
Last Updated: March 6, 2026
Introduction AMP-Activated Protein Kinase (AMPK)
AMP-activated protein kinase (AMPK) was first discovered as an activity that inhibited preparations of acetyl-CoA carboxylase (ACC) and 3-hydroxy-3-methylglutaryl-CoA reductase (HMG-CoA reductase, HMGR) and was induced by AMP. AMPK induces a cascade of events within cells in response to the ever changing energy charge of the cell. The role of AMPK in regulating cellular energy charge places this enzyme at a central control point in maintaining energy homeostasis. More recent evidence has shown that AMPK activity can also be regulated by physiological stimuli, independent of the energy charge of the cell, including hormones and nutrients.
Once activated, AMPK-mediated phosphorylation events switch cells from active ATP consumption (e.g. fatty acid and cholesterol biosynthesis) to active ATP production (e.g. fatty acid and glucose oxidation). These events are rapidly initiated and are referred to as short-term regulatory processes. The activation of AMPK also exerts long-term effects at the level of both gene expression and protein synthesis. Other important activities attributable to AMPK are regulation of insulin synthesis and secretion in pancreatic islet β-cells and modulation of hypothalamic functions involved in the regulation of satiety. How these latter two functions impact obesity and diabetes will be discussed below.
Structure of AMPK
The human AMPK is a trimeric enzyme composed of a catalytic α subunit and the non-catalytic β and γ subunits. There are two genes encoding isoforms of both the α and β subunits (α1, α2, β1 and β2) and three genes encoding isoforms of the γ subunit (γ1-γ3). The gene encoding the α1 protein is PRKAA1, the α2 protein is PRKAA2, the β1 protein is PRKAB1, the β2 protein is PRKAB2, the γ1 protein is PRKAG1, the γ2 protein is PRKAG2, and the γ3 protein is PRKAG3.
The PRKAA1 gene is located on chromosome 5p13.1 and is composed of 12 exons that generate eight alternatively spliced mRNAs that collectively encode six protein isoforms.
The PRKAA2 gene is located on chromosome 1p32.2 and is composed of 10 exons that encode a 552 amino acid protein.
The PRKAB1 gene is located on chromosome 12q24.23 and is composed of 8 exons that encode a 270 amino acid protein.
The PRKAB2 gene is located on chromosome 1q21.1 and is composed of 9 exons that generate three alternatively spliced mRNAs, only one of which is protein coding generating a protein of 272 amino acids. The other two PRKAB2 derived mRNAs are likely candidates for nonsense-mediated decay, NMD.
The PRKAG1 gene is located on chromosome 12q13.12 and is composed of 13 exons that generate three alternatively spliced mRNAs encoding γ1 isoform 1 (331 amino acids), γ1 isoform 3 (340 amino acids), and γ1 isoform 4 (299 amino acids).
The PRKAG2 gene is located on chromosome 7q36.1 and is composed of 22 exons that generate 24 alternatively spliced mRNAs encoding 21 distinct isoforms.
The PRKAG3 gene is located on chromosome 2q35 and is composed of 14 exons that encode a 489 amino acid protein.
Given that there are multiple genes encoding each of the subunits of AMPK it is possible that 12 different isoforms of the hetertrimeric enzyme can be formed. The α2 isoform is the subunit of AMPK found predominantly within skeletal and cardiac muscle, whereas, approximately equal distribution of both the α1 and α2 isoforms are present in hepatic AMPK. Within pancreatic islet β-cells the α1 isoform predominates which is also the case for white adipose tissue. The expression of specific γ subunits also exhibits tissue specificity with the γ3 subunit found almost exclusively in glycolytic skeletal muscle.
The N-terminal half of the α subunits contains a typical serine/threonine kinase catalytic domain. Interaction with the β and γ subunits occurs via the C-terminal half of the α subunits. The yeast AMPK β subunits are lipid modified with myristic acid. Myristoylation may account for the membrane association of mammalian AMPK. The core of the β subunits have a glycogen-binding domain (GBD). This domain is closely related to the isoamylase N domain subfamily and weakly related to domains in the glycogen-targeting phosphatase subunits and several starch-binding proteins. The close proximity of AMPK to cellular glycogen stores allows it to rapidly effect changes in glycogen metabolism in response to changes in metabolic demands.
The γ subunits of AMPK have been shown to contain four tandem repeats that form nucleotide binding sites with similarity to cystathionine β-synthase which are, therefore, referred to as CBS domains. Indeed, direct AMP-binding studies have shown that AMP is bound to the γ subunits by a pair of CBS domains. The CBS domains also bind ATP and the binding of AMP and ATP to AMPK occurs in a mutually exclusive manner. Binding of ATP keeps the activity of AMPK low and when AMP levels rise the exchange of AMP for ATP results in a 5-fold increase in kinase activity (see below).
Of clinical significance is the observation that mutations in the CBS domains of the γ2 subunit (gene symbol PRKAG2) are associated with Wolff-Parkinson-White syndrome and familial hypertrophic cardiomyopathy. An additional inherited disorder associated with mutations in the PRKAG2 gene is a severe cardiac condition called lethal congenital glycogen storage disease of the heart. This disorder is caused by a single amino acid substitution of glutamine for arginine at position 531 (R531Q).
Regulation of AMPK Activation
In the presence of AMP the activity of AMPK is increased approximately 5-fold. However, more importantly is the role of AMP in regulating the level of phosphorylation of AMPK. An increased AMP to ATP ratio leads to a conformational change in the γ-subunit leading to increased phosphorylation and decreased dephosphorylation of AMPK. The phosphorylation of AMPK results in activation by at least 100-fold. AMPK is phosphorylated by at least two different upstream AMPK kinases (AMPKK).
Phosphorylation of AMPK occurs in the α subunits at a critical threonine residue that resides in the activation loop of this subunit. The designation of this regulatory phosphorylation site as threonine 172 (T172) stems from its location in the rat AMPK α-subunit but does not precisely occur at this location in the human α-subunit proteins.
One kinase activator of AMPK is Ca2+-calmodulin-dependent kinase kinase β (CaMKKβ) which phosphorylates and activates AMPK in response to increased calcium. The distribution of CaMKKβ expression is primarily in the brain with detectable levels also found in the testes, thymus, and T cells. As described for the Ca2+-mediated regulation of glycogen metabolism, increased release of intracellular stores of Ca2+ create a subsequent demand for ATP. Activation of AMPK in response to Ca2+ fluxes provides a mechanism for cells to anticipate the increased demand for ATP.
Evidence has also demonstrated that the serine-threonine kinase, LKB1 (liver kinase B1) is required for activation of AMPK in response to stress. LKB1 is also known as serine-threonine kinase 11 (STK11) and as the Peutz-Jeghers syndrome (PJS) tumor suppressor. The gene encoding LKB1/STK11/PJS is identified as the STK11 gene. The active LKB1 kinase is actually a complex of three proteins: LKB1, Ste20-related kinase adaptor alpha (STRADA) and calcium-binding protein 39 (CAB39; also known as mouse protein 25, MO25). Thus, the enzyme complex is sometimes referred to as LKB1-STRAD-MO25.
Phosphorylation of AMPK by LKB1 also occurs on the critical threonine residue (T172). Unlike the limited distribution of CaMKKβ, LKB1 is widely expressed, thus making it the primary AMPK-regulating kinase. Loss of LKB1 activity in adult mouse liver leads to near complete loss of AMPK activity and is associated with hyperglycemia. The hyperglycemia is due, in part, to an increase in the transcription of gluconeogenic genes. Of particular significance is the increased expression of the peroxisome proliferator-activated receptor-γ (PPAR-γ) coactivator 1α (PGC-1α) which drives gluconeogenesis. Reduction in PGC-1α activity results in normalized blood glucose levels in LKB1-deficient mice.
A third kinase that is believed to play a role in the regulation of AMPK activity through phosphorylation is transforming growth factor-β (TGFβ)-activated kinase 1 (TAK1). AMPK and TAK1 have been shown to directly interact with one another resulting in reciprocal regulation of their respective kinase activities.
As the name implies, AMPK is also regulated by AMP. The effects of AMP are two-fold. One effect is a direct allosteric activation and the other effect is in making AMPK a poorer substrate for dephosphorylation. AMP binds to the γ-subunits of AMPK resulting in a conformational change that leads to modulation of the phosphorylation of the α-subunits. The binding of AMP to the γ-subunits occurs within four tandemly repeated domains referred to as cystathionine β-synthase (CBS) domains due to their original identification in the enzyme involved in methionine and cysteine metabolism.
Because AMP affects both the rate of AMPK phosphorylation in the positive direction and dephosphorylation in the negative direction, the cascade is ultrasensitive. This means that a very small rise in AMP levels can induce a dramatic increase in the activity of AMPK. The activity of adenylate kinase, catalyzing the reaction shown below, ensures that AMPK is highly sensitive to small changes in the intracellular [ATP]/[ADP] ratio.
2 ADP → ATP + AMP
Negative allosteric regulation of AMPK also occurs and this effect is exerted by phosphocreatine. As indicated above, the β subunits of AMPK have a glycogen-binding domain, GBD. In muscle, a high glycogen content represses AMPK activity and this is likely the result of interaction between the GBD of the β-subunit and glycogen, although this has not been shown directly. As suggested above, the GBD of the β-subunit of AMPK allows association of the enzyme with the regulation of glycogen metabolism by placing AMPK in close proximity to one of its substrates glycogen synthase.
AMPK has also been shown to be activated by receptors that are coupled to phospholipase C-β (PLC-β) and by hormones secreted by adipose tissue (termed adipokines) such as leptin and adiponectin (discussed below).
Targets of AMPK
The signaling cascades initiated by the activation of AMPK exert effects on glucose and lipid metabolism, gene expression and protein synthesis. These effects are most important for regulating metabolic events in the liver, skeletal muscle, heart, adipose tissue, and pancreas.
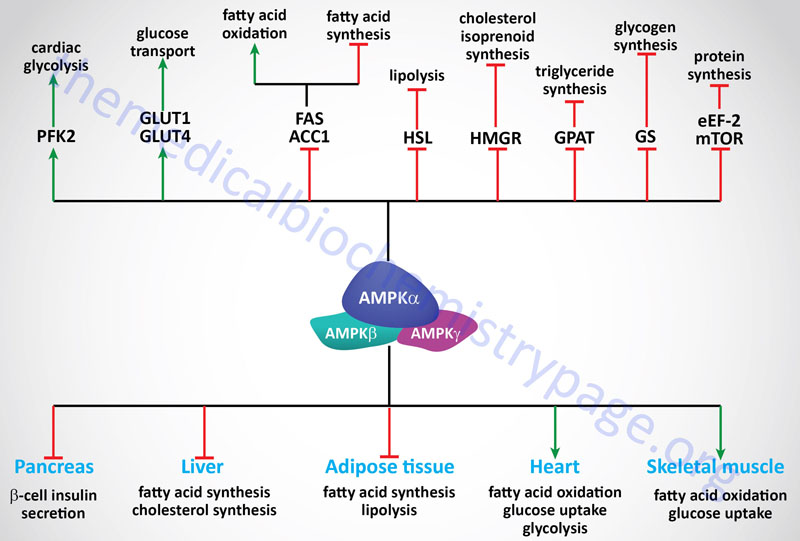
The uptake, by skeletal muscle, accounts for >70% of the glucose removal from the serum in humans. Therefore, it should be obvious that this event is extremely important for overall glucose homeostasis, keeping in mind, of course, that glucose uptake by cardiac muscle and adipocytes cannot be excluded from consideration. An important fact related to skeletal muscle glucose uptake is that this process is markedly impaired in individuals with type 2 diabetes. The uptake of glucose increases dramatically in response to stress (such as ischemia) and exercise and is stimulated by insulin-induced recruitment of glucose transporters to the plasma membrane, primarily GLUT4.
Insulin-independent recruitment of glucose transporters also occurs in skeletal muscle in response to contraction (exercise). The activation of AMPK plays an important, albeit not an exclusive, role in the induction of GLUT4 recruitment to the plasma membrane. In fact, the ability of AMPK to stimulate GLUT4 translocation to the plasma membrane in skeletal muscle occurs via a mechanism distinct from that stimulated by insulin since together insulin and AMPK effects are additive. AMPK activation also results in increased expression of the GLUT4 gene through enhanced binding of the transcription factor MEF-2 (myocyte enhancer factor-2) to promoters in the GLUT4 gene. In addition, there is some demonstration that AMPK may regulate glucose transport through GLUT1. Increased glucose uptake will result in an increase in glycolysis and ATP production.
Under ischemic/hypoxic conditions in the heart the activation of AMPK leads to the phosphorylation and activation of the kinase activity of phosphofructokinase-2, PFK-2 (6-phosphofructo-2-kinase). The product of the kinase activity of PFK-2 (fructose-2,6-bisphosphate, F2,6BP) is one of the most potent regulators of the rate of flux through glycolysis and gluconeogenesis. In liver the PKA-mediated phosphorylation of PFK-2 results in conversion of the enzyme from a kinase that generates F2,6BP to a phosphatase that removes the 2-phosphate thus, reducing the levels of the potent allosteric activator of the glycolytic enzyme 6-phosphfructo-1-kinase, PFK-1 and the potent allosteric inhibitor of the gluconeogenic enzyme fructose-1,6-bisphosphatase (F1,6BPase).
It is important to note that like many enzymes, there are multiple isoforms of PFK-2 (at least four) and neither the liver nor the skeletal muscle isoforms contain the AMPK phosphorylation sites that are found in the cardiac and inducible (iPFK2) isoforms of PFK-2. For more detailed information on the various forms of PFK-2 go to the Glycolysis and the Regulation of Blood Glucose page. Inducible PFK-2 is expressed in the monocyte/macrophage lineage in response to pro-inflammatory stimuli. The ability to activate the kinase activity by phosphorylation of PFK-2 in cardiac tissue and macrophages in response to ischemic conditions allows these cell to continue to have a source of ATP via anaerobic glycolysis. This phenomenon is recognized as the Pasteur effect: an increased rate of glycolysis in response to hypoxia. Of pathological significance is the fact that the inducible form of PFK-2 is commonly found expressed in many tumor cells and this may allow AMPK to play an important role in protecting tumor cells from hypoxic stress. Indeed, techniques for depleting AMPK in tumor cells have shown that these cells become sensitized to nutritional stress upon loss of AMPK activity.
Whereas, stress and exercise are powerful inducers of AMPK activity in skeletal muscle, additional regulators of its activity have been identified. Insulin-sensitizing drugs of the thiazolidinedione family (activators of PPAR-γ, see below) as well as the hypoglycemia drug metformin exert a portion of their effects through regulation of the activity of AMPK. As indicated above, the activity of the AMPK activating kinase, LKB1, is critical for regulation of gluconeogenic flux and consequent glucose homeostasis. The action of metformin in reducing blood glucose levels requires the activity of LKB1 in the liver for this function. Also, several adipokines (hormones secreted by adipocytes) either stimulate or inhibit AMPK activation: leptin and adiponectin have been shown to stimulate AMPK activation, whereas, resistin inhibits AMPK activation.
Within skeletal muscle and heart, activation of AMPK leads to the phosphorylation and inhibition of both isoforms of acetyl-CoA carboxylase (ACC1 and ACC2). This inhibition results in a drop in the level of malonyl-CoA which itself is an inhibitor of carnitine palmitoyltransferase 1 (CPT1). With a drop in the inhibition of CPT1 a concomitant increase in β-oxidation of fatty acids will occur within the mitochondria. An increase in fatty acid oxidation, like increases in glycolysis, will lead to increases in ATP production.
In addition to ACC, AMPK has been shown to phosphorylate and thus regulate the activities of HMG-CoA reductase (HMGR), hormone-sensitive lipase (HSL), glycerol-3-phosphate acyltransferase 1 (GPAT1 which is encoded by the GPAM gene); malonyl-CoA decarboxylase (MCD), glycogen synthase (both GYS1 and GYS2), and creatine kinase (CK). Therefore, the effects of AMPK activation are exerted not only on glucose homeostasis and fatty acid metabolism but overall energy homeostasis including glycogen metabolism, cholesterol metabolism, and phosphocreatine metabolism.
When HSL is phosphorylated in response to PKA activating signals it actively removes fatty acids from triglycerides. Therefore, the phosphorylation and inhibition of HSL by AMPK may seem paradoxical since the release of fatty acids stored in triglycerides would seem necessary to promote the production of ATP via fatty acid oxidation. This paradigm can be explained if one considers that if the fatty acids that are released from triglycerides are not consumed they will be recycled back into triglycerides at the expense of ATP consumption. Thus, it has been proposed that inhibition of HSL by AMPK mediated-phosphorylation is a mechanism to ensure that the rate of fatty acid release does not exceed the rate at which they are utilized either by export or oxidation.
Cardiac effects exerted by activation of AMPK also include phosphorylation of endothelial nitric oxide synthase, eNOS in cardiac endothelium. AMPK-mediated phosphorylation of eNOS leads to increased activity and consequent NO production and provides a link between metabolic stresses and cardiac function. In platelets, insulin action leads to an increase in eNOS activity that is due to its phosphorylation by AMPK. Activation of NO production in platelets leads to a decrease in thrombin-induced aggregation, thereby, limiting the pro-coagulant effects of platelet activation. The response of platelets to insulin function clearly indicates why disruption in insulin action is a major contributing factor in the development of the metabolic syndrome.
Not only does activation of AMPK exert effects on enzyme activity through phosphorylation, there are marked effects on the expression of a number of glycolytic and lipogenic enzymes in the liver and adipose tissue. Included in this list are the genes for the liver isoform of pyruvate kinase (L-PK), fatty acid synthase (FAS), and ACC. Activation of AMPK leads to a reduction in the level of SREBP a transcription factor that is a key regulator of the expression of numerous lipogenic enzymes. Another transcription factor reduced in response to AMPK activation is hepatocyte nuclear factor 4α, HNF4α which is a member of the steroid/thyroid hormone superfamily. HNF4α is known to regulate the expression of several liver and pancreatic β-cell genes such as GLUT2, L-PK and preproinsulin. Of clinical significance is that mutations in HNF4α are responsible for maturity-onset diabetes of the young, MODY-1. Recent evidence indicates that the gene for the carbohydrate-response-element-binding protein (ChREBP) is a target for AMPK-mediated transcriptional regulation in the liver. ChREBP is considered a master regulator of lipid metabolism in liver, in particular in response to glucose uptake.
The target of the thiazolidinedione (TZD) class of drugs used to treat type 2 diabetes is the peroxisome proliferator-activated receptor γ, PPARγ which itself may be a target for the action of AMPK. The transcription co-activator, p300, is phosphorylated by AMPK which inhibits interaction of p300 with not only PPARγ but also the retinoic acid receptor, retinoid X receptor, and thyroid hormone receptor. PPARγ is primarily expressed in adipose tissue and thus it was at first difficult to reconcile how a drug that was apparently acting only in adipose tissue could lead to improved insulin sensitivity of other tissues. The answer to this question came when it was discovered that the TZDs stimulated the expression and release of the adipocyte hormone (adipokine), adiponectin. Adiponectin stimulates glucose uptake and fatty acid oxidation in skeletal muscle. In addition, adiponectin stimulates fatty acid oxidation in liver while inhibiting expression of gluconeogenic enzymes in this tissue. These responses to adiponectin are exerted via activation of AMPK. Another transcription factor target of AMPK is the forkhead protein, FKHR (now identified as FoxO1). FoxO1 is involved in the activation of glucose-6-phosphatase expression and, therefore, loss of FoxO1 activity in response to AMPK activation will lead to reduced hepatic output of glucose.
AMPK activation in response to hypoxia exerts effects on rates of protein synthesis. Hepatic translation elongation factor 2 (eEF2) is a target for phosphorylation in response to AMPK activation. AMPK phosphorylates and activates the kinase that phosphorylates eEF2 (eEF2K) leading to inhibition of protein synthesis. Another direct substrate for AMPK that plays a role in the regulation of protein synthesis is the mechanistic target of rapamycin, mTOR.
AMPK Function in Nutrient Deprivation: Regulation of Autophagy
A central regulator of the signaling pathways that trigger autophagy is the Ser/Thr kinase mTOR (mechanistic target of rapamycin) which is the principal component of both the mTOR complex 1 (mTORC1) and mTORC2 protein complexes. The mTORC1 is the major mTOR-containing complex that regulates cellular responses to growth factor signaling, nutrient deprivation, and various forms of cellular stress.
When mTORC1 is activated by a variety of different growth factor receptors autophagy is suppressed. Upstream signaling proteins that stimulate the activity of mTOR include the Ser/Thr kinases, AKT/PKB, phosphatidylinositol-3-kinase (PI3K), and several members of the MAPK family of kinases. AKT (AK strain transforming)/PKB (protein kinase B) was originally identified as the tumor inducing gene in the AKT8 retrovirus found in the AKR strain of mice. Humans express three genes in the AKT family identified as AKT1 (PKBα), AKT2 (PKBβ), and AKT3 (PKBγ). Under conditions of energy depletion, such as nutrient starvation, the master metabolic regulatory kinase, AMPK, is activated which phosphorylates and inhibits mTOR, thereby promoting autophagy induction. The tumor suppressor p53 can also activate autophagy under conditions of genotoxic stress through repression of mTOR activity.
Amino acid deprivation can be sensed by the cell as a result of reduced levels of charged tRNAs. Uncharged tRNAs are known to bind to the eIF-2α kinase family kinase identified as GCN2 GCN2 is the mammalian homolog of the General Control Non-derepressible-2 gene first identified in yeast. Mammalian GCN2 is encoded by the EIF2AK4 gene. Binding of uncharged tRNAs to GCN2 activates its kinase activity resulting in the phosphorylation of AMPK. Specifically, GCN2 phosphorylates Thr 172 (T172) of the AMPKα2 catalytic protein of the heterotrimeric AMPK enzyme. The net effect of the phosphorylation of AMPKα2 is suppression of protein synthesis, particularly within the brain. Clinical significance of T172 phosphorylation of the AMPKα2 is that this regulatory phosphorylation is significantly reduced in the brains of Alzheimer disease patients.
Within the context of autophagy there are two related downstream kinases whose activities are regulated by activated mTORC1. These Ser/Thr kinases are known as UNC-51-like autophagy activating kinases -1, and -2, which are encoded by the ULK1 and ULK2 genes. Humans express a third related gene, UKL3, whose encoded kinase is primarily involved in the regulation of the activity of the sonic hedgehog (SHH) protein. However, evidence does indicate that ULK3 can function in some capacity in the regulation of autophagy. The UNC genes represent a large family of genes identified in the round worm C. elegans that, when mutated, result in uncoordinated motor activity. The human ULK1 protein functions similarly to the yeast ATG1 protein. ULK1 (or its close family member ULK2) interacts with several other proteins to form a large complex that is required for the formation of an autophagosome. The other proteins that complex with ULK1/ULK2 include ATG13, ATG101, and RB1CC1 (FIP200/ATG17). As indicated, under conditions of nutrient deprivation AMPK is activated, thereby phosphorylating and inhibiting the mTORC1. When the mTORC1 is active it phosphorylates ULK1 on a regulatory Ser residue (S757) thereby, inhibiting the activation of ULK1. Thus, AMPK-mediated inhibition of mTORC1 activity allows ULK1 to induce autophagy. AMPK also directly phosphorylates ULK1 on two different Ser residues (S317 ans S777) which results in activation of the autophagy induction properties of ULK1.
In addition to the role of AMPK in the regulation of ULK1/ULK2 activity via its effects on mTORC1 activity, this master metabolic regulatory kinase also regulates the autophagy complex downstream of ULK1/ULK2 at the level of the beclin 1 complex, also called the class III PI3K complex. The beclin 1 complex consists of several proteins including, but not limited to beclin 1 (the mammalian homolog of yeast ATG6), ATG14 (often referred to as ATG14-like, ATG14L), ultraviolet irradiation resistance-associated gene (UVRAG), AMBRA1 (autophagy and beclin 1 regulator 1), RUBICON (RUN and cysteine-rich domain containing beclin 1 interacting protein), and SH3 domain containing GRB2 like endophilin B1 (encoded by the SH3GLB1 gene; also known as BIF-1). The primary function of the activation of the beclin 1 complex is to activate the class III PI3K encoded by the PIK3C3 gene. PIK3C3 encodes a p110 catalytic subunit which forms a heterodimer with the p150 regulatory subunit encoded by the PIK3R4 gene. Activation of the class III PI3K results in the production of phosphatidylinositol 3-phosphate (PI3P) at the site of phagophore formation.
One important role of PI3P is the recruitment of the WIPI proteins (human homologs of the yeast ATG18 protein) to the phagophore membrane. In addition, PI3P may recruit other downstream effector proteins of the autophagy process such as the ATG5-ATG12-ATG16L1 complex. The function of the beclin 1 protein within the complex can be inhibited via interaction with the pro-survival (anti-apoptotic) proteins BCL-2 and BCL-XL (see Mitochodrial Apoptosis section in the Protein, Organelle, and Cell Turnover page). BCL-2 and BCL-XL bind to the BH3 domain of beclin 1 thereby inhibiting its activity. The interaction of beclin 1 with either of the two pro-survival proteins can be inhibited by phosphorylation of beclin 1 or BCL-2 or by the ubiquitylation of beclin 1. Therefore, it is clear that the beclin 1 complex plays an important role in promoting mitochondrial apoptosis as well. Mitochondria localized beclin 1 induces the translocation of the BAX protein which forms a mitochondial outer membrane (MOM) pore complex allowing pro-apoptotic factors, such as cytochrome c to be released setting off the intrinsic apoptotic cascade.
The nucleation of membrane into the initial pre-autophagosome (phagophore) requires several ATG proteins and lipids in what is referred to as the conjugation step of autophagosome formation. Phosphatidylethanolamine (PE) is known to be a critical lipid component of the conjugation process and it is believed that the PI3P derived via the action of the class III PI3K is also an important lipid. Phagophore formation begins when ATG12 is activated via the ubiquitin conjugating E1-like activity of ATG7. Activated ATG12 is then transferred to ATG5 via the ubiquitin conjugating E2-like activity of ATG10. The ATG12-ATG5 conjugate then interacts with ATG16L1 forming an ATG12-ATG5-ATG16L1 complex that possesses ubiquitin conjugating E3-like activity. The ATG12-ATG5-ATG16L1 complex is functionally coupled to proteins of the human ATG8 family. The ATG8 proteins are considered members of the ubiquitin-like modifier family of proteins.
In yeast there is but a single ATG8 protein involved in autophagy, however, in humans there are at least seven ATG8 family members. The human ATG8 family proteins are GABA type A receptor-associated protein (encoded by the GABARAP gene; considered ATG8A), GABA type A receptor-associated protein like 1 (encoded by the GABARAPL1 gene; considered ATG8B), GABA type A receptor-associated protein like 2 (encoded by the GABARAPL2 gene; considered ATG8C), microtubule associated protein 1 light chain 3 alpha (encoded by the MAP1LC3A gene; considered ATG8E; also called simply LC3), microtubule associated protein 1 light chain 3 beta (encoded by the MAP1LC3B gene; considered ATG8F), microtubule associated protein 1 light chain 3 beta2 (encoded by the MAP1LC3B2 gene; considered ATG8G), and microtubule associated protein 1 light chain 3 gamma (encoded by the MAP1LC3C gene; considered ATG8J).
AMPK and Hypothalamic Functions
The activity of AMPK in the hypothalamus indicates that within the central nervous system this important metabolic regulating enzyme is also critical in the control of food intake and other neuroendocrine functions. The level of active AMPK in the hypothalamus rises during periods of fasting and then decreases in response to re-feeding. In animal studies the pharmacologic activation of hypothalamic AMPK by the purine nucleotide intermediate AICAR (5-aminoimidazole-4-carboxamide ribotide) results in an increase in food intake.
Within the hypothalamus, leptin, GLP-1, and insulin (anorexigenic hormones) actions result in inhibition of AMPK activation, whereas, adiponectin and ghrelin, (orexigenic hormones) actions result in activation of AMPK activity. Of significance to the tissue specific distribution of AMPK isoforms, as well as to the differing roles of AMPK in various tissues, the actions of leptin in adipose tissue and liver result in activation of AMPK while in the heart leptin inhibits AMPK activation as is the case in the hypothalamus.
The ultimate effects of AMPK regulation in the hypothalamus are complex and represent a focal point for major regulation of whole body energy homeostasis. Hypothalamic AMPK activation in response to low blood glucose and gastric release of ghrelin during fasting results in increased release of the hypothalamic releasing factors, CRH (corticotropin releasing hormone) and TRH (thyrotropin releasing hormone) which stimulate the pituitary to release ACTH and TSH (thyroid stimulating hormone), respectively. The actions of TSH on the thyroid gland result in an increased production and release of the thyroid hormones, T4 and T3. The metabolic effects of T3 are diverse resulting in increased carbohydrate digestion and absorption in the gut, increased hepatic gluconeogenesis, and increased protein metabolism in skeletal muscle. As the level of T3 increases it eventually feeds back and inhibits hypothalamic AMPK. The ACTH that is released stimulates the adrenal cortex to produce cortisol which in turn stimulates lipid mtabolism and hepatic gluconeogenesis.
Adipose tissue also plays a central role in the metabolic status-induced regulation of hypothalamic AMPK function. Lean adipose tissue, representative of a fasted state, releases adiponectin which stimulates the activation of hypothalamic AMPK. The activated AMPK then leads to the activation of the pituitary and thyroid responses described above. Conversely, increased fat mass in adipose tissue, indicative of the well fed state, results in the release of leptin. Leptin then exerts inhibitory effects on the hypothalamus leading to reduced activation of AMPK.
Relevance of AMPK to Type 2 Diabetes
An impairment in fuel metabolism occurs in obesity and this impairment is a leading pathogenic factor in type 2 diabetes. The insulin resistance associated with type 2 diabetes is most profound at the level of skeletal muscle as this is the primary site of glucose and fatty acid utilization. Therefore, an understanding of how to activate AMPK in skeletal muscle would offer significant pharmacologic benefits in the treatment of type 2 diabetes. As indicated above, it has already been shown that metformin and the thiazolidinedione drugs exert some of their effects via activation of AMPK. In the non-pharmacologic context, activation of AMPK occurs in response to exercise, an activity known to have significant benefit for type 2 diabetics.
As this above discussion has made clear, AMPK serves as a master sensor of energy status whose activity is triggered in response to changes in nutritional status in order to modulate tissue-specific metabolic pathways. Another protein (that was originally discovered in yeast responding to caloric restriction) that plays an important role in response to nutrient deprivation is SIRT1. SIRT1 is a member of the sirtuin family of proteins that function as NAD+-dependent protein deacetylases or ADP-ribosyltransferases. There are seven mammalian sirtuins which are homologues of the yeast protein silent information regulator 2 (Sir2). SIRT1 catalyzes lysine deacetylation of target proteins coupled to the hydrolysis of NAD+. Major targets of SIRT1 (as well as SIRT3) are the histones, deacetylation of which results in altered chromatin structure and changes in gene expression. The sirtuins also deacetylate transcription factors and transcription co-regulators leading to changes in gene expression.
As already discussed, the primary function of AMPK is to phosphorylate and, thereby regulate the activity of numerous metabolic enzymes. However, if ATP depletion remains protracted AMPK will phosphorylate a number of transcription factors and transcription co-regulators such as HNF-4α, FoxO3A (formerly identified as FKHR-L1), PGC1α (peroxisome proliferator-activated receptor-γ co-activator 1α), and CBP/p300. It turns out that these factors are also targets for regulation by SIRT1. Like SIRT1, AMPK has been proposed to be one of several molecules involved in the regulation of mammalian longevity. AMPK can increase the life span of the round worm C. elegans and in aging rats the level of AMPK phosphorylation is decreased relative to young animals.
In skeletal muscle, AMPK phosphorylates PGC1α leading to increased mitochondrial biogenesis and fatty acid oxidation. Both of these effects of PGC1α require SIRT1. In addition, AMPK-activated PGC1α results in increased GLUT4 presentation in skeletal muscle cell membranes resulting in increased glucose uptake, another effect that requires SIRT1 activity. Indeed, it is postulated that many of the gene regulatory effects of AMPK in skeletal muscle are in fact the result of PGC1α phosphorylation. SIRT1 has been shown to deacetylate and activate PGC1α at the promoters of several genes involved in fatty acid utilization and mitochondrial respiration. Therefore, it is likely that SIRT1 and AMPK actions that regulate lipid oxidation and mitochondrial biogenesis converge through the action of PGC1α.
In the liver AMPK activates fatty acid oxidation via activation of PGC1α as well as its effects on other enzymes of lipogenesis described above (e.g. SREBP and FAS). In addition, hepatic cholesterol synthesis is inhibited by AMPK actions at the level of SREBP and HMGR. SIRT1 also regulates hepatic cholesterol metabolism via deacetlyation and activation of the LXR nuclear receptors. In this context one target of LXR is the ATP-binding cassette transporter A1 (ABCA1) which is involved in the transport of cholesterol from tissues to the circulation and its subsequent interaction with HDLs (this is reverse cholesterol transport). SIRT1 also decreases lipogenesis via reductions in SREBP levels. The ability of AMPK and SIRT1 to effect reductions in SREBP suggests that these two proteins may be involved in the same pathway whereby AMPK activates SIRT1 and SIRT1 feeds-back to activate AMPK.
Whereas, there is a convergence of AMPK and SIRT1 action on lipid oxidation in skeletal muscle, there is a divergence of their actions in the liver at the level of gluconeogenesis. In the nutrient deprived state (e.g. fasting) SIRT1 activates hepatic PGC1α leading to increased fatty acid oxidation and gluconeogenesis. However, fasting results in AMPK-mediated inhibition of gluconeogenesis while simultaneously increasing fatty acid oxidation. The negative effect of AMPK on gluconeogenesis does not involve PGC1α regulation.
Under fasting conditions the pancreas releases glucagon which exerts numerous effects on hepatic glucose metabolism. Glucagon induces nuclear translocation of the protein TORC2 (transducer of regulated CREB activity 2) which then binds and activates CREB (cAMP response element-binding protein) CREB is encoded by the CREB1 gene. CREB stimulates gluconeogenesis and fatty acid oxidation via induction of PGC-1α binding to the promoters of the genes encoding FOXO1 and HNF-4α leading to increased transcription of the gluconeogenic enzymes, pyruvate carboxylase (PC), phosphoenolpyruvate carboxykinase (PEPCK), glucose 6-phosphatase, and fructose-1,6-bisphosphatase.
Humans express seven genes in the CREB subfamily of the basic leucine zipper (bZIP) family of transcription factors. The seven CREB gene are identified as CREB1, CREB3, CREB5, and CREB3-like 1, 2, 3, and 4 (CREB3L1, CREB3L2, CREB3L3, and CREB3L4). The CREB3L3 encoded protein is commonly identified as CREBH. The CREB1 encoded proteins are most closely related in structure and function to two add itional transcription factors called cAMP response element modulator (CREM) and activating transcription factor 1 (ATF-1). Another member of the activating transcription factor (ATF) family, ATF-4, was originally identified as CREB2.
When ATP levels are low or under stress conditions activation of AMPK inhibits hepatic gluconeogenesis because AMPK phosphorylates TORC2 which sequesters it in the cytoplasm resulting in inactivation of CREB. AMPK has also been proposed to phosphorylate and inhibit HNF4α. These effects of hepatic AMPK activation override the inductive signals elicited through glucagon.
Newer therapeutic approaches to the intervention of type 2 diabetes include small molecule activators of SIRT1. In studies in mice overexpression of SIRT1 results in improved glucose tolerance and enhanced insulin excretion in response to glucose administration. Both of these effects of increased SIRT1 action would be beneficial in the treatment of type 2 diabetes. AMPK is already the target of several classes of drugs used in the treatment of type 2 diabetes including metformin and the thiazolidinediones, TZDs. Given the effects of AMPK and SIRT1 on metabolism, and their convergence in many of these processes, it seems logical to assume that future diabetes therapies will combine both AMPK and SIRT1 activators to lower blood glucose and increase insulin sensitivity.
AMPK-Related Kinases
Recent evidence has identified at least 12 enzymes that are related to AMPK at the level of homology in the catalytic domain. The AMPK-related kinase family of kinase is referred to as the AMPK-RK family. These AMPK-RK family enzymes are BRSK1, BRSK2, NUAK1, NUAK2, SIK1, SIK2, SIK3, MARK1, MARK2, MARK3, MARK4 and MELK.
BRSK1 and BRSK2 are brain-specific serine/threonine kinases 1 and 2, respectively.
NUAK1 refers to nuclear AMPK-related kinase. The NUAK1 gene is also called AMPK-related kinase 5 (ARK5). NUAK2 is also called SNF1/AMPK-related kinase (SNARK). The SNF1 gene was originally identified as a mutant in yeast that were defective in sucrose utilization and hence given the name sucrose nonfermenting. The SNF1 gene is related to the mating-type switching gene of yeast. There are several SNF and SWI family members in eukaryotes. These genes, referred to as SNF/SWI, are factors that control gene transcription via modulation of chromatin structure.
SIK refers to salt-induced kinase which was originally found to be expressed in the adrenal glands of rats fed a high-salt diet. SIK2 was identified as a SIK1-related gene expressed in adipose tissue and also shown to be the same as the QIK gene (for Qin-induced kinase where Qin is the name of an oncogene identified in avian sarcoma virus 31). SIK3 is a third member of the SIK family and is also known as QSK.
MARK1–4 are Microtubule Affinity-Regulating Kinases 1 through 4. These four kinases were first identified in brain based upon their ability to phosphorylate and regulate the dissociation of microtubule-associated proteins (MAPs) from microtubules.
MELK is maternal embryonic leucine zipper kinase.
Like regulation of AMPK by the LKB1 kinase, 11 of these AMPK-related family members are also subject to LKB1-mediated regulation. MELK is the only family member not shown to be a substrate for LKB1. Several of these AMPK-RK family members have also been shown to have important roles in the regulation of metabolic processes (as well as other roles that are not defined in this discussion).
The activity of NUAK2 has been shown to be regulated by glucose deprivation, ER stress, oxidative stress, and elevation in cellular AMP levels. These results suggest that NUAK2 and AMPK are regulated by many of the same stimuli. However, unlike AMPK which is activated by the diabetes drug metformin (Glucophage®), NUAK2 activity is down-regulated. In NUAK2 knock-out mice there is an increased rate of metabolic disorder and they display a maturity-onset type of diabetes.
The tissue with the highest levels of SIK2 expression is adipose tissue. SIK2 has been shown to phosphorylate the insulin receptor substrate-1 protein (IRS1) on the same serine residue that is phosphorylated by AMPK (serine 794). This suggests that SIK2 may be important in the regulation of insulin-mediated signal transduction events. In addition, SIK2 inhibits CREB-mediated gene expression by phosphorylating TORC2 at the same site as AMPK (see section above).