, cell-binding sites, and antiangiogenic properties. The binding of HMWK to negatively charged surfaces occurs through a histidine region of the light chain which is in domain 5. Domain 6 contains the prekallikrein and factor XI-binding sites. By being able to bind to charged surfaces via domain 5 and simultaneously bind factor XI and prekallikrein via domain 6, HMWK can serve as the cofactor in contact activation of plasma.
LMWK is considered a β-globulin and has a molecular weight of 50–68 kDa. The structure of LMWK is similar to that of HMWK, however, the light chain is only 4–5 kDa and has no contact activation nor prekallikrein-binding sites.
Bradykinin
Bradykinin is a 9-amino acid vasoactive peptide that induces vasodilation and increases vascular permeability. Activated tissue kallikrein cleaves lysyl-bradykinin (also called kallidin) from LMWK. Lysyl-bradykinin is bradykinin with a lysine residue at the N-terminus making it a 10-amino acid vasoactive peptide. Its activities are essentially identical to those of bradykinin. Bradykinin is metabolized by three different enzymes of the metalloprotease family. The activity of angiotensin-converting enzyme (ACE) on bradykinin represents the main degradation pathway that transforms bradykinin into its final inactive metabolite bradykinin(1–5). Aminopeptidase P (APP) is the next most import enzyme involved in bradykinin degradation which converts bradykinin into an inactive peptide called bradykinin(2–9). Bradykinin(2-9) is further degraded by dipeptidyl peptidase IV into bradykinin(4–9). Various carboxypeptidases (collectively referred to as kininase I), converts bradykinin into des-Arg(9)-bradykinin.
Bradykinin and des-Arg(9) bradykinin function by binding to either of two receptors identified as bradykinin type 1 receptor (B1, B1R, B1) and bradykinin type 2 receptor (B2, B2R, B2).
The B1 receptor is encoded by the BDKRB1 gene. The BDKRB1 gene is located on chromosome 14q32.2 and is composed of 3 exons that encode a 353 amino acid protein.
The B2 receptor is encoded by the BDKRB2 gene. The BDKRB2 gene is also located on chromosome 14q32.2 and is composed of 3 exons that encode a 391 amino acid protein.
The primary receptor for bradykinin responses is the B2 receptor. The primary ligand for the B1 receptor is des-Arg(9) bradykinin. Both the B1 receptor and the B2 receptor have been shown to activate Gi-type and a Gq-type G-proteins. However, the predominate result of activation of the B1 receptor is Gi activation and that for activation of the B2 receptor is Gq activation.
Active Gq results in the activation of PLCβ and the subsequent production of DAG and IP3 (Ins-1,4,5-P3) from membrane-associated PIP2 (PtdIns-4,5-P2). Within endothelial cells the resultant IP3 binds to receptors in the endoplasmic reticulum leading to release of stored Ca2+. The increase in cytoplasmic Ca2+ activates the calmodulin subunits of endothelial nitric oxide synthase (eNOS). Active eNOS produces nitric oxide (NO) which diffuses to the underlying smooth muscle leading to their relaxation and vasodilation. When the B1 (or B2 receptor) activates the associated Gi-type G-protein the result is the activation of PLA2 but the net effect is also smooth muscle relaxation and vasodilation.
The plasma kinin forming system is called the contact system of plasma and is composed of factor XII, factor XI, prekallikrein and HMWK. Factor XII, prekallikrein, and HMWK saturably and reversibly bind to endothelial cells, platelets, and granulocytes in a zinc-dependent reaction. When plasma makes contact with a negatively charged surface factor XII binds and is autoactivated to factor XIIa (the “a” signifies the activated factor). Factor XIIa then activates prekallikrein to kallikrein and kallikrein cleaves HMWK releasing bradykinin. There is also reciprocal activation of factor XII by kallikrein resulting in amplification of the system. The actual surface that leads to factor XII autoactivation is unknown however, several physiologic substances support the process. These substances include hematin, skin, fatty acids, sodium urate crystals, protoporphyrin, sulfatides, heparins, chondroitin sulfates, articular cartilage, endotoxin, L-homocysteine, and amyloid β-protein. Once the contact system is activated the intrinsic pathway (described below) is initiated.
The Intrinsic Clotting Cascade
The intrinsic pathway (also called the contact activation pathway) is much less significant to hemostasis under normal physiological conditions than is the extrinsic pathway. However, abnormal physiology such as hyperlipidemic states or bacterial infiltration can lead to activation of thrombosis via the intrinsic clotting cascade.
The intrinsic pathway requires the clotting factors VIII, IX, X, XI, and XII. Also required are the proteins prekallikrein (PK) and high-molecular-weight kininogen (HK or HMWK), as well as calcium ions and phospholipids secreted from platelets. The role of PK and HMWK is described in the above section. Each of these pathway constituents leads to the conversion of factor X (inactive) to factor Xa. Initiation of the intrinsic pathway occurs when prekallikrein, high-molecular-weight kininogen, factor XI and factor XII are exposed to a negatively charged surface. This is termed the contact phase and can occur as a result of interaction with the phospholipids (primarily phosphatidylethanolamine, PE) of circulating lipoprotein particles such as chylomicrons, VLDLs, and oxidized LDLs. This is the basis of the role of hyperlipidemia in the promotion of a pro-thrombotic state and the development of atherosclerosis.
Contact activation of the intrinsic pathway can also occur on the surface of bacteria, and through the interaction with urate crystals, fatty acids, protoporphyrin, amyloid β, and homocysteine. Indeed, elevated levels of homocysteine in the blood have been shown to correlate with cardiovascular dysfunction. Therefore, it is important to ensure that proper function of the methionine synthase reaction is maintained. Although it would be assumed that increased intake of vitamin B12 should lead to increased conversion of homocysteine to methionine, and thus reduced levels of circulating homocysteine, controlled studies have shown that this does not occur.
The assemblage of contact phase components results in conversion of prekallikrein to kallikrein, which in turn activates factor XII to factor XIIa. Factor XIIa then activates factor XI to factor XIa. Factor XIIa will also hydrolyze more prekallikrein to kallikrein, establishing a reciprocal activation cascade. Kallikrein acts upon HMWK leading to the release of bradykinin, a potent vasodilator.
In the presence of Ca2+, factor XIa activates factor IX to factor IXa. Factor IX is a proenzyme that contains vitamin K-dependent γ-carboxyglutamate (gla) residues, whose serine protease activity is activated following Ca2+ binding to these gla residues. Several of the serine proteases of the cascade (II, VII, IX, and X) are gla-containing proenzymes. Active factor IXa cleaves factor X at an internal arg-ile (R-I) bond leading to its activation to factor Xa.
The activation of factor Xa requires assemblage of the tenase complex (Ca2+ and factors VIIIa, IXa and X) on the surface of activated platelets. One of the responses of platelets to activation is the presentation of phosphatidylserine (PS) and phosphatidylinositol (PI) on their surfaces. The exposure of these phospholipids allows the tenase complex to form. The role of factor VIII in this process is to act as a receptor, in the form of factor VIIIa, for factors IXa and X. Factor VIIIa is termed a cofactor in the clotting cascade. The activation of factor VIII to factor VIIIa (the actual receptor) occurs in the presence of minute quantities of thrombin. As the concentration of thrombin increases, factor VIIIa is ultimately cleaved by thrombin and inactivated. This dual action of thrombin, upon factor VIII, acts to limit the extent of tenase complex formation and thus the extent of the coagulation cascade.
Extrinsic Clotting Cascade
Activated factor Xa is the site at which the intrinsic and extrinsic coagulation cascades converge. The extrinsic pathway is initiated at the site of injury in response to the release of tissue factor (factor III) and thus, is also known as the tissue factor pathway. Tissue factor is a cofactor in the factor VIIa-catalyzed activation of factor X. Factor VIIa, a gla residue containing serine protease, cleaves factor X to factor Xa in a manner identical to that of factor IXa of the intrinsic pathway. The activation of factor VII occurs through the action of thrombin or factor Xa. The ability of factor Xa to activate factor VII creates a link between the intrinsic and extrinsic pathways. An additional link between the two pathways exists through the ability of tissue factor and factor VIIa to activate factor IX. The formation of complex between factor VIIa and tissue factor is believed to be a principal step in the overall clotting cascade. Evidence for this stems from the fact that persons with hereditary deficiencies in the components of the contact phase of the intrinsic pathway do not exhibit clotting problems.
A major mechanism for the inhibition of the extrinsic pathway occurs at the tissue factor-factor VIIa-Ca2+-Xa complex. The protein, lipoprotein-associated coagulation inhibitor, LACI specifically binds to this complex. LACI is also referred to as extrinsic pathway inhibitor, EPI or tissue factor pathway inhibitor, TFPI and was formerly named anticonvertin. LACI is composed of 3 tandem protease inhibitor domains. Domain 1 binds to factor Xa and domain 2 binds to factor VIIa only in the presence of factor Xa.
Activation of Prothrombin to Thrombin
The common point in both coagulation pathways is the activation of factor X to factor Xa. Factor Xa activates prothrombin (factor II) to thrombin (factor IIa). Thrombin, in turn, converts fibrinogen to fibrin. The activation of thrombin occurs on the surface of activated platelets and requires formation of a prothrombinase complex. This complex is composed of the platelet phospholipids, phosphatidylinositol and phosphatidylserine, Ca2+, factors Va and Xa, and prothrombin. Factor V is a cofactor in the formation of the prothrombinase complex, similar to the role of factor VIII in tenase complex formation. Like factor VIII activation, factor V is activated to factor Va by means of minute amounts and is inactivated by increased levels of thrombin. Factor Va binds to specific receptors on the surfaces of activated platelets and forms a complex with prothrombin and factor Xa.
Prothrombin is encoded by the coagulation factor II gene (F2) which is located on chromosome 11p11.2 and is composed of 14 exons that encode a 622 amino acid preproprotein. Expression of the F2 gene is exclusive to the liver.
Prothrombin is a 72 kDa, single-chain protein containing ten gla residues in its N-terminal region. Within the prothrombinase complex, prothrombin is cleaved at two sites by factor Xa. This cleavage generates a 2-chain active thrombin molecule containing an A and a B chain which are held together by a single disulfide bond.
In addition to its role in activation of fibrin clot formation, thrombin plays an important regulatory role in coagulation. Thrombin combines with thrombomodulin present on endothelial cell surfaces forming a complex that converts protein C to protein Ca. The cofactor protein S and protein Ca degrade factors Va and VIIIa, thereby limiting the activity of these 2 factors in the coagulation cascade (see details below).
Thrombin binds to a class of G-protein-coupled receptors (GPCRs) called protease activated receptors (PARs), specifically PAR-1, -3 and -4. PARs utilize a unique mechanism to convert the result of extracellular proteolytic cleavage into an intracellular signaling event. PARs carry their own ligand which remains inactive until protease cleavage, such as by thrombin, “unmasks” the ligand. Following thrombin cleavage the unmasked ligand is still a part of the intact PAR but is now capable of interacting with the ligand-binding domain of the PAR resulting in the activation of numerous signaling cascades. Because the activation of PARs requires proteolytic cleavage the activation process is irreversible. The signaling cascades activated by thrombin-activated PARs include release of the interleukins, (ILs), IL-1 and IL-6, increased secretion of intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1). The thrombin-induced signaling also leads to increased platelet activation and leukocyte adhesion.
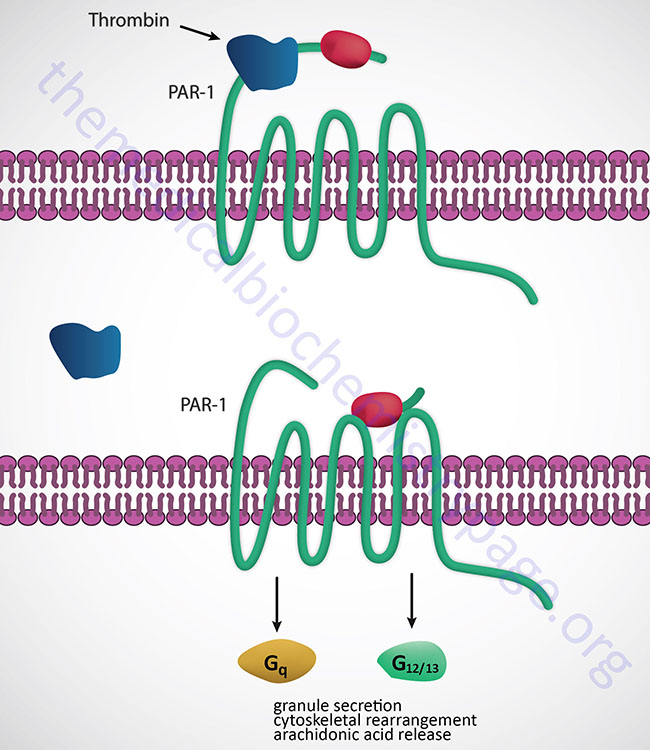
Thrombin also activates thrombin-activatable fibrinolysis inhibitor (TAFI) thus modulating fibrinolysis (degradation of fibrin clots). TAFI is also known as carboxypeptidase U (CPU) whose activity leads to removal of C-terminal lysines from partially degraded fibrin. This leads to an impairment of plasminogen activation, thereby reducing the rate of fibrin clot dissolution (i.e. fibrinolysis).
Control of Thrombin Levels
The inability of the body to control the circulating level of active thrombin would lead to dire consequences. There are 2 principal mechanisms by which thrombin activity is regulated. The predominant form of thrombin in the circulation is the inactive prothrombin, whose activation requires the pathways of proenzyme activation described above for the coagulation cascade. At each step in the cascade, feedback mechanisms regulate the balance between active and inactive enzymes.
The activation of thrombin is also regulated by four specific thrombin inhibitors. Antithrombin III is the most important since it can also inhibit the activities of factors IXa, Xa, XIa and XIIa, plasmin, and kallikrein. The activity of antithrombin III is potentiated in the presence of heparin. Heparin binds to a specific site on antithrombin III, producing an altered conformation of the protein, and the new conformation has a higher affinity for thrombin as well as its other substrates. This effect of heparin is the basis for its clinical use as an anticoagulant. The naturally occurring heparin activator of antithrombin III is present as heparan and heparan sulfate on the surface of vessel endothelial cells. It is this feature that controls the activation of the intrinsic coagulation cascade. Antithrombin III is a member of the serpin (serine protease inhibitor) superfamily of serine protease inhibitors and as such, is encoded by the SERPINC1 gene.
However, thrombin activity is also inhibited by α2-macroglobulin, heparin cofactor II and α1-antitrypsin. Although a minor player in thrombin regulation α1-antitrypsin is the primary serine protease inhibitor of human plasma. Its physiological significance is demonstrated by the fact that lack of this protein plays a causative role in the development of emphysema.
Protein C: Control of Coagulation and Sepsis
Protein C (PC) is a trypsin-like serine protease that serves as a major regulator of the coagulation process. Protein S (PS) serves as a co-factor for the functions of activated protein C (abbreviated aPC, and also APC). The human protein C gene (gene symbol: PROC) is located on chromosome 2q14.3 and is composed of 8 exons that encode a 461 amino acid preproprotein. Protein C undergoes a series of post-translational modifications that include several N-linked glycosylation sites and γ-carboxylation of nine glutamate residues (gla residues) in the amino terminus. These gla residues in the amino terminus of PC constitute the “gla domain” of the protein. In addition to the gla domain, PC contains two epidermal growth factor-like regions (EGF domains), the serine protease domain, and an activation peptide.
Thrombin cleavage of PC removes the activation peptide generating aPC. When activated through cleavage by thrombin, aPC cleaves both factor Va and factor VIIIa into inactive enzymes. This results in the termination of the role of VIIIa as the scaffold for the formation of the tenase complex and Va as a co-factor in the conversion of prothrombin to thrombin in the prothrombinase complex. The net effect, at the level of coagulation, of the activation of PC is termination of further increases in thrombin production and a halt to further fibrin clot formation. The activation of PC by thrombin occurs on the surface of the endothelium when thrombin binds to thrombomodulin and “captures” circulating PC. Following activation, aPC interacts with PS and cleaves VIIIa and Va.
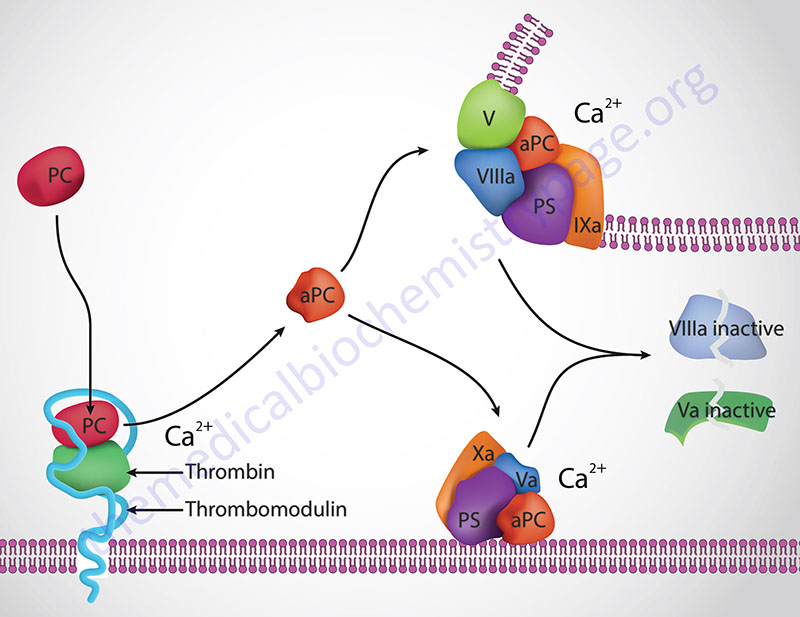
The importance of aPC in controlling Va activity can be seen in the hypercoagulopathy (thrombophilia) referred to as factor V Leiden. This thrombophilia is caused by a mutation in the factor V gene resulting in a protein that is not effectively degraded by aPC. Factor V Leiden is the most common inherited thrombophilia in Caucasian populations of European descent. Overall, 5% of the world population harbors the factor V Leiden mutation. The symptoms of factor V Leiden are deep vein thrombosis (DVT) and pulmonary embolism, both of which can be fatal. In fact it is estimated that in as many as 30% of patients who experience DVT and/or pulmonary embolisms are carriers of the factor V Leiden mutation. Loss of protein C results in massive and usually lethal thrombotic complications in infants with homozygous PC deficiency. In individuals who are heterozygous for PC deficiencies there is an increased risk for venous thrombosis.
Although the role of aPC in the termination of coagulation is extremely important it also serves many additional functions that alter the inflammatory processes occurring in the vasculature. Activated PC binds to the endothelial protein C receptor (EPCR; encoded by the PROCR gene) and leads to the activation of PAR-1 (see above for the role of thrombin in PAR activation) which elicits cytoprotective and anti-inflammatory responses within endothelial cells. The EPCR functions as a co-receptor for aPC-mediated cleavage and activation of PAR-1. The EPCR is also found on monocytes, neutrophils, fibroblasts, and keratinocytes. The cytoprotective effects elicited via aPC activation of PAR-1 include protection of the endothelial cell barrier and induction of anti-apoptotic signaling pathways as well as expression of eNOS. Additional endothelial responses to aPC activation of PAR-1 occur via inhibition of the expression and actions of the potent pro-inflammatory transcription factor NF-κB (nuclear factor kappa-B). The suppression of NF-κB action results in downregulation of the synthesis of endothelial pro-inflammatory cytokines such as IL-6 and IL-8, the chemokine MCP-1 (monocyte chemoattractant protein-1), and the cell adhesion molecule ICAM-1 (intercellular adhesion molecule-1).
The binding of aPC to the EPCR on monocytes leads to inhibition of the synthesis and release of pro-inflammatory cytokines (e.g. IL-1, IL-6 and TNFα) from these cells which results from inhibition of the actions of NF-κB. Additional monocyte effects of aPC include decreased tissue factor (factor III) expression and inhibition of the release of the chemokines MIP-1α (macrophage inflammatory protein-1α) and MCP-1.
The anti-inflammatory and cytoprotective effects of aPC were the basis for the development of the sepsis drug Xigris® (dotrecogin alpha). Although used for several years, this drug has been removed from the market due to insignificant reductions in sepsis-induced mortality. Sepsis is initiated by infection whereby microbes and/or microbial toxins released into the blood trigger a systemic and uncontrolled activation of both the coagulation cascade and inflammatory pathways. Sepsis is the leading cause of death in intensive care patients who are not coronary patients. Severe sepsis afflicts more than 700,000 people in the United States each year with a mortality rate of 30% to 50%.
In addition to its demonstrated efficacy in the treatment of sepsis, aPC is currently being investigated for the treatment of numerous conditions. These include the treatment of stoke since in mouse models the anti-inflammatory and anticoagulant actions of aPC have the additional benefit of exerting neuroprotective effects. Ischemia-reperfusion injury (I/R) results when tissues are temporarily deprived of oxygenated blood (ischemia) and the return of blood flow (reperfusion) results in additional tissue damage. The secondary damage of I/R is a consequence of the intense inflammatory reactions that are initiated in response to anoxia. The damage from reperfusion can occur in tissues or organs that were not affected by the initial ischemic episode. In mouse models it has been shown that infusion of aPC attenuates the oxidative tissue damage of ischemia and this effect may be due to direct aPC-mediated inhibition of neutrophil activation. Additional conditions that may be treated with aPC infusion include acute lung injury, asthma, and acute pancreatitis. Studies have also demonstrated that aPC may promote wound healing and angiogenesis.
Activation of Fibrinogen to Fibrin
Fibrinogen (factor I) consists of 3 pairs of polypeptides ([Aα][Bβ][γ])2. The 6 chains are covalently linked near their N-terminals through disulfide bonds. The A and B portions of the Aα and Bβ chains comprise the fibrinopeptides, A and B, respectively. The fibrinopeptide regions of fibrinogen contain several glutamate and aspartate residues imparting a high negative charge to this region and aid in the solubility of fibrinogen in plasma. Active thrombin is a serine protease that hydrolyses fibrinogen at four arg-gly (R-G) bonds between the fibrinopeptide and the a and b portions of the protein.
Thrombin-mediated release of the fibrinopeptides generates fibrin monomers with a subunit structure (αβγ)2. These monomers spontaneously aggregate in a regular array, forming a somewhat weak fibrin clot. In addition to fibrin activation, thrombin converts factor XIII to factor XIIIa, a highly specific transglutaminase that introduces cross-links composed of covalent bonds between the amide nitrogen of glutamines and ε-amino group of lysines in the fibrin monomers.
Dissolution of Fibrin Clots
Degradation of fibrin clots is the function of plasmin, a serine protease that circulates as the inactive proenzyme, plasminogen. Any free circulating plasmin is rapidly inhibited by α2-antiplasmin. Plasminogen binds to both fibrinogen and fibrin, thereby being incorporated into a clot as it is formed. Tissue plasminogen activator (tPA) and, to a lesser degree, urokinase are serine proteases which convert plasminogen to plasmin. Inactive tPA is released from vascular endothelial cells following injury; it binds to fibrin and is consequently activated. Urokinase is produced as the precursor, prourokinase by epithelial cells lining excretory ducts. The role of urokinase is to activate the dissolution of fibrin clots that may be deposited in these ducts.
Active tPA cleaves plasminogen to plasmin which then digests the fibrin; the result is soluble degradation product to which neither plasmin nor plasminogen can bind. Following the release of plasminogen and plasmin they are rapidly inactivated by their respective inhibitors. The inhibition of tPA activity results from binding to specific inhibitory proteins. At least 4 distinct inhibitors have been identified, of which the plasminogen activator-inhibitors type 1 (PAI-1) and type 2 (PAI-2) are of greatest physiological significance.
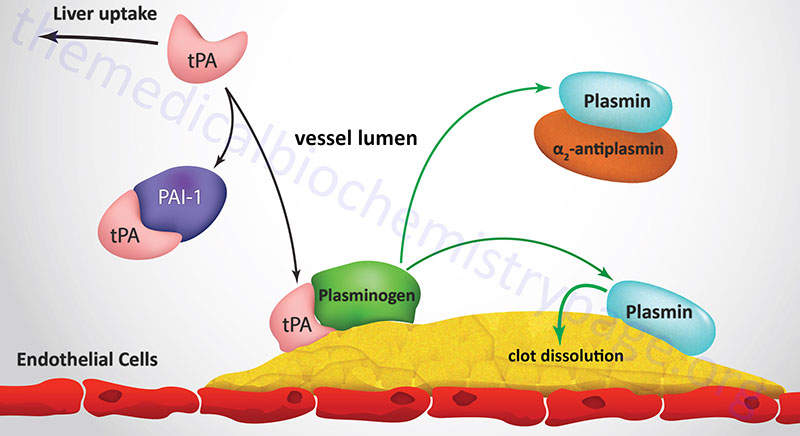
PAI-1 is encoded by the SERPINE1 gene. SERPINE1 refers to serine protease inhibitor (SERPIN) family E member 1. The SERPINE1 gene is located on chromosome 7q22.1 and is composed of 9 exons that generate 12 alternatively spliced mRNAs that each encode a distinct precursor protein.
PAI-2 is encoded by the SERPINB2 gene. The SERPINB2 gene is located on chromosome 18q21.33-q22.1 and is composed of 9 exons that generate two alternatively spliced mRNAs, both of which encode the same 415 amino acid protein.
Blood Coagulation Tests and Interpretations
Bleeding time
Bleeding time assays are used to evaluate the vascular and platelet responses that are associated with hemostasis. The bleeding time is a frequent assay performed on preoperative patients to ensure there is an adequate response to vessel injury prior to surgery. As indicated above, the rapid responses to vascular injury (occurring within seconds) are vessel constriction and platelet adhesion to the vessel wall. The Ivy method for determining the bleeding time involves the use of a blood pressure cuff (sphygmomanometer) which is placed on the forearm and inflated to 40mmHg. A superficial incision is then made on the forearm and the time it takes for bleeding to stop is recorded. With the Ivy method bleeding should stop within 1–9 minutes. Any bleeding time greater than 15 minutes would be indicative of a defect in the initial responses of vessels and platelets to vascular injury. A less invasive bleeding time assay involves the use of a lancet or special needle and a 3–4mm deep prick is made on the fingertip or earlobe. This bleeding time assay is referred to as the Duke method and in this assay bleeding should cease within 1–3 minutes. The bleeding time is affected (prolonged) by any defect in platelet function, by vascular disorders, and in von Willebrand disease but is not affected by other coagulation factors. Disorders that are commonly associated with an increased bleeding time include thrombocytopenia, disseminated intravascular coagulation (DIC), Bernard-Soulier syndrome and Glanzmann thrombasthenia. Abnormal bleeding times are also found in patients with Cushing syndrome, severe liver disease, leukemia, and bone marrow failure.
Prothrombin time (PT)
Defects associated with factors of the pathways of blood coagulation can also be assessed with specific assays. The prothrombin time (PT) is an assay designed to screen for defects in fibrinogen, prothrombin, and factors V, VII, and X and thus measures activities of the extrinsic pathway of coagulation. When any of these factors is deficient then the PT is prolonged. A normal PT is 11.0–12.5 seconds. A PT greater than 20 seconds is indicative of coagulation deficit. The PT is measured using plasma after the blood cells are removed. A blood sample is collected in a tube containing citrate or EDTA to chelate any calcium and thus inhibit coagulation and then the cells are removed by centrifugation. After the cells are removed excess calcium is added to the plasma to initiate coagulation. The most common measure of PT is to divide the time of coagulation of a patients blood by that of a known standard and this value is referred to as the international normalized ratio (INR). Normal INR values range from 0.8–1.2. PT is used to determine the correct dosage of the warfarin class of anti-coagulation drugs (e.g. Coumadin), for the presence of liver disease or damage, and to evaluate vitamin K status.
Partial thromboplastin time (PTT)
The partial thromboplastin time (PTT) is used to assay for defects in the intrinsic pathway of coagulation. The PTT assay has been modified by the addition of activators that shorten the normal clotting time and this form of the assay is referred to as the activated partial thromboplastin time (aPTT). The PTT is normally prescribed in patients with unexplained bleeding or clotting. The assay will evaluate the function of fibrinogen, prothrombin, and factors V, VIII, IX, X, XI, and XII. A defect in any of these factors will result in a prolonged PTT (or aPTT). A normal PTT is 60–70 seconds, whereas for the aPTT the normal range is 30–40 seconds. The PTT is a standard assay used to assess the efficacy of heparin anticoagulant therapy. Prolonged PTTs are associated with acquired or congenital bleeding disorders associated with coagulation factor deficiency, vitamin K deficiency, liver disease, DIC, von Willebrand disease, leukemia, hemophilia, and during heparin administration.
The Bleeding Disorders
Defects in the process of hemostasis, leading to bleeding disorders, have been identified at the level of the proteins of the clotting cascades, platelet activation and function, contact activation and antithrombin function This list is not all inclusive and for more details click on the Diseases and Disorders link in the left panel.
Hemophilia A
Hemophilia A is classic hemophilia (a disease referring to the inability to clot blood). It is an X-linked disorder resulting from a deficiency in factor VIII, a key component of the coagulation cascade. There are severe, moderate and mild forms of hemophilia A that reflect the level of active factor VIII in the plasma.
Hemophilia A arises from a variety of mutations. Some 150 different point mutations have been characterized in the factor VIII gene in hemophilia A. Inheritance of the disorder occurs with a frequency of 1:5,000 to 1:10,000 males in all populations. Factor VIII is a cofactor in the activation of factor X to factor Xa in a reaction catalyzed by factor IXa. Activation of factor VIII occurs via proteolytic cleavage by thrombin and factor Xa. Inactivation of factor VIIIa occurs by limited proteolysis by factor Xa or activated protein C.
Individuals with deficiencies in factor VIII suffer joint and muscle hemorrhage, easy bruising and prolonged bleeding from wounds. Treatment of hemophilia A is accomplished by infusion of factor VIII concentrates prepared from either human plasma or by recombinant DNA technology.
Hemophilia B
Hemophilia B results from deficiencies in factor IX. The prevalence of hemophilia B is approximately one-tenth that of hemophilia A. All patients with hemophilia B have prolonged coagulation time and decreased factor IX clotting activity. Like hemophilia A, there are severe, moderate and mild forms of hemophilia B and reflect the factor IX activity in plasma.
At least 300 unique factor IX mutations have been identified, 85% are point mutations, 3% are short nucleotide deletions or insertions and 12% are gross gene alterations.
Disorders of Fibrinogen and Factor XIII
Several cardiovascular risk factors are associated with abnormalities in fibrinogen. As a result of the acute-phase response or through other poorly understood mechanisms, elevated plasma fibrinogen levels have been observed in patients with coronary artery disease, diabetes, hypertension, peripheral artery disease, hyperlipoproteinemia and hypertriglyceridemia. In addition, pregnancy, menopause, hypercholesterolemia, use of oral contraceptives and smoking lead to increased plasma fibrinogen levels.
Although rare, there are inherited disorders in fibrinogen. These disorders include afibrinogenemia (a complete lack of fibrinogen), hypofibrinogenemia (reduced levels of fibrinogen) and dysfibrinogenemia (presence of dysfunctional fibrinogen). Afibrinogenemia is characterized by neonatal umbilical cord hemorrhage, ecchymoses, mucosal hemorrhage, internal hemorrhage, and recurrent abortion. The disorder is inherited in an autosomal recessive manner. Hypofibrinogenemia is characterized by fibrinogen levels below 100mg/dL (normal is 250-350mg/dL) and can be either acquired or inherited. Symptoms of hypofibrinogenememia are similar to, but less severe than, afibrinogenemia. Dysfibrinogenemias are extremely heterogeneous affecting any of the functional properties of fibrinogen. Clinical consequences of dysfibrinogenemias include hemorrhage, spontaneous abortion and thromboembolism.
Factor XIII is the proenzyme form of plasma transglutaminase and is activated by thrombin in the presence of calcium ions. Active factor XIII catalyzes the cross-linking of fibrin monomers. Factor XIII is a tetramer of two different peptides, A and B (forming A2B2). Hereditary deficiencies (autosomal recessive) occur resulting in the absence of either subunit. Clinical manifestation of factor XIII deficiency is delayed bleeding although primary hemostasis is normal. Deficiency leads to neonatal umbilical cord bleeding, intracranial hemorrhage and soft tissue hematomas.
von Willebrand Disease
von Willebrand disease (vWD) is due to inherited deficiency in von Willebrand factor (vWF). vWD is the most common inherited bleeding disorder of humans. Using sensitive laboratory testing, abnormalities in vWF can be detected in approximately 8000 people per million. Clinically significant vWD occurs in approximately 125 people per million. This is a frequency at least twice that of hemophilia A.
Deficiency of vWF results in defective platelet adhesion and causes a secondary deficiency in factor VIII. The result is that vWF deficiency can cause bleeding that appears similar to that caused by platelet dysfunction or hemophilia. vWD is an extremely heterogeneous disorder that has been classified into several major subtypes. Type I vWD is the most common and is inherited as an autosomal dominant trait. This variant is due to simple quantitative deficiency of all vWF multimers. Type 2 vWD is also subdivided further dependent upon whether the dysfunctional protein has decreased or paradoxically increased function in certain laboratory tests of binding to platelets. Type 3 vWD is clinically severe and is characterized by recessive inheritance and virtual absence of vWF.
Factor XI and Contact Activation
When blood makes contact with negatively charged surfaces it triggers a series of interactions that involve factor XI, prekallikrein and high molecular weight kininogen leading to blood coagulation. This process is referred to as contact activation. Deficiency in factor XI confers an injury-related bleeding tendency. This deficiency was identified in 1953 and originally termed hemophilia C. Factor XI deficiency is very common in Ashkenazic Jews and is inherited as an autosomal disorder with either homozygosity or compound heterozygosity. Three independent point mutations in factor XI have been identified.
Antithrombin III Deficiency
Antithrombin III functions to inhibit several activated coagulation factors including thrombin, factor IXa and factor Xa, by forming a stable complex with the various factors. Heparin and heparan sulfates increase the activity of antithrombin III at least 1000 fold.
Deficiency in antithrombin III is seen in approximately 2% of patients with venous thromboembolic disease. Inheritance occurs as an autosomal dominant trait. The prevalence of symptomatic antithrombin III deficiency ranges from 1 per 2000 to 1 per 5000 in the general population. Deficiencies results from mutations that affect synthesis or stability of antithrombin III or from mutations that affect the protease and/or heparin binding sites of antithrombin III.
Clinical manifestations of antithrombin III deficiency include deep vein thrombosis and pulmonary embolism. Arterial thrombosis is rare in antithrombin III deficiency. Thrombosis may occur spontaneously or in association with surgery, trauma or pregnancy. Treatment of acute episodes of thrombosis is by infusion of heparin (for 5-7 days) followed by oral anticoagulant therapy.
Pharmacological Intervention in Bleeding
Inhibitors of Vitamin K-Dependent gla Residue Formation
The coumarin drugs (based on the chemical benzopyrone), such as warfarin (trade name Coumadin®) inhibit coagulation by inhibiting the vitamin K-dependent γ-carboxylation reactions necessary to the function of thrombin, and factors VII, IX, and X as well as proteins C and S. These drugs act by inhibiting the reduction of the quinone derivatives of vitamin K to their active hydroquinone forms. Because of the mode of action of coumarin drugs, it takes several days for their maximum effect to be realized. For this reason, heparin is normally administered first followed by warfarin or warfarin-related drugs.
Activation of Antithrombin III
The glycosaminoglycans, heparin and heparan sulfate, are useful as anticoagulants. Heparin functions as an anticoagulant because it binds to, and activates, antithrombin III which then inhibits the serine proteases of the coagulation cascade. Heparin is abundant in granules of the mast cells that line the vasculature. In response to injury, the heparin is released and inhibits coagulation.
Plasminogen Activators
The plasminogen activators also are useful for controlling coagulation. Because tPA is highly selective for the degradation of fibrin in clots, it is extremely useful in restoring the patency of the coronary arteries following thrombosis, in particular during the short period following myocardial infarct. Streptokinase (an enzyme from the Streptococci bacterium) is another plasminogen activator useful from a therapeutic standpoint. However, it is less selective than tPA, being able to activate circulating plasminogen as well as that bound to a fibrin clot.
Inhibition of Eicosanoid Synthesis
Aspirin is an important inhibitor of platelet activation. By virtue of inhibiting the activity of cyclooxygenase, aspirin reduces the production of TXA2 by platelets. Aspirin also reduces endothelial cell production of prostacyclin (PGI2), an inhibitor of platelet aggregation and a vasodilator. Localized to the site of coagulation is a balance between the levels of platelet derived TXA2 and endothelial cell derived PGI2. This allows for platelet aggregation and clot formation but preventing excessive accumulation of the clot, thus maintaining blood flow around the site of the clot. Endothelial cells regenerate active cyclooxygenase faster than platelets because mature platelets cannot synthesize the enzyme, requiring new platelets to enter the circulation (platelet half-life is approximately 4 days). Therefore, PGI2 synthesis is greater than that of TXA2. The net effect of aspirin is more in favor of endothelial cell-mediated inhibition of the coagulation cascade. This reflects one of the cardiovascular benefits to low dose administration of aspirin. Aspirin also has important effects on inflammatory processes that impact cardiovascular systems (see the Bioactive Lipid Mediators of Inflammation page for more details).
Inhibition of Platelet Activation / Function
Newer classes of anticoagulation drugs are being developed that function by inhibiting the activation of platelets and their subsequent aggregation. The drug clopidogrel (Plavix) is an irreversible inhibitor of the ADP receptors on platelet membranes. The primary platelet ADP receptor is a member of the G-protein coupled (GPCR) purinergic receptor family identified as the P2Y12 receptor. In addition to P2Y12, platelets express the P2Y1 receptor , however, the role of P2Y1 in platelet activation is not as significant as it is for the P2Y12 receptor. When ADP binds to the P2Y12 receptor on platelets they are activated and aggregate leading to amplification of the coagulation response, thus clopidogrel interferes with this process. Clopidogrel is prescribed for the treatment of peripheral vascular and cerebrovascular disease as well as coronary artery disease to prevent the formation of thrombotic plaques.
Additional P2Y12 receptor antagonists include prasugrel (Effient), ticagrelor (Brilinta), ticlopidine (Ticlid), and cangrelor (Kengreal). Ticagrelor functions as an allosteric antagonist of the P2Y12 receptor. Prasugrel and ticlopidine are irreversible inhibitors of the P2Y12 receptor like clopidogrel. Cangrelor is a reversible inhibitor of the P2Y12 receptor and, unlike clopidogrel, it is an active drug not a prodrug.
Another target of pharmacologic intervention in coagulation involving platelets is the role of GPIIb-GPIIIa in fibrinogen-induced platelet aggregation. Glanzmann thrombasthenia is an inherited platelet function defect characterized by a lack of platelet aggregation in response to all physiological agonists. This disorder is due to a lack of the GPIIb-GPIIIa receptor complex on platelets. Patients with this disorder present with significantly increased bleeding times. Although a rare disorder, study of the pathophysiology of the disease led to the development of anticoagulant drugs that inhibit platelet aggregation regardless of the agonist. Therefore, the GPIIb-GPIIIa antagonists more completely inhibit platelet aggregation than do aspirin or Plavix. The current family of these drugs includes ReoPro® (abciximab: a human monoclonal antibody), Integrilin® (eptifibatide: a cyclic hexapeptide derived from a protein found in the venom of the southeastern pygmy rattlesnake) and Aggrastat® (tirofiban: a synthetic organic non-peptide molecule).
Inhibition of Thrombin
Individuals that suffer from atrial fibrillation (AF) are at increased risk of strokes, in particular if other vascular conditions are present such as hypertension. AF is the most common cause of cardiac arrhythmia which is associated with palpitations, chest pain, and fainting. The direct reversible inhibitor of thrombin, dabigatran (Pradaxa®) is now prescribed to AF patients to prevent the occurrence of stroke in these patients. The primary use of dabigatran is in patients being switched from warfarin.
Inhibition of Factor Xa
Another mechanism to prevent the increased risk of stroke in patients suffering from atrial fibrillation is the inhibition of factor Xa. The drug rivaroxaban (Xarelto®) is a direct inhibitor of factor Xa either free or within the prothrombinase complex. In addition to prevention of stroke risk in AF patients, rivaroxaban is useful in the prevention of venous thromboembolism. Another recently approved drug in the direct factor Xa inhibitor family is apixaban (Eliquis®).