
Last Updated: February 13, 2026
Bile Acid Synthesis and Utilization
The end products of cholesterol utilization are the bile acids. Indeed, the synthesis of the bile acids is the major pathway of cholesterol catabolism in mammals. Although several of the enzymes involved in bile acid synthesis are active in many cell types, the liver is the only organ where their complete biosynthesis can occur. Synthesis of bile acids is one of the predominant mechanisms for the excretion of excess cholesterol. However, the excretion of cholesterol in the form of bile acids is insufficient to compensate for an excess dietary intake of cholesterol. Although bile acid synthesis constitutes the route of catabolism of cholesterol, these compounds are also important in the solubilization of dietary cholesterol, lipids, fat soluble vitamins, and other essential nutrients, thus promoting their delivery to the liver.
Synthesis of a full complement of bile acids requires 17 individual enzymes and occurs in multiple intracellular compartments that include the cytosol, endoplasmic reticulum (ER), mitochondria, and peroxisomes. The genes encoding several of the enzymes of bile acid synthesis are under tight regulatory control to ensure that the necessary level of bile acid production is coordinated to changing metabolic conditions. Given the fact that many bile acid metabolites are cytotoxic it is understandable why their synthesis needs to be tightly controlled. Several inborn errors in metabolism are due to defects in genes of bile acid synthesis and are associated with liver failure in early childhood to progressive neuropathies in adults.
Classic (Neutral) Pathway of Bile Acid Synthesis
The major pathway for the synthesis of the bile acids is initiated via hydroxylation of cholesterol at the 7 position via the action of cholesterol 7α-hydroxylase (encoded by the CYP7A1 gene) which is an ER-localized enzyme. The product of the CYP7A1 catalyzed reaction is 7α-hydroxycholesterol.
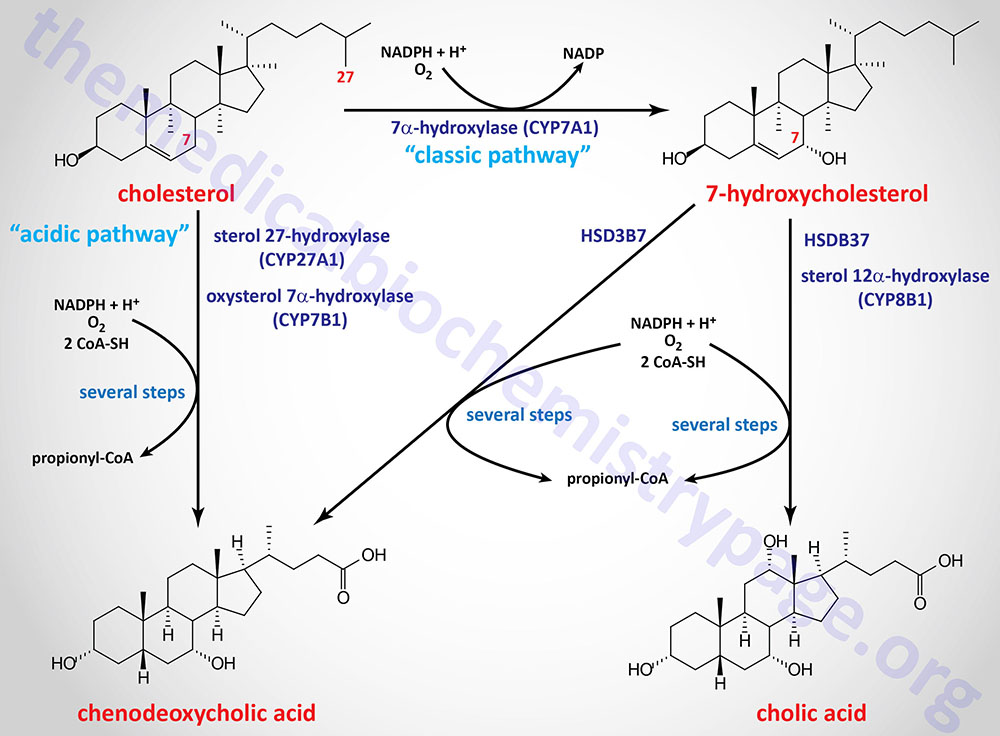
The CYP7A1 encoded enzyme is a member of the cytochrome P450 family of metabolic enzymes. This pathway is depicted in highly abbreviated fashion in the Figure below. The pathway initiated by CYP7A1 is referred to as the “classic” or “neutral” pathway of bile acid synthesis.
The CYP7A1 gene is located on chromosome 8q12.1 and is composed of 6 exons that encode a 504 amino acid protein. Expression of the CYP7A1 gene is essentially exclusive to the liver.
The hydroxyl group on cholesterol at the 3 position is in the β-orientation and must be epimerized to the α-orientation during the synthesis of the bile acids. This epimerization is initiated by conversion of the 3β-hydroxyl to a 3-oxo group catalyzed by 3β-hydroxy-Δ5-C27-steroid oxidoreductase (encoded by the HSD3B7 gene).The product of the action of the HSD3B7 catalyzed reaction on 7α-hydroxycholesterol is 7α-hydroxycholest-4-en-3-one.
That HSD3B7 catalyzed reaction is critical for bile acid synthesis and function is demonstrated in patients harboring mutations in the HSD3B7 gene. These individuals develop progressive liver disease that is characterized by cholestatic jaundice.
The HSD3B7 gene is located on chromosome 16p11.2 and is composed of 7 exons that generate three alternatively spliced mRNAs. These three mRNAs encode proteins of 369 amino acids (isoform a) and 196 amino acids (isoform b). The HSD3B7 encoded enzyme is a member of the short-chain dehydrogenase/reductase superfamily of enzymes that is composed of 77 proteins.
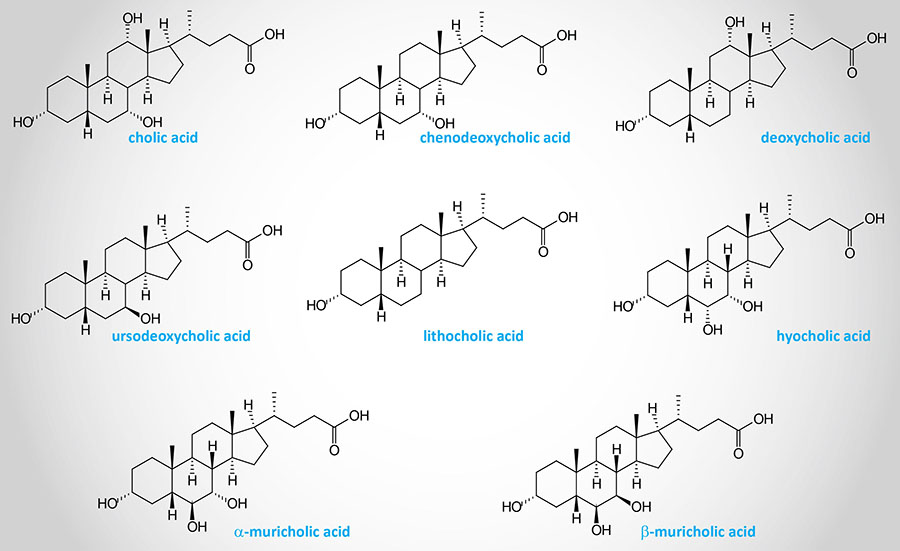
Following the action of HSD3B7 the bile acid intermediates can proceed via two pathways whose end products are the most abundant bile acids in human bile: chenodeoxycholic acid, CDCA (45%) and cholic acid, CA (31%). These are referred to as the primary bile acids. Chemically cholic acid is 3α,7α,12α-trihydroxy-5β-cholanoic acid and chenodeoxycholic acid is 3α,7α-dihydroxy-5β-cholanoic acid.
An additional primary bile acid is hyocholic acid, HCA. HCA is found at high concentrations in porcine species but is only found in humans at any significant level following gastric bypass surgery or in individuals with cholestasis.
Cholic Acid Pathway
The pathway to cholic acid involves the CYP8B1 encoded enzyme which is member of the CYP monooxygenase family of enzymes. The common name for the CYP8B1 encoded enzyme is sterol 12α-hydroxylase (also identified as 7α-hydroxy-4-cholesten-3-one 12α-hydroxylase). The product of this reaction is 4-cholesten-7α,12α-diol-3-one.
The distribution of CA and CDCA is determined by the activity of CYP8B1 gene. The product of the CYP8B1 encoded enzyme becomes CA, deoxycholic acid (DCA), and the conjugates of CA and DCA. When the 7α-hydroxycholest-4-en-3-one product of the HSD3B7 reaction is a substrate for the AKR1D1 encoded enzyme it will become CDCA, muricholic acid (MCA), and the conjugates of these bile acids. The activity of the CYP8B1 gene will, therefore, determine the ratio of CA to CDCA. As discussed below, the CYP8B1 gene is subject to regulation by bile acids themselves via their ability to regulate the action of the nuclear receptor, FXR.
Muricholic acid (MCA), in humans, exists primarily in two conformations, α-muricholic acid (αMCA) and β-muricholic acid (βMCA). Whereas muricholic acid is abundant in rodents it is not an abundant bile acid in humans. The sites of hydroxylation in αMCA are 3α, 6β, and 7α and in βMCA they are 3α, 6β, and 7β.
The activity of CYP8B1 is important for absorption of dietary lipids as demonstrated in CYP8B1 knockout mice. Lack of CYP8B1 results in impaired uptake of dietary cholesterol and triglycerides. In addition, these mice have reduced food intake resulting in decreased body weight.
The CYP8B1 gene is located on chromosome 3p22.1 and is the only CYP superfamily member gene that is intronless. The gene encodes an endoplasmic reticulum (ER)-localized enzyme composed of 501 amino acids.
Following the CYP8B1 reaction, the pathway to cholic acid involves at least 12 more reactions that are catalyzed by 9 different enzymes. These enzymes are encoded, in order, by the AKR1D1, AKR1C4 (aldo-keto reductase family 1 member C4), CYP27A1, SLC27A5, AMACR (alpha-methylacyl-CoA racemase), ACOX2 (acyl-CoA oxidase 2), HSD17B4 (hydroxysteroid 17β-dehydrogenase 4), SCP2 (sterol carrier protein 2) and ACOT8 (acyl-CoA thioesterase 8) genes.
The AKR1C4 encoded enzyme is commonly referred to as 3α-hydroxysteroid dehydrogenase type 1. The HSD17B4 encoded enzyme is more commonly identified as D-bifunctional protein, DBP. The SLC27A5 encoded protein is commonly known as acyl-CoA synthetase very long-chain family, member 6 (ACSVL6). The ACOX2 encoded enzyme is also involved in the process of peroxisomal β-oxidation of fatty acids.
Chenodeoxycholic Acid Pathway
The pathway to chenodeoxycholic acid involves the AKR1D1 (aldo-keto reductase family 1 member D1) encoded enzyme converting 7α-hydroxycholest-4-en-3-one to 7α-hydroxy-5β-cholestan-3-one. The AKR1D1 encoded enzyme is commonly called steroid 5β-reductase (also identified as 3-oxo-5β-steroid 4-dehydrogenase).
The pathway to cholic acid also involves the AKR1D1 encoded enzyme, which functions to convert the product of the CYP8B1 encoded enzyme (4-cholesten-7α,12α-diol-3-one) to 5β-cholesten-7α,12α-diol-3-one.
Following the AKR1D1 reaction, the pathway to chenodeoxycholic acid involves at least 11 more reactions that are catalyzed by 8 different enzymes that are the same as those involved in the cholic acid pathway. These enzymes are encoded, in order, by the AKR1C4 (aldo-keto reductase family 1 member C4), CYP27A1, SLC27A5, AMACR (alpha-methylacyl-CoA racemase), ACOX2 (acyl-CoA oxidase 2), HSD17B4 (hydroxysteroid 17β-dehydrogenase 4), SCP2 (sterol carrier protein 2) and ACOT8 (acyl-CoA thioesterase 8) genes.
Secondary and Tertiary Bile Acids
When the primary bile acids are excreted into the intestines they are acted upon by gut microbiota generating the secondary bile acids. In humans the secondary bile acid derived from CA is deoxycholic acid (DCA) and from CDCA is lithocholic acid (LCA).
Humans also produce a tertiary bile acid from the secondary bile acid, deoxycholic acid (DCA), identified as ursodeoxycholate (UDCA). UDCA is also referred to as ursodiol. Administration of UDCA to patients with primary biliary cirrhosis aids in the dissolution of gallstones that cannot be surgically removed and also helps delay the potential for liver damage in these patients.
Acidic Pathway of Bile Acid Synthesis
There is an alternative pathway of bile acid synthesis that involves hydroxylation of cholesterol at the 27 position by the mitochondrial enzyme, sterol 27-hydroxylase, which is encoded by the CYP27A1 gene. This alternative pathway is referred to as the “acidic” pathway of bile acid synthesis. Although in rodents the acidic pathway can account for up to 25% of total bile acid synthesis, in humans it accounts for no more than 6%.
Although the acidic pathway is not a major route for human bile acid synthesis it is an important one as demonstrated by the phenotype presenting in a newborn harboring a mutation in the CYP7B1 gene encoding the second enzyme of the acidic pathway. This infant presented with severe cholestasis (blockage in bile flow from liver) with cirrhosis and liver dysfunction.
The CYP27A1 gene is located on chromosome 2q35 and is composed of 9 exons that encode a 531 amino acid protein.
The primary product of the sterol 27-hydroxylase reaction, acting on cholesterol, is cholest-5-ene-3β,26-diol which is commonly designated (25R)-26-hydroxycholesterol (also erroneously referred to as 27-hydroxycholesterol). The CYP27A1 encoded enzyme can also generate 25-hydroxycholesterol (cholest-5-ene-3β,25-diol) and 24S-hydroxycholesterol (cholest-5-ene-3β,24S-diol; also known as cerebrosterol) from cholesterol.
The next reaction in the acidic pathway is catalyzed by the CYP7B1 encoded enzyme. This enzyme is commonly called oxysterol 7α-hydroxylase. The product of the CYP7B1 catalyzed reaction is 7α,27-dihydroxy-cholesterol. The rapid 7α-hydroxylation of the products of the CYP27A1 catalyzed reaction reduces their cytotoxicity as well as their regulatory effects on cholesterol and lipid homeostasis.
The CYP7B1 gene is located on chromosome 8q12.3 and is composed of 10 exons that generate two alternatively spliced mRNAs encoding proteins of 506 amino acids (isoform 1) and 460 amino acids (isoform 2).
The products of the oxysterol 7α-hydroxylase catalyzed reaction, primarily 7α,27-dihydroxy-cholesterol, are further converted to 3β,7α-dihydroxy-5-cholestenoic acid. The further conversion of 3β,7α-dihydroxy-5-cholestenoic acid to CDCA involves enzymes of the aldo-keto reductase family 1 family that includes AKR1D1 (steroid 5β-reductase) and AKR1C4 (3α-hydroxysteroid dehydrogenase).
Deficiency in sterol 27-hydroxylase (CYP27A1) is associated with a rare disorder, cerebrotendinous xanthomatosis (CTX), that is associated with excessive sterol deposition in tissue macrophages. This disorder is associated with an increased risk of developing premature atherosclerosis, despite normal circulating levels of cholesterol.
Although the CYP27A1 encoded enzyme was originally characterized in the liver within the context of bile acid synthesis, evidence has demonstrated a role for the enzyme in the sterol elimination pathway in macrophages and endothelial cells. Accumulation of excess cholesterol within intimal macrophages leads to the generation of lipid-laden foam cells. This process is a key early event in the development of atherosclerosis and helps explain the pathology associated with CTX.
Structures of Several Human Bile Acids
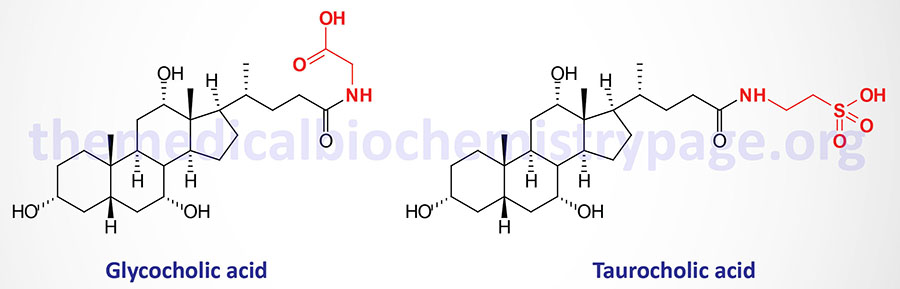
Bile Acids Conjugation
Before the primary bile acids are secreted into the canalicular lumen they are conjugated via an amide bond at the terminal carboxyl group with either of the amino acids glycine or taurine. These conjugation reactions yield the glycoconjugates and the tauroconjugates, respectively. This conjugation process increases the amphipathic nature of the bile acids making them much less toxic as well as more easily secreted into the bile canaliculi.
Conjugation of bile acids involves the sequential action of bile acid-CoA synthetase (BACS) and bile acid-CoA:amino acid N-acetyltransferase (encoded by the BAAT gene). The BACS enzyme is encoded by the SLC27A5 gene. The SLC27A5 encoded protein is a member of the very long-chain acyl-CoA synthetase family. The SLC27A5 encoded enzyme catalyzes the activation of bile acids via formation of bile acid-CoA thioesters which is the necessary first step for their subsequent conjugation with glycine or taurine.
Both primary bile acids [cholic acid (CA) and chenodeoxycholic acid (CDCA)] and secondary bile acids [deoxycholic acid (DCA) and lithocholic acid (LCA)] are the principal substrates for SLC27A5.
The SLC27A5 gene is located on chromosome 19q13.43 and is composed of 11 exons that generate two alternatively spliced mRNAs encoding proteins of 690 amino acids (isoform 1) and 606 amino acids (isoform 2). Expression of the SLC27A5 gene is essentially exclusive to the liver.
Conjugation of a bile acid-CoA with glycine or taurine is then catalyzed by the BAAT encoded enzyme. The BAAT gene is located on chromosome 9q31.1 and is composed of 6 exons that generate three alternatively spliced mRNAs, all of which encode the same 418 amino acid protein. Expression of the BAAT gene is highest in the liver with low levels of expression seen in the gallbladder as well.
Expression of both SLC27A5 gene and the BAAT genes is regulated by the nuclear receptor, FXR.
Humans generate several glycine and taurine conjugated bile acids derived from the primary, secondary, and tertiary bile acids. The glycine conjugated bile acids identified in humans include glycocholic acid (GCA or GlyCA), glycochenodeoxycholic acid (GCDCA or GlyCDCA), glycodeoxycholic acid (GDCA or GlyDCA), glycolithocholic acid (GLCA or GlyLCA), and glycoursodeoxycholic acid (GUDCA or GlyUDCA). The taurine conjugated bile acids are TCA (or TaurCA), TCDCA (or TaurCDCA), TDCA (or TaurDCA), TLCA (or TaurLCA), and TUDCA (or TaurUDCA).
Hepatic Secretion of Bile Acids
Bile salts are secreted from hepatocytes, into the bile canaliculi, via the action of the bile salt export protein (BSEP) in conjunction with MRP2 (multidrug resistance associated protein 2, MRP2). BSEP is a member of the ATP-binding cassette (ABC) family of transporters and as such is encoded by the ABCB11 gene. MRP2 is also a member of the ABC family of transporters and as such is encoded by the ABCC2 gene.
The ABC11 gene is located on chromosome 2q31.1 and is composed of 30 exons that encode a 1321 amino acid protein.
The ABCC2 gene is located on chromosome 10q24.2 and is composed of 34 exons that encode a 1545 amino acid protein.
Transport of phospholipids into the canaliculi requires the transporter ABCB4. ABCB4 is also known as multi-drug resistance protein 3 (MDR3, a member of the P-glycoprotein family of transporters).
The ABCB4 gene is located on chromosome 7q21.12 and is composed of 34 exons that generate three alternatively spliced mRNAs, each of which encode distinct protein isoforms.
Some free cholesterol is also transported out of hepatocytes into the canaliculi via the action of the obligate heterodimeric transporter ABCG5/ABCG8. The transport of cholesterol via this complex also requires ABCB4. The ABCB4 requirement is consistent with the known actions of phospholipids in the bile canaliculi functioning as a sink to accept cholesterol transported out by ABCG5/ABCG8. Each of these hepatic lipid transporters is critical for normal hepato-biliary function since mutations in any of the genes encoding the transporters have been shown to be associated with cholestatic liver diseases. These transport defects result in the accumulation of toxic levels of bile salts within the hepatocytes resulting in liver failure.
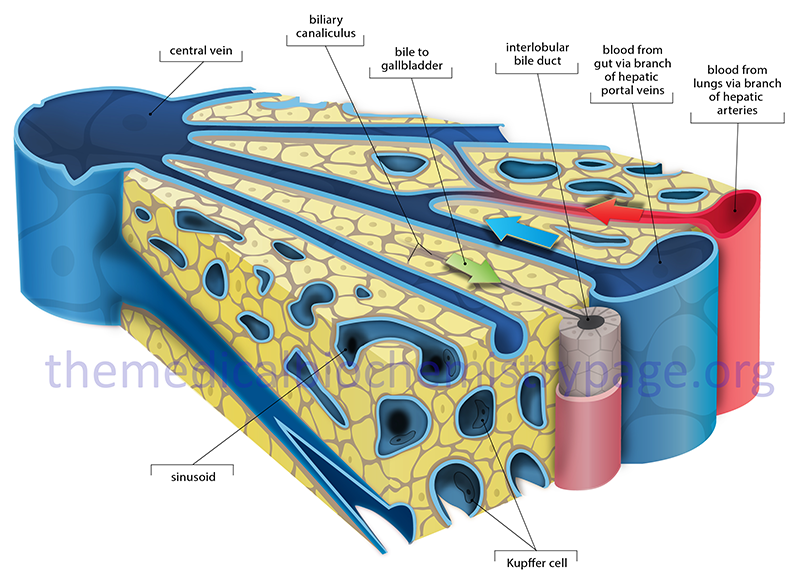
The mixture of bile salts, phospholipids and cholesterol is then transported, via the canaliculi, into the gallbladder, where they are concentrated to form bile. The composition of bile is 85% water, 67% bile salts, 22% phospholipids, and 4% cholesterol. In addition, bile contains electrolytes, minerals, minor levels of proteins, plus bilirubin and biliverdin pigments. The bilirubin and biliverdin are what impart the yellow-green or even orange hue to bile. The primary role of bile salt in the bile contained in the gallbladder is to solubilize cholesterol, thereby preventing cholesterol crystallization and the formation of cholesterol calculi (gallstones).
Gallbladder Excretion of Bile
Following the consumption of lipid in the diet, enteroendocrine I cells in the duodenum secrete the hormone cholecystokinin (CCK) into the circulation. The release of CCK, and its subsequent binding to receptors on the gallbladder, promotes contraction of smooth muscle cells of the gallbladder and relaxation of the sphincter of Oddi, resulting in the pulsatile secretion of bile into the duodenum.
Within the lumen of the duodenum the bile salt-containing mixed micelles facilitate absorption of the fat-soluble vitamins A, D, K, and E and the digestion of dietary lipids by pancreatic enzymes. Although the gallbladder stores bile, as an organ it is not essential since patients who have undergone a cholecystectomy (gallbladder removal), are still able to absorb lipids from the diet as a result of direct secretion of bile into the duodenum.
Bile Reabsorption: Enterohepatic Circulation
Once bile salts are secreted into the duodenum and carry out their emulsification role, around 95% are reabsorbed into the distal ileum. Bile salt reabsorption occurs via the apical sodium-dependent bile transporter (ASBT) present in the brush border (apical) membrane of the intestinal enterocyte. The ASBT protein is a member of the solute carrier (SLC) family of transporters and is encoded by the SLC10A2 gene.
Ileal bile acid-binding protein (IBABP) is thought to be involved in the transport of bile salts across the enterocyte cytosol to the basolateral membrane. IBABP is a member of the fatty acid-binding (FABP) family of proteins and as such is encoded by the FABP6 (fatty acid-binding protein subclass 6) gene.
Once bile salts reach the basolateral membrane of enterocytes they are transported (effluxed) out to the blood by the heterodimeric transporter composed of the proteins identified as OSTα and OSTβ (organic solute transporters). Both of these proteins are members of the solute carrier (SLC) family of transporters. The OSTα protein is encoded by the SLC51A gene and the OSTβ protein is encoded by the SLC51B gene.
A percentage of the bile salts are not reabsorbed and undergo deconjugation by intestinal microbiota before either being absorbed or converted into secondary and tertiary bile acids.
Anaerobic bacteria present in the colon modify the primary bile acids converting them to the secondary bile acids, where the major secondary bile acids are identified as deoxycholate (DCA from cholate) and lithocholate (LCA, from chenodeoxycholate). These secondary bile acids are either passively absorbed from the colon or they are excreted in the feces. Intestinal bacteria also generate the major tertiary bile acid from deoxycholic acid called ursodeoxycholate (UDCA).
The absorbed primary and secondary bile acids, as well as other bile salts, are transported from the portal circulation back to the liver where most, but not all, are actively transported into hepatocytes by sodium (Na+)-taurocholate co-transporting polypeptide (NTCP) and organic anion transporters (OATP) that mediate the uptake of bile salts (conjugated) and bile acids (unconjugated), respectively.
The NTCP transporter is encoded by the SLC10A1 gene.
The OATP proteins are encoded by genes of the SLC21 family, all of which are designated by the SLCO acronym. The liver-specific OATP transporters are encoded by the SLCO1B1 and SLCO1B3 genes whose encoded proteins are identified as OAT1B1 and OAT1B3, respectively. The OAT1B1 protein was originally identified as OATPC.
Once in the liver the bile acids are re-conjugated and then re-secreted together with newly synthesized bile acids. This overall process constitutes one cycle of what is called the enterohepatic circulation. When LCA is returned to the liver it undergoes a sulfation reaction and is subsequently excreted in the feces.
The bile acid pool contains about 2–4 gm of bile acids and this pool is recycled via the enterohepatic circulation on the order of six to ten times each day. Of the total bile salt pool, around 0.2–0.6 gm are excreted in the feces each day. This lost fraction of bile salts is replenished via de novo hepatic bile acid synthesis from cholesterol.
Regulation of Bile Acid Homeostasis
Bile acids, in particular chenodeoxycholic acid (CDCA) and cholic acid (CA), can regulate the expression of genes involved in their synthesis, thereby, creating a feed-back loop. The elucidation of this regulatory pathway came about as a consequence of the isolation of a class of receptors called the farnesoid X receptors, FXR. The FXRs belong to the superfamily of nuclear receptors that includes the steroid/thyroid hormone receptor family as well as the liver X receptors (LXR), retinoid X receptors (RXR), and the peroxisome proliferator-activated receptors (PPAR).
There have been two distinct FXR characterized and they are identified as FXRα and FXRβ. Since these are nuclear receptors the genes encoding these receptors utilize the nuclear receptor nomenclature with FXRα being NR1H4 (nuclear receptor subfamily 1, group H, member 4) gene. In humans, the gene (NR1H5P) encoding FXRβ is likely to be a pseudogene.
The human NR1H4 gene encodes for at least four FXRα isoforms as a result of activation from different promoters and the use of alternative splicing. These four isoforms are identified as FXRα1, FXRα2, FXRα3, and FXRα4. The NR1H4 gene is expressed at highest levels in the intestines and the liver.
Like all receptors of this superfamily, ligand binds the receptor in the cytoplasm and then the complex migrates to the nucleus and forms a heterodimer with other members of the family. FXR forms a heterodimer with members of the RXR family. Following heterodimer formation the complex binds to specific sequences in target genes called FXR response elements (FXREs) resulting in regulated expression. One major target of FXR is the small heterodimer partner (SHP) gene. Activation of SHP expression by FXR results in inhibition of transcription of SHP target genes. Of significance to bile acid synthesis, SHP represses the expression of the cholesterol 7α-hydroxylase gene (CYP7A1). CYP7A1 is the rate-limiting enzyme in the synthesis of bile acids from cholesterol via the classic pathway.
In the Ayurvedic tradition of medicine, any resin that is collected by tapping the trunk of a tree is called guggul (or guggal). The cholesterol lowering action of the guggul from the Mukul myrrh tree (Commiphora mukul) of India is that a lipid component of this extract called guggulsterone (also called guggul lipid) is an antagonist of FXR. However, in addition to its effects on FXR function, guggulsterone has been shown to activate the pregnane X receptor (PXR) which is another member of the nuclear receptor superfamily. PXR is a recognized receptor for lithocholic acid and other bile acid precursors. PXR activation leads to repression of bile acid synthesis due to its physical association with hepatocyte nuclear factor 4α (HNF-4α) causing this transcription factor to no longer be able to associate with the transcriptional co-activator PGC-1α (PPARγ co-activator 1α) which ultimately leads to loss of transcription factor activation of CYP7A1.
The expression of other genes involved in bile acid synthesis is also regulated by FXR action. The action of FXR can either be to induce or repress the expression of these genes. Genes that are repressed in addition to CYP7A1 include SREBP-1c, CYP8B1 (encoding sterol 12α-hydroxylase1), and SLC10A1 ([encoding solute carrier family 10 (sodium/bile acid cotransporter family), member 1]. This latter gene is identified as the Na+-taurocholate co-transporting polypeptide (NTCP). NTCP is involved in hepatic uptake of bile acids through the sinusoidal/basolateral membrane. Thus bile acid-mediated repression of NTCP gene expression would reduce uptake of bile acids and protect the liver from the toxic effects of excess bile acid accumulation.
Bile acids repress the transcription of another bile acid transporter that is expressed in the sinusoidal/basolateral membrane. This transporter is Na+-independent and is called the organic anion transporting polypeptide 1B1 (OATP1B1). OATP1B1 is encoded by the SLCO1B1 gene. OATP1B1 was formerly identified as OATP-C. The effect of bile acids on OATP1B1 expression is indirect and involves SHP-mediated reduction in HNF-4α activity which in turn reduces the expression of another liver-enriched transcription factor HNF-1α which is the major activator of the SLO1B1 gene.
Genes that, in addition to SHP, are induced by FXR include ABCB11 (encoding liver bile salt export pump, BSEP), ABCB4 (encoding multidrug resistance protein 3, MDR3), and ABCC2 (encoding multidrug resistance associated protein 2, MRP2). The latter two genes are involved in export of organic compounds and were identified based upon their ability to transport drugs out of cells thereby, allowing the cells to resist the intended effects of the administered drug.
The normal function of MDR3, which is a member of the ATP-binding cassette (ABC) family of transporters, is the translocation of phospholipids through the canalicular membrane of hepatocytes. Thus, it is inferred that the bile acid-mediated increase in MDR3 expression is necessary to allow hepatocytes to respond to bile acid toxicity via the formation of cholesterol, phospholipid, and bile acid containing micelles.
BSEP is also a member of the ABC family of transporters and it is involved in the process of exporting bile acids out of hepatocytes thus reducing their toxicity to these cells. Although guggulsterones antagonize the actions of FXR, which would lead to a reduction in bile acid export, these lipids have been shown to activate the expression of BSEP through an FXR-independent mechanism. This latter effect likely explains the cholesterol lowering benefits attributed to these compounds.
Of major clinical significance is that many of the FXR target genes have been implicated in several inherited cholestatic liver disorders. Mutations in the genes encoding BSEP and MDR3 are associated with progressive familial intrahepatic cholestasis type 2 and 3 (PFIC2 and PFIC3), respectively. Mutations in the gene encoding MRP2 are associated with Dubin-Johnson syndrome, a form of inherited hyperbilirubinemia.
Bile Acids and FGF19
The fibroblast growth factor family of growth factors and hormones currently consists of 18 members in humans that function by binding to proteins of the FGF receptor family. These proteins are divided into six subfamilies with subfamily 6 containing FGF19, FGF21 and FGF23 (commonly referred to as the FGF19 subfamily). All three members of the FGF19 subfamily function as endocrine hormones. Some confusion can arise given the fact that the mouse ortholog of the human FGF19 gene is identified as FGF15.
FGF19 subfamily members have weak affinity for heparan sulfate proteoglycans (HSPG) which allows them to escape from the extracellular compartment into circulation enabling them to function as endocrine hormones. Other members of the FGF family utilize their interactions with HSPG to form high-affinity interactions with FGF receptors (FGFR). However, since the FGF19 subfamily members have low affinity for HSPG they interact with the single-transmembrane containing Klotho proteins to facilitate their interactions with FGFR. There are two related Klotho proteins, αKlotho and βKlotho. FGF19 can interact with both Klotho co-receptor proteins.
FGF19 shows preference for activation of FGF receptor 4 (FGFR4) but via its Klotho interactions can also activate FGFR1. FGF21 selectively use βKlotho as its co-receptor, whereas, FGF23 uses αKlotho. Expression of the FGF19 gene occurs in numerous tissues but the highest levels of expression are found in the small intestine, gallbladder, and brain.
FGF19 Regulation of and by Bile Acids
Intestinal FGF19 plays a critical role in the overall processes of the enterohepatic circulation. It carries out this role by regulation of hepatic bile acid synthesis and bile acid delivery to the gallbladder. Expression of FGF19 in the gallbladder is important in the regulation of overall gallbladder function. The ability of intestinal FGF19 to regulate overall bile acid homeostasis is controlled by bile acids themselves. This process represents an intricate mechanism for feedback regulation of bile acid synthesis involving communication from the small intestine to the liver through the actions of FGF19.
When bile acids are secreted by the gallbladder, these compounds can be absorbed by ileal enterocytes. Within the enterocytes the bile acids bind to, and activate, the nuclear receptor, farnesoid X receptor: FXR. Bile acid activation of FXR leads to the enhanced expression of numerous genes in the intestinal enterocyte, with the FGF19 gene being one of these FXR targets. There are at least four FXR-binding sites in the promoter region of the FGF19 gene allowing it to be induced on the order of 300-fold via FXR activation. Activation of the FGF19 gene results in the protein being secreted into the enterohepatic circulation.
When the FGF19 reaches the liver, it interacts with the βKlotho/FGFR4 complex and triggers activation of a signal transduction cascade that ultimately alters the expression of several genes. The net result of FGF19 signaling in the liver is transcriptional repression of the CYP7A1 gene, which encodes the rate-limiting enzyme in the “classic pathway” of bile acid synthesis.
In addition to the regulation of bile acid synthesis through repression of CYP7A1, FGF19 signaling in the liver evokes changes in a wide range of other metabolic pathways, including cholesterol, lipid, glucose, amino acid, and nucleotide metabolism as well as affecting RNA metabolism and inflammatory processes.
FGF19 signaling prevents lipid and glucose accumulation in the liver by inducing fatty acid oxidation, in part by decreasing expression of the acetyl-CoA carboxylase 2 (ACC2) gene. The ACC2 protein is normally closely associated with, and inhibits, the outer mitochondrial membrane associated enzyme, carnitine palmitoyl transferase 1 (CPT1) which is involved in long-chain fatty acid oxidation.
With respect to hepatic lipid homeostasis, FGF19 inhibits lipid synthesis by counteracting the insulin-induced increase of sterol regulatory element-binding protein-1c (SREBP-1c) expression. SREBP-1c, a member of the SREBP family of transcription factors, is a key transcriptional activator of numerous genes involved in lipogenesis.
Regulation of hepatic glucose homeostasis by FGF19 signaling involves stimulation of glycogen synthesis and inhibition of gluconeogenesis via inactivation of the transcription factor, cAMP response element-binding protein (CREB; encoded by the CREB1 gene). Decreased CREB activity results in decreased expression of a key transcriptional co-activator, peroxisome proliferator-activated receptor gamma (PPARγ) coactivator-1 alpha (PGC-1α) The effects of FGF19 on hepatic carbohydrate homeostasis have significant clinical implications in the control of glucose homeostasis and insulin sensitivity in obese and type 2 diabetes.
Bile Acids as Metabolic Regulators
Bile acids were originally identified as being involved in four primary physiologically significant functions:
- their synthesis and subsequent excretion in the feces represent the only significant mechanism for the elimination of excess cholesterol.
- bile acids and phospholipids solubilize cholesterol in the bile, thereby preventing the precipitation of cholesterol in the gallbladder.
- they facilitate the digestion of dietary triacylglycerols by acting as emulsifying agents that render fats accessible to pancreatic lipases.
- they facilitate the intestinal absorption of fat-soluble vitamins.
However, over the past several years new insights into the biological activities of the bile acids have been elucidated. Recent findings have demonstrated that bile acids are involved in the control of their own metabolism and transport via the enterohepatic circulation, regulate lipid metabolism, regulate glucose metabolism, control signaling events in liver regeneration, and the regulation of overall energy expenditure.
Following the isolation and characterization of the farnesoid X receptors (FXR), for which the bile acids are physiological ligands, the functions of bile acids in the regulation of lipid and glucose homeostasis has begun to emerge. As indicated above, the binding of bile acids to FXR results in the attenuated expression of several genes involved in overall bile acid homeostasis (e.g. FGF19). Chenodeoxycholic acid (CDCA) exhibits the highest affinity for FXR followed by DCA, LCA, and CA.
However, genes involved in bile acid metabolism are not the only ones that are regulated by FXR action as a consequence of binding bile acid. In the liver, FXR action is known to regulate the expression of genes involved in lipid metabolism (e.g. SREBP-1c), lipoprotein metabolism (e.g. apoC-II), glucose metabolism (e.g. PEPCK), and hepatoprotection (e.g. CYP3A4, which was originally identified as nifedipine oxidase; nifedipine being a member of the calcium channel blocker drugs).
In addition to their roles in lipid emulsification in the intestine and activating FXR, the bile acids participate in various signal transduction processes via activation of the c-JUN N-terminal kinase (JNK) as well as the mitogen-activated protein kinase (MAPK) pathways.
Other members of the nuclear receptor family that are activated by bile acids are the pregnane X receptor (PXR), the constitutive androstane receptor (CAR), and the vitamin D receptor (VDR). Lithocholic acid (LCA) is the primary bile acid that binds to and activates these three nuclear receptors.
An additional receptor activated in response to bile acids, that may have implications for control of obesity, is the G-protein coupled bile acid receptor 1, GPBAR1 (originally identified as TGR5 and also known as GPR131). Activation of GPBAR1 in brown adipose tissue results in activation of uncoupling protein 1, UCP1 (thermogenin) leading to enhanced energy expenditure. Lithocholic acid (LCA) exhibits the highest affinity for GPBAR1 binding followed by DCA, CDCA, and CA.
Bile acids have also been shown to activate one of the sphingosine-1-phosphate (S1P) receptors, specifically S1PR2. The S1PR2 binds taurocholic acid (TCA) which enhances insulin-mediated signaling processes.
Given that bile acids are released to the circulation, similar to many other classical steroid hormones, coupled with the fact that these circulating bile acids bind to cell surface receptors, as well as intracellular receptors of the nuclear receptor family, has led to the classification of these molecules as bilokines.
Bile acids have also been shown to regulate the activity of certain enzymes in a manner suggestive of allosteric regulation. Deoxycholic acid (DCA), lithocholic acid (LCA), and chenodeoxycholic acid (CDCA) have been shown to bind to ac regulate the activity of the enzyme, N-acyl phosphatidylethanolamine-specific phospholipase D (NAPE-PLD). LCA binding inhibits the enzyme whereas DCA and CDCA binding activates the enzyme. NAPE-PLD is involved in the synthesis of the endocannabinoid, anandamide (N-arachidonoylethanolamine) and also in the synthesis of oleoylethanolamide (OEA; also identified as N-oleoylethanolamine) from oleic acid. Both anandamide and OEA are considered bioactive lipids and both are involved in the regulation of feeding behaviors.
Role of Bile Acids in White Adipose Tissue
White adipose tissue (WAT) represents a major target for the bilokine-related activity of bile acids. The effects of bilokines on adipose tissue involves both the GPBAR1 encoded GPCR as well as the nuclear receptor, FXR.
Bile acid activation of FXR in adipose tissue plays an important role in overall adipocyte differentiation. This has been identified in mice in which the FXR gene has been knocked out. FXR-deficient mice have an overall reduced mass of WAT and reduced levels of leptin production. As a result of these effects of FXR-deficiency of WAT development there are numerous systemic effects that includes elevated levels of plasma free fatty acid (FFA), impaired glucose tolerance and reduced whole-body glucose disposal as well as increased insulin resistance.
Bile acid effects, specifically CDCA effects, in WAT that are dependent on binding to and activation of GPBAR1 include the stimulation of the expression of leptin and fatty acid binding protein 4 (FABP4). In contrast, CDCA induces expression of PPARγ and lipoprotein lipase (LPL) independent of GPBAR1 signaling.
Additional effects of bile acids on adipose tissue include modulation of inflammatory processes. For example, in adipocytes in culture CDCA has been shown to suppress the expression of adipokines that exert proinflammatory
effects. These adipokines include resistin, MCP-1, vaspin, RBP4, PAI, chemerin, IL-6, and TNF. In contrast, CDCA has been shown to induce the anti-inflammatory adipokine, adiponectin.
Inborn Errors in Bile Acids Synthesis
Metabolic disorders associated with bile acid synthesis and metabolism are broadly classified as primary or secondary disorders. Primary disorders involve inherited deficiencies in enzymes responsible for catalyzing key reactions in the synthesis of cholic and chenodeoxycholic acids.
The enzyme encoded by the HSD17B4 (hydroxysteroid 17β dehydrogenase 4) gene, which is commonly called D-bifunctional protein (DBP), is involved in not only bile acid synthesis but also in peroxisomal β-oxidation of certain fatty acids. Mutations in the HSD17B4 gene are associated with the disorder referred to as D-bifunctional protein deficiency.
Bile acid disorders classified as secondary refer to metabolic defects that impact primary bile acid synthesis but that are not due to defects in the enzymes responsible for synthesis. Secondary disorders of bile acid metabolism include peroxisomal disorders such as Zellweger syndrome and related peroxisomal biogenesis disorders and Smith-Lemli-Opitz syndrome which results from a deficiency of 7-dehydrocholesterol reductase (DHCR7). Shown in the Table below are six of the known primary disorders of bile acid metabolism.
Table of Primary Disorders of Bile Acid Synthesis
| Affected Enzyme | Gene Symbol | Phenotype / Comments |
| cholesterol 7α-hydroxylase | CYP7A1 | catalyzes the rate-limiting reaction of the classic pathway of bile acid synthesis; no liver dysfunction; clinical phenotype manifests with markedly elevated total cholesterol as well as LDL, premature gallstones, premature coronary and peripheral vascular disease; elevated serum cholesterol is not responsive to statin drug therapy |
| sterol 27-hydroxylase | CYP27A1 | catalyzes the initial reaction of the acidic pathway of bile acid synthesis; deficiency results in cerebrotendinous xanthomatosis (CTX) which is associated with progressive neurological dysfunction, neonatal cholestasis, bilateral cataracts, and chronic diarrhea |
| oxysterol 7α-hydroxylase | CYP7B1 | second enzyme of the acidic pathway of bile acid synthesis; a single case was reported in 1998 of a 10-week-old boy presenting with severe progressive cholestasis, hepatosplenomegaly, cirrhosis, and liver failure; serum ALT and AST were markedly elevated; recently several individuals suffering from autosomal recessive hereditary spastic paraplegia 5A (SPG5A) have been shown to harbor mutations in the CYP7B1 gene although the number of cases only represents around 1% of all SPG cases |
| 3β-hydroxy-Δ5-C27-steroid oxidoreductase | HSD3B7 | functions in both the classic and the acidic pathways of bile acid synthesis; most commonly reported defect in bile acid synthesis; heterogeneous clinical presentation that includes progressive jaundice, hepatomegaly, pruritus, malabsorption with resultant steatorrhea (fatty diarrhea), fat soluble vitamin deficiency, and rickets |
| Δ4-3-oxosteroid 5β-reductase | AKR1D1 | AKR1D1 refers to aldo-keto reductase family 1 member D1; encoded protein is often referred to as simply 5β-reductase; functions in the synthesis of both cholic and chenodeoxycholic acid; similar clinical manifestation to HSD3B7 deficiency although with earlier presentation; afflicted infants will have a more severe liver disease with rapid progression to cirrhosis and death if no clinical intervention is undertaken; liver function tests will show marked elevation in AST and ALT; serum tests show elevated conjugated bilirubin, coagulopathy will also be evident |
| 2-methylacyl-CoA racemase | AMACR | enzyme is also called alpha-methylacyl-CoA racemase; functions in the synthesis of both cholic and chenodeoxycholic acid; first reported in 3 adults who presented with a sensory motor neuropathy; symptoms can include migraines, seizures, visual failure, and spasticity; defect was also found in a 10-week-old infant who had severe fat-soluble vitamin deficiencies, hematochezia (passage of bright red stool), and mild cholestatic liver disease |