

Last Updated: July 13, 2026
Introduction to Andersen Disease
Glycogen storage disease type 4 (GSD4) is more commonly known as Andersen disease or also as amylopectinosis. Andersen disease is inherited as an autosomal recessive disorder. This disease was originally described by the American pediatrician and pathologist, Dorothy Hansine Andersen in 1956, hence the association of her name with the disease.
The disease was seen in a patient exhibiting progressive hepatosplenomegaly along with the storage of an abnormal glycogen that had poor solubility in the liver. The abnormal glycogen had few branch points with long outer chains containing more α-1,4-linked glucose than the normal polysaccharide which contains both α-1,4- and α-1,6-glycosidic linkages. This resultant structure was similar to that of amylopectin (the structure of starch in plants), thus the associated name of amylopectintosis. Because of the structure of the glycogen it was suspected that there was a deficiency in glycogen branching enzyme activity. This was indeed found to be true in 1966.
Molecular Biology of Andersen Disease
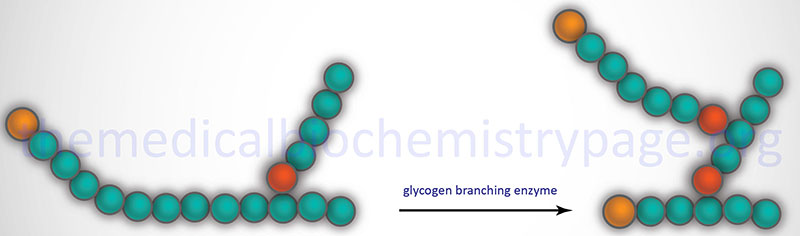
Glycogen branching enzyme, also called amylo-(1,4 to 1,6) transglycosylase, is a monomeric protein. The α-1,6 branches in glucose are produced by this enzyme through a process involving the transfer of a terminal fragment of 6-7 glucose residues (from a polymer at least 11 glucose residues long) to an internal glucose residue at the C-6 hydroxyl position.
Glycogen branching enzyme is encode by the GBE1 (1,4-alpha-glucan branching enzyme 1) gene. The GBE1 gene is located on chromosome 3p12.2 and is composed of 17 exons that encode a protein of 702 amino acids.
Several missense and nonsense variants have been identified in the GBE1 gene in patients with Andersen disease. A lethal form of Andersen disease has been associated with a deletion of 210 nucleotides from the GBE1 gene.
Clinical Features of Andersen Disease
Analysis of pathogenic variants in the branching enzyme gene demonstrated that both hepatic and neuromuscular forms of GSD4 were the result of defects in the same gene. Classic Andersen disease is a rapidly progressive disorder that will result in early lethality as a result of liver failure. Liver transplantation is the only means of survival with this form of the disease.
Other pathogenic variants in the GBE1 gene have been identified in patients with the milder non-progressive hepatic form of the disease. In addition to the classic lethal form of Andersen disease and the milder hepatic form, several other variant forms of the disease have been reported. There is a variant form that consists of multi-system involvement including skeletal and cardiac muscle, nerve and liver. There is a juvenile polysaccharidosis form that manifests with multi-system involvement but patients with this form have normal GBE1 enzyme activity. There is a fetal neuromuscular form that results in death within the first few months of life that results from a splice site variant in the GBE1 gene.
The clinical presentation of the classic form of Andersen disease usually occurs in the first few months of life and is characterized by hepatosplenomegaly and failure to thrive. The disease progresses to liver cirrhosis, portal vein hypertension, esophageal varices (dilated sub-mucosal veins in the lower third of the esophagus), and ascites. Death will usually ensue by 5 years of age due to liver failure.
Because of the similarities in symptoms between Andersen disease and other causes of cirrhosis in infancy it is necessary to carry out a biopsy of the liver and examine for the presence of the associated abnormal glycogen. In addition, assay for branching enzyme deficiency in muscle, leukocytes, erythrocytes, or fibroblasts can be carried out to determine the exact defect resulting in the hepatomegaly.
Treatment of Andersen Disease
Treatment of Andersen disease normally involves maintenance of normal blood glucose along with adequate nutrient intake, both of which will improve liver function and muscle strength. In cases of progressive liver failure, liver transplant is the only effective option.