

Last Updated: May 31, 2026
Introduction to Amino Acid Derivatives
Amino acids play critical roles as the building blocks of proteins, as intermediates in numerous metabolic processes, and as reservoirs of energy through their catabolism. Equally important roles for amino acids relates to their serving as substrates for the synthesis of various bioactive derivatives such as neurotransmitters and biochemical mediators such as nitric oxide.
Tyrosine-Derived Neurotransmitters / Hormones
Much of the tyrosine that does not get incorporated into proteins is catabolized for energy production. Another significant fate of tyrosine is conversion to the catecholamines. The catecholamines are dopamine, norepinephrine, and epinephrine. All three catecholamines exert effects in numerous locations in the body as either a neurotransmitter or as a hormone. Within the central and peripheral nervous systems (CNS and PNS, respectively) the catecholamines exert their effects as neurotransmitters, in the periphery they do so as hormones. The details of catecholamine effects, exerted via activation of specific receptors, are only covered briefly in this section. For greater detail go to the Biochemistry of Nerve Transmission page.
Tyrosine is transported into catecholamine-secreting neurons and adrenal medullary chromaffin cells (which are functionally similar to sympathetic postganglionic neurons) where catecholamine synthesis takes place. The first step in the process involves the enzyme tyrosine hydroxylase which, like phenylalanine hydroxylase (of tyrosine synthesis), requires tetrahydrobiopterin (BH4, or also written H4B) as a cofactor.
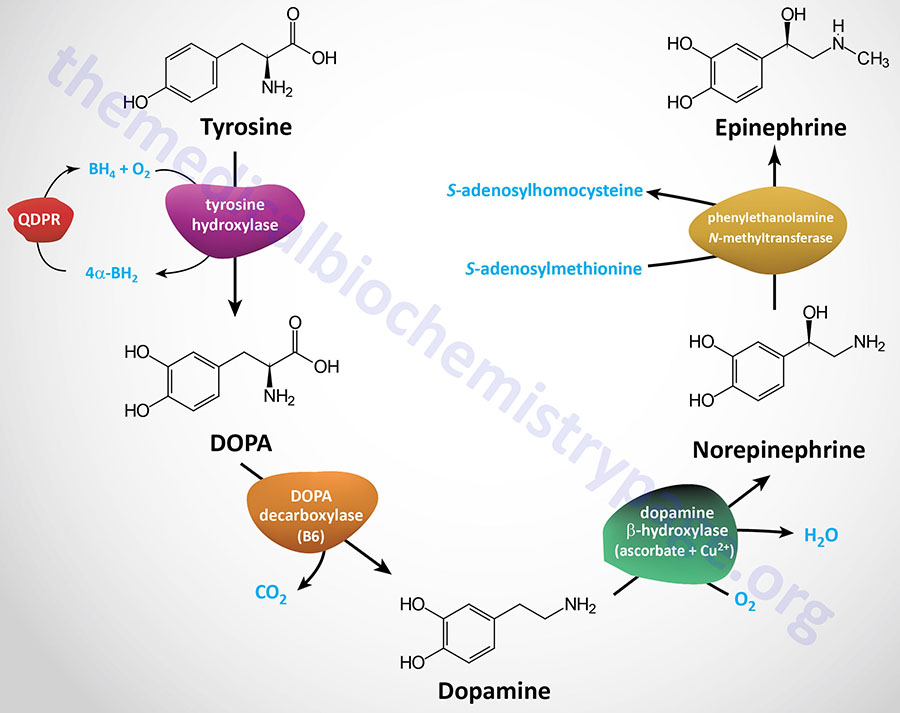
The tyrosine hydroxylase reaction represents the rate-limiting reaction of catecholamine biosynthesis. The dependence of tyrosine hydroxylase on BH4 necessitates the coupling to the action of dihydropteridine reductase (encoded by the QDPR gene: quinoid dihydropteridine reductase) as is the situation for tryptophan hydroxylase (see below) and phenylalanine hydroxylase. The product of the tyrosine hydroxylase reaction is L-3,4-dihydroxyphenylalanine (L-DOPA; more commonly just DOPA). Clinically L-DOPA is referred to as levodopa.
Tyrosine hydroxylase is encoded by the TH gene. The TH gene is located on chromosome 11p15.5 and is composed of 14 exons that generate three alternatively spliced mRNAs that encode tyrosine hydroxylase isoform a (528 amino acids), isoform b (497 amino acids), and isoform c (524 amino acids).
Mutations in the TH gene are associated with the development of Segawa syndrome. Segawa syndrome is an autosomal recessive disorder that manifests in early infancy as a DOPA-responsive dystonia. Two distinct phenotypes are associated with Segawa syndrome, one that manifests very early and presents with symptoms of greater severity, and a later onset less severe type that responds better to L-DOPA (levodopa) therapy.
The enzyme DOPA decarboxylase converts DOPA to dopamine. DOPA decarboxylase, which is also known as aromatic L-amino acid decarboxylase (AADC), is encoded by the DDC gene. The DDC encoded enzyme is dependent upon pyridoxal phosphate (PLP: vitamin B6-derived cofactor) for activity.
The DDC gene is located on chromosome 7p12.2–p12.1 and is composed of 18 exons that generate seven alternatively spliced mRNAs that collectively encode six isoforms of the enzyme. The DDC encoded enzyme is also responsible for the conversion of 5-hydroxytryptophan to serotonin (see below) and in the biosynthesis of several of the trace amine neurotransmitters.
The enzyme dopamine β-hydroxylase then converts dopamine to norepinephrine. Dopamine β-hydroxylase is a major vitamin C and copper (Cu2+)-dependent enzyme whose activity is negatively affected in Menkes disease.
Dopamine β-hydroxylase is encoded by the DBH gene. The DBH gene is located on chromosome 9q34.2 and is composed of 12 exons that encode a precursor protein of 617 amino acids. The primary site of expression of the DBH gene is in the adrenal glands. Dopamine β-hydroxylase is found as both a soluble and a membrane-bound enzyme dependent upon whether or not the signal peptide from the precursor protein is present.
The last step of catecholamine biosynthesis is the conversion of norepinephrine to epinephrine which involves a methylation reaction. The enzyme phenylethanolamine N-methyltransferase catalyzes this methylation reaction utilizing S-adenosylmethionine (SAM or AdoMet) as a methyl donor. In addition to epinephrine synthesis, the last reaction generates S-adenosylhomocysteine (SAH).
Phenylethanolamine N-methyltransferase is encoded by the PNMT gene. The PNMT gene is located on chromosome 17q12 and is composed of 4 exons that generate two alternatively spliced mRNAs, one of which is non-coding, the other encodes the functional protein of 282 amino acids. The PNMT encoded enzyme is also involved in the synthesis of several of the trace amine neurotransmitters.
Within the substantia nigra (largest of four nuclei in the basal ganglia of the midbrain), and some other regions of the brain, synthesis proceeds only to dopamine. Within the locus coeruleus (a brainstem nucleus in the dorsal pontine tegmentum) the end product of the pathway is norepinephrine. Within adrenal medullary chromaffin cells, tyrosine is converted to norepinephrine and epinephrine.
Secretory Vesicle Incorporation of Monoaminergic Neurotransmitters
Once synthesized, dopamine, norepinephrine, and epinephrine are packaged in granulated vesicles for secretion in response to the appropriate nerve impulse. Incorporation of these neurotransmitters into synaptic vesicles requires the activity of one of two vesicular monoamine transporters, VMAT. The VMAT transporters are antiporters where H+ export is coupled to monoaminergic neurotransmitter import. Within these synaptic vesicles, norepinephrine and epinephrine are bound to ATP and a protein called chromogranin A.
Humans express two VMAT encoding genes, SLC18A1 which encodes VMAT1 and SLC18A2 which encodes VMAT2. VMAT2 exhibits higher affinity for monoamine substrates than does VMAT1, and VMAT2 is expressed in all monoaminergic neurons in the brain. Indeed, evidence indicates that VMAT2 is the only monoaminergic neurotransmitter expressed in CNS neurons.
The energy necessary for the transport of these neurotransmitters into the secretory vesicles is provided by the proton gradient generated by the vacuolar H+-ATPase (V-ATPase). The vacuolar H+-ATPase is a multisubunit complexes that are members of the larger V-ATPase family of proton transporters and are related to the F-type ATPases exemplified by the F0F1-ATPase (complex V) of the oxidative phosphorylation process.
In addition to dopamine, norepinephrine, and epinephrine, the VMAT transporters, in particular VMAT2, are responsible for vesicular import of serotonin (5-HT), histamine, and several trace amines such as the tyramines [para-tyramine (p-tyramine), meta-tyramine (m-tyramine), 3-methoxytyramine, and N-methyltyramine], β-phenylethylamine (β-PEA), and thyronamine (3-iodothyronamine).
The clinical significance of the VMAT transporters is evidenced by the fact that variants in VMAT1 have been found to be associated with susceptibility for schizophrenia and bipolar depression. Mutations in VMAT2 are also associated with schizophrenia.
Catecholamine Functions
Norepinephrine is the principal neurotransmitter of sympathetic postganglionic nerves. Both norepinephrine and epinephrine are stored in synaptic knobs of neurons that secrete it, however, epinephrine is not a mediator at postganglionic sympathetic nerve impulses.
The major location, within the brain, for norepinephrine synthesis is the locus coeruleus of the brainstem. The major brain region for the synthesis of dopamine is the substantia nigra which is located below the posterior hypothalamus and next to the ventral tegmental area.
The presence of high concentrations of tyrosine in the locus coeruleus and the substantia nigra leads to increased melanin synthesis which confers on these brain regions a dark bluish coloration observable in brain sections. Indeed, these brain regions are so-called due to the dark bluish-black pigmentation. The Latin term, substantia nigra, means “black substance”. The Latin word coeruleus means “dark blue, blue, or blue-green”.
Outside the brain, the major site of norepinephrine and epinephrine synthesis is in adrenal medullary chromaffin cells. Outside the brain, dopamine is synthesized in several tissues including the gastrointestinal system where its actions reduce gastrointestinal motility, the pancreas where its actions inhibit insulin synthesis, and in the kidneys where its actions increase sodium excretion and urinary output.
The actions of norepinephrine and epinephrine are exerted via receptor-mediated signal transduction events. The receptors to which epinephrine and norepinephrine bind are referred to as adrenergic receptors. The adrenergic receptors are members of the G-protein coupled receptor (GPCR) family. There are two distinct classes of adrenergic receptor identified as the α (alpha) and β (beta) receptors. There are two distinct subclasses of α adrenergic receptor identified as the α1 and α2 sub-classes. For greater detail on the adrenergic receptors go to the Biochemistry of Nerve Transmission page.
Within each class of adrenergic receptor there are several sub-classes. The α1 class contains the α1A, α1B, and α1D receptors. The α1 receptor class are coupled to Gq-type G-proteins that activate PLCβ resulting in increases in IP3 and DAG release from membrane PIP2. The α2 class contains the α2A, α2B, and α2C receptors. The α2 class of adrenergic receptors are coupled to Gi-type G-proteins that inhibit the activation of adenylate cyclase and therefore, receptor activation results in reduced levels of cAMP and consequently reduced levels of active PKA.
The β class of receptors is composed of three subtypes: β1, β2, and β3 each of which couple to Gs-type G-proteins resulting in activation of adenylate cyclase and increases in cAMP with concomitant activation of PKA. However, the β2 class of receptor can switch from Gs to Gi/o signaling following phosphorylation of the receptor by PKA.
Dopamine binds to dopaminergic receptors identified as D-type receptors and there are five subclasses identified as D1, D2, D3, D4, and D5. All five dopamine receptors belong to the G-protein coupled receptor (GPCR) family. The D1 and D5 dopamine receptors are coupled to the activation of Gs-type G-proteins and, therefore, receptor activation results in activation of adenylate cyclase. The D2, D3, and D4 dopamine receptors are coupled to Gi-type G-proteins and, therefore, receptor activation results in the inhibition of adenylate cyclase. For more details on the dopamine receptors go to the Biochemistry of Nerve Transmission page.
Catecholamine Catabolism
The catecholamine, DOPA, dopamine, epinephrine, and norepinephrine are catabolized to inactive compounds through the sequential actions of enzymes that include catecholamine-O-methyltransferase (COMT) and monoamine oxidase (MAO). Humans express two monoamine oxidase encoding genes identified as MAOA and MAOB. Both of these genes are localized to the same region of the X chromosome (Xp11.13) and both genes are composed of 16 exons. The major MAO responsible for catecholamine catabolism is the MAOA encoded enzyme.
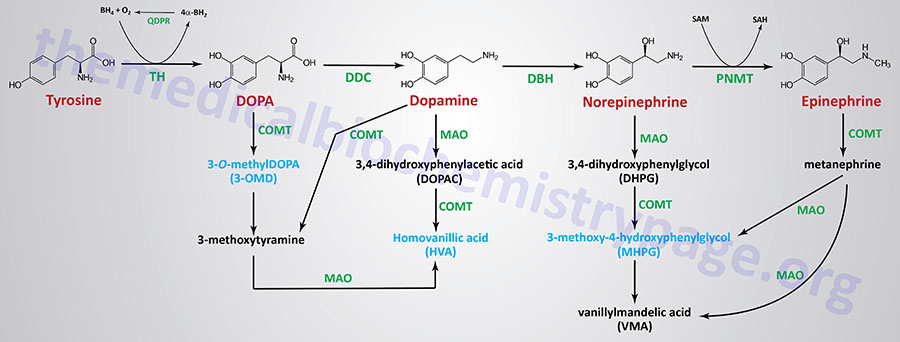
Compounds that inhibit the action of MAO exhibited beneficial effects in the treatment of clinical depression, even when tricyclic antidepressants are ineffective. The utility of MAO inhibitors was discovered serendipitously when patients treated for tuberculosis with isoniazid showed signs of an improvement in mood. This effect of isoniazid was subsequently found to be the result of its inhibition of MAO. The use of MAO inhibitors and tricyclic antidepressants are no longer common practice in the treatment of depressive disorders.
Tryptophan-Derived Neurotransmitters
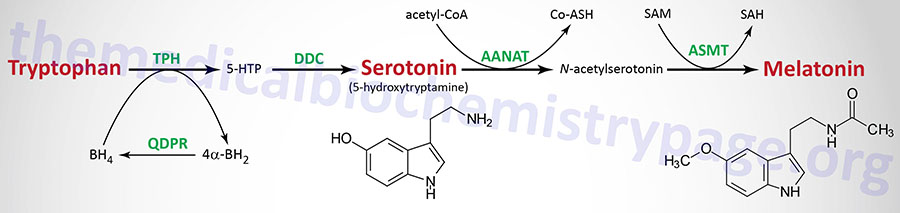
Tryptophan serves as the precursor for the synthesis of serotonin (5-hydroxytryptamine, 5-HT, see also Biochemistry of Nerve Transmission) and melatonin (N-acetyl-5-methoxytryptamine).
Serotonin
Serotonin is synthesized through a two-step process involving a tetrahydrobiopterin-dependent hydroxylation reaction (catalyzed by tryptophan-5-monooxygenase, also called tryptophan hydroxylase) and then a decarboxylation catalyzed by DOPA decarboxylase (also known as aromatic L-amino acid decarboxylase). Tryptophan hydroxylase is normally not saturated and as a result, an increase in dietary uptake of tryptophan will lead to increased brain serotonin content. Tryptophan hydroxylase represents the rate-limiting step in serotonin and melatonin synthesis.
Humans express two tryptophan hydroxylase genes identified as TPH1 and TPH2.
The TPH1 gene is located on chromosome 11p15.1 and is composed of 11 exons that encode a protein (tryptophan 5-hydroxylase 1) of 444 amino acids. The TPH1 gene encodes the predominant tryptophan hydroxylase responsible for serotonin synthesis in enterochromaffin cells of the intestines.
The TPH2 gene is located on chromosome 12q21.1 and is composed of 13 exons that encode a protein (tryptophan 5-hydroxylase 2) of 490 amino acids. Expression of TPH2 in the brain is responsible for synthesis of serotonin in the central nervous system, CNS.
Serotonin is present at highest concentrations in platelets and in the gastrointestinal tract. Indeed, over 90% of total body serotonin is derived from intestinal synthesis. Lesser amounts are found in the brain and the retina. Serotonin containing neurons (serotonergic neurons) have their cell bodies in the midline raphe nuclei of the brain stem and project to portions of the hypothalamus, the limbic system, the neocortex, and the spinal cord.
After release from serotonergic neurons, most of the released serotonin is recaptured by an active reuptake mechanism. The re-uptake of serotonin is catalyzed by the SLC family transporter, SCL6A4. The function of the antidepressant, Prozac®, and related drugs called selective serotonin re-uptake inhibitors (SSRIs), is to inhibit this reuptake process, thereby, resulting in prolonged serotonin presence in the synaptic cleft.
Mutations in the SLC6A4 gene affect the rate of serotonin re-uptake and have been shown to be correlated with obsessive-compulsive disorder, anxiety-related traits, and are suspected to be involved in sudden infant death syndrome, aggressive behavior in Alzheimer disease patients, and depression-susceptibility in persons experiencing emotional trauma.
The function of serotonin is exerted upon its interaction with specific receptors. Several serotonin receptors have been cloned and are identified as 5HT1, 5HT2, 5HT3, 5HT4, 5HT5, 5HT6, and 5HT7. Within the 5HT1 group there are subtypes 5HT1A, 5HT1B, 5HT1D, 5HT1E, and 5HT1F. There are three 5HT2 subtypes, 5HT2A, 5HT2B, and 5HT2C as well as two 5HT5 subtypes, 5HT5a and 5HT5B. Most of these receptors are coupled to G-proteins that affect the activities of either adenylate cyclase or phospholipase Cβ (PLCβ). The 5HT3 class of receptors are ion channels, referred to as ionotropic receptors.
Some serotonin receptors are presynaptic and others postsynaptic. The 5HT2A receptors mediate platelet aggregation and smooth muscle contraction. The 5HT2C receptors are suspected in control of food intake as mice lacking this gene become obese from increased food intake and are also subject to fatal seizures. The 5HT3 receptors are present in the gastrointestinal tract and are involved in the regulation of emesis (vomiting). Also present in the gastrointestinal tract are 5HT4 receptors where they function in secretion and peristalsis. The 5HT6 and 5HT7 receptors are distributed throughout the limbic system of the brain and the 5HT6 receptors have high affinity for antidepressant drugs.
Melatonin
Melatonin is derived from serotonin within the pineal gland and the retina, where the rate-limiting enzyme for melatonin synthesis is expressed. The pineal parenchymal cells secrete melatonin into the blood and cerebrospinal fluid. Synthesis and secretion of melatonin increases during the dark period of the day and is maintained at a low level during daylight hours.
As outlined in the Figure above the pathway of melatonin synthesis from serotonin involves two reactions. Serotonin is acetylated by aralkylamine N-acetyltransferase (AANAT) forming N-acetyl serotonin. N-acetyl serotonin is then methylated by the S-adenosylmethionine (SAM)-dependent enzyme, acetyl-serotonin-O-methyltransferase which is derived from the ASMT gene that is localized to the pseudoautosomal region 1 (PAR1) of the X and Y chromosomes.
The AANAT gene is located on chromosome 17q25.1 and is composed of 8 exons that generate two alternatively spliced mRNA that encode proteins of 252 amino acids (isoform 1) and 207 amino acids (isoform 2).
The ASMT gene contains 9 exons that generate three alternatively spliced mRNAs encoding proteins of 373 amino acids (isoform 1), 298 amino acids (isoform 2), and 345 amino acids (isoform 3). The ASMT encoded enzyme is also known as hydroxyindole-O-methyltransferase (HIOMT).
This diurnal variation in melatonin synthesis is brought about by norepinephrine secreted by the postganglionic sympathetic nerves that innervate the pineal gland. The effects of norepinephrine are exerted through interaction with β-adrenergic receptors. This leads to increased levels of cAMP, which in turn activates PKA. PKA phosphorylates aralkylamine N-acetyltransferase activating this enzyme required for melatonin synthesis.
Melatonin functions by inhibiting the synthesis and secretion of other neurotransmitters such as dopamine and GABA.
Humans express two genes encoding receptors for melatonin, MTNR1A and MTNR1B, encoding the MT1 (initially called Mel1a) and MT2 (initially called Mel1b) receptors, respectively. The melatonin receptors are members of the G-protein coupled receptor (GPCR) superfamily. Expression of the MTNR1A gene is seen in the suprachiasmatic nucleus (SCN), the pars tuberalis of the pituitary gland, the retina, and the skin. Expression of the MTNR1B gene is seen in the retina and the skin.
The MT1 and MT2 receptors are coupled to both Gi-type and Gq-type G-proteins. Coupling to Gi-type G-proteins occurs when the receptors are in the monomeric state. When the receptors heterodimerize with the serotonin 5-HT2c receptor they activate a Gq-type G-protein.
The sleep inducing effects of melatonin are the result of the activation of MT1 in the suprachiasmatic nuclei (SCN) within the hypothalamus. Activation of the MT2 receptor is associated with induction of sleep and the reduction of anxiety. The most significant effect of the MT1 and MT2 receptors to phase shift local and overt circadian rhythms.
Trace Amines
The trace amines represent a family of biogenic amines that are found in the brain at much lower concentrations than the more classical biogenic monoamines such as dopamine, norepinephrine, epinephrine, serotonin, and histamine. The trace amines are derived via the actions of biosynthetic and catabolic enzymes that are also utilized in the synthesis and catabolism of the classic biogenic amines.
The amino acids phenylalanine, tyrosine, and tryptophan serve as the major substrates for the synthesis of several trace amines. The synthesis and actions of numerous trace amines are described in greater detail in the Biochemistry of Nerve Transmission page.
(Gamma) γ-Aminobutyric Acid (GABA) from Glutamate
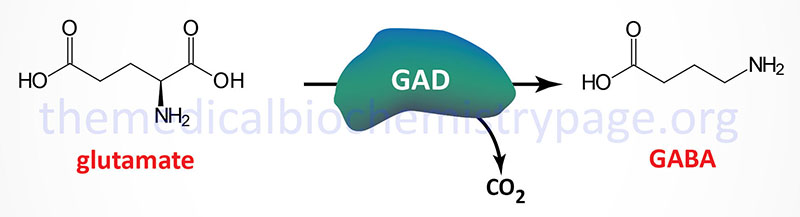
The amino acid derivative, γ-aminobutyrate (GABA; also called 4-aminobutyrate) is a major inhibitory neurotransmitter responsible for the regulation of presynaptic transmission in the CNS, and also in the retina. Neurons that secrete GABA are termed GABAergic.
GABA cannot cross the blood-brain-barrier and as such must be synthesized within neurons in the CNS. The synthesis of GABA in the brain occurs via a metabolic pathway referred to as the GABA shunt. Glucose is the principal precursor for GABA production via its conversion to α-ketoglutarate in the TCA cycle.
Within the context of the GABA shunt 2-oxoglutarate (α-ketoglutarate) is transaminated to the amino acid glutamate by GABA α-oxoglutarate transaminase (GABA-T). Glutamic acid decarboxylase (GAD) catalyzes the decarboxylation of glutamic acid to form GABA. There are two GAD genes in humans identified as GAD1 and GAD2.
The GAD1 gene is located on chromosome 2q31.1 and is composed of 21 exons that generate two alternatively spliced mRNAs. One of these mRNAs encodes a 594 amino acid protein (GAD67). The other mRNA encodes a 224 amino acid protein (GAD25).
The GAD2 gene is located on chromosome 10p12.1 and is composed of 16 exons that generate two alternatively spliced mRNAs, both of which encode the same 585 amino acid protein (GAD65).
The GAD designations for the major proteins produced by these two genes are reflective of their molecular weights. Both the GAD1 and GAD2 genes are expressed in the brain and GAD2 expression also occurs in the pancreas. The activity of GAD requires pyridoxal phosphate (PLP) as a cofactor. PLP is generated from the B6 vitamins (pyridoxine, pyridoxal, and pyridoxamine) through the action of pyridoxal kinase. Pyridoxal kinase itself requires zinc for activation.
A deficiency in zinc or defects in pyridoxal kinase can lead to seizure disorders, particularly in seizure-prone pre-eclamptic patients (hypertensive condition in late pregnancy). The presence of anti-GAD antibodies (both anti-GAD65 and anti-GAD67) is a strong predictor of the future development of type 1 diabetes in high-risk populations.
GABA exerts its effects by binding to two distinct receptors, GABA-A (GABAA) and GABA-B (GABAB) that are members of the ion channel (ionotropic) receptors. GABA-A receptors are chloride channels that in response to GABA binding increases chloride influx into the neuron. The GABA-B receptors are potassium channels that when activated by GABA leads to potassium efflux from the cell. For more details on the synthesis and function of GABA go to the Biochemistry of Nerve Transmission page.
Histamine from Histidine
Histamine is a potent neurotransmitter that binds to specific histamine receptors. Histamine is synthesized by the enzymatic decarboxylation of the amino acid histidine by the enzyme L-histidine decarboxylase (HDC). Within the gastrointestinal tract bacteria also produce histamine via a similar decarboxylation reaction. The principal cells that synthesize and release histamine are mast cells and basophils of the immune system, enterochromaffin-like cells of the gastrointestinal system, and neurons. The synthesis and storage of histamine by mast cells and basophils represents the greatest store (>90%) of the neurotransmitter. Within the brain the neurons that synthesize histamine are within the tuberomammillary nucleus of the hypothalamus.
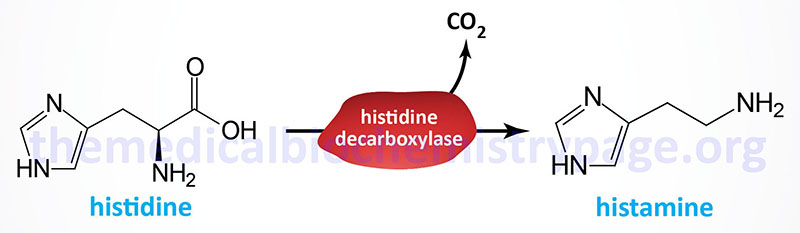
Histidine decarboxylase is encoded by the HDC gene. The HDC gene is located on chromosome 15q21.2 and is composed of 14 exons that generate two alternatively spliced mRNAs encoding two distinct isoforms of the enzyme. The isoform 1 protein is composed of 662 amino acids and the isoform 2 protein is composed of 629 amino acids.
Histamine Receptors
Humans express four distinct histamine receptors identified as H1R, H2R, H3R, and H4R. All four histamine receptors are members of the G-protein coupled receptor superfamily. The H1R protein is encoded by the HRH1 gene, the H2R protein is encoded by the HRH2 gene, the H3R protein is encoded by the HRH3 gene, and the H4R protein is encoded by the HRH4.
The HRH1 gene is located on chromosome 3p25 and is composed of 8 exons that generate four alternatively spliced mRNAs all of which encode the same 487 amino acid protein.
The HRH2 gene is located on chromosome 5q35.2 and is composed of 8 exons that generate two alternatively spliced mRNAs. HRH2 isoform 1 is 397 amino acids and HRH2 isoform 2 is 359 amino acids.
The HRH3 gene is located on chromosome 20q13.33 and is composed of 4 exons that encode a 445 amino acid protein.
The HRH4 gene is located on chromosome 18q11.2 and is composed of 5 exons that generate three alternatively spliced mRNAs. Isoform 1 is 390 amino acids. Isoform 2 is 302 amino acids. Isoform 3 is 67 amino acids.
The H1R protein is coupled to a Gq-type G-protein. These receptors are expressed in a wide variety of tissues including the gastrointestinal tract, central nervous system (specifically cells of the tuberomammillary nucleus of the hypothalamus), both airway and vascular smooth muscle cells, endothelial cells, chondrocytes, monocytes, neutrophils, dendritic cells, T and B lymphocytes, adrenal medulla, and the cardiovascular and genitourinary systems. Expression of H1R in lymphocytes and monocytes is critical to the role of this receptor in mediating the modulatory effects of histamine within the immune system. With its distributed expression the H1R participates in ileum contraction, modulates circadian cycle, systemic vasodilatation, bronchoconstriction (allergy-induced asthma), and mediates pruritus (sensation of itchy skin).
The H2R protein is coupled to a Gs-type G-protein. Most immune cells that express the HRH1 gene also express the HRH2 gene. In addition to immune cells, the HRH2 gene is expressed in gastric parietal cells where the receptor plays a critical role in the regulation of gastric acid production in response to histamine. Indeed, histamine is the major molecule responsible for increased gastric acid production. Acetylcholine (ACh) and gastrin also contribute to gastric acid production but not to the degree of histamine. The HRH2 gene is also expressed in additional cells of the gastrointestinal tract, cells of the central nervous system, smooth muscle cells, endothelial cells, and cardiomyocytes. The principal responses to histamine binding to the H2R are stimulation of gastric acid secretion, increased sinus rhythm (cardiac rhythm initiated via depolarization from the sinus node), smooth muscle relaxation, inhibition of antibody synthesis, inhibition of T-cell proliferation and inhibition of cytokine production.
The H3R protein is coupled to a Gi-type G-protein. Expression of the HRH3 gene occurs nearly exclusively within the central nervous system, CNS. Regions of the brain that contain cell types expressing H3R include basal ganglia, cortex, hippocampus and strial area. Within the CNS the effects of histamine binding to H3R include decreases in acetylcholine (ACh), serotonin and norepinephrine production and release. Outside the CNS low level HRH3 expression has been found in the gastrointestinal and cardiovascular systems and in bronchial cells of the lungs.
The H4R protein is coupled to a Gi-type G-protein. Expression of the HRH4 gene is seen at high levels in the gastrointestinal tract, spleen, thymus, medullary cells, bone marrow and peripheral hematopoietic cells, including eosinophils, basophils, mast cells, T-cells, leukocytes and dendritic cells. Lower levels of expression of HRH4 are detected in the brain, heart, liver and lung.
For more details on histamine function go to the Biochemistry of Nerve Transmission page.
Nitric Oxide Synthesis and Function
Vasodilators, such as acetylcholine, bradykinin, and adenosine do not exert their effects upon the vascular smooth muscle cells in the absence of the overlying endothelium. When acetylcholine (or bradykinin or adenosine) binds its receptor on the surface of endothelial cells, a signal cascade, coupled to the activation phospholipase C-β (PLCβ), is initiated. The activation of PLCβ, via acetylcholine binding to vascular endothelial cells, results from the activation of the M3 (muscarinic) receptor. The activation of PLCβ via bradykinin results from its binding to the B2 bradykinin receptor. The activation of PLCβ via adenosine results from the activation of the adenosine A2a receptor subtype in the coronary vascular endothelium.
All three receptor subtypes (muscarinic acetylcholine M3, bradykinin B2, and adenosine A2a) are coupled to Gq-type G-proteins that exert their effects through activation of PLCβ. The activation of PLCβ results in the release of inositol-1,4,5-trisphosphate (IP3) from membrane associated phosphatidylinositol-4,5-bisphosphate (PIP2), which then binds to specific IP3 receptors in the endoplasmic reticulum/sarcoplasmic reticulum (ER/SR) resulting in the release of intracellular stores of Ca2+. Original observations demonstrated that the elevation in cytosolic Ca2+ led to the production of a factor that was termed endothelium-derived relaxing factor (EDRF). EDRF was shown to then diffuse from the endothelium to the underlying smooth muscle resulting in smooth muscle relation and vasodilation.
Quite unexpectedly, EDRF was found to be the free radical diatomic gas, nitric oxide, NO. So stunning was the elucidation of the pathway to, and actions of, NO that Drs. Murad, Ignarro, and Furchgott were awarded the Nobel Prize in 1998 for their work on this system.
Endogenous NO is formed by the action of NO synthase, (NOS) on the amino acid arginine. The activation of NOS in endothelial cells by acetylcholine, bradykinin, or adenosine is the result of the increased cytosolic Ca2+ binding to the calmodulin subunit of the NOS complex. The half-life of NO is extremely short, lasting only 2-4 seconds. This is because it is a highly reactive free radical and interacts with oxygen and superoxide anion. NO is also inhibited by hemoglobin and other heme proteins which bind it tightly.
Within smooth muscle cells (SMC), NO reacts with the heme moiety of a soluble guanylate cyclase (sGC), resulting in activation of the latter and a consequent elevation of intracellular levels of cGMP. The activity of NO as a biological messenger classifies it as a gasotransmitter. The net effect is the activation of cGMP-dependent protein kinase (PKG; specifically the PKGI isoforms) and the phosphorylation of various protein substrates.
Significant targets and the effects of PKGI in SMC are activation of myosin light chain phosphatase, inhibition of PLCβ, inhibition of the IP3 receptor, inhibition of plasma membrane localized L-type Ca2+ channels, and activation of plasma membrane localized calcium-activated K+ channels (commonly called the big potassium, BK, channels). The net effect of all these phosphorylation events is smooth muscle cell relaxation.
In addition to activating PKGI, cGMP itself contributes to smooth muscle relaxation through direct inhibition of the plasma membrane localized L-type Ca2+ channels. Inhibition of the plasma membrane L-type Ca2+ channels results in reduced intracellular Ca2+ concentration which reduces the capacity to activate myosin light chain kinase, a key enzyme in the contractile activity of smooth muscle.
NO and S-Nitrosylation of Proteins
Although many of the effects of NO are exerted via the activation of soluble guanylate, NO can also directly modify the sulfhydryl group of cysteine residues in proteins, a process termed S-nitrosylation. Protein S-nitrosylation involves the attachment of NO to the reactive thiol group of a cysteine in a target protein generating an S-nitrosothiol (SNO). In addition to the role of the NOS enzymes in generating NO, S-nitrosylation of proteins involves enzymes called SNO synthases and tansnitrosylases. SNO synthases and transnitrosylases can both be classified as protein S-nitrosylases. The SNO synthases convert NO into SNO and then the transnitrosylases transfer NO to the target cysteine generating the S-nitrosylated protein (SNO-protein). Proteins that undergo S-nitrosylation directly interact with NOS. The cysteine that is S-nitrosylated is found to reside in a consensus motif I/L-X-C-X2-D/E, where X refers to any amino acid. Proteins undergo removal of the S-nitrosylation via the action of denitrosylases.
Important proteins in smooth muscle that are targets for NO-mediated S-nitrosylation are the L-type Ca2+ channels. As indicated, the inhibition of the plasma membrane L-type Ca2+ channels results in reduced intracellular Ca2+ concentration and a consequent reduction in the contractile activity of smooth muscle.
Within cardiac myocytes nNOS is localized to the sacroplasmic reticulum (SR) whereas eNOS is localized to caveolae in the plasma membrane. With its localization to the SR, nNOS regulates the S-nitrosylation of the calcium release channels, the type 2 ryanodine receptors (RYR2) resulting in release of stored calcium, The localization of nNOS in the plasma membrane allows for direct S-nitrosylation of L-type Ca2+ channels resulting in increased influx of calcium. The net effect of these concerted actions of nNOS and eNOS is increased inotropy (contractile force) of the heart.
Mammalian Isoforms of NOS
Neuronal NOS (nNOS), also called NOS-1. The NOS1 gene is located on chromosome 12q24.22 and is composed of 33 exons the generate four alternatively spliced mRNAs that collectively encode three distinct protein isoforms.
Inducible or macrophage NOS (iNOS), also called NOS-2. The NOS2 gene is located on chromosome 17q11.2 and is composed of 27 exons that encode a protein of 1153 amino acids.
Endothelial NOS (eNOS), also called NOS-3. The NOS3 gene is located on chromosome 7q36.1 and is composed of 28 exons that generate four alternatively spliced mRNAs, each of which encode distinct protein isoforms.
Nitric oxide synthases are very complex enzymes, employing five redox cofactors: NADPH, FAD, FMN, heme, and tetrahydrobiopterin (H4B; also written BH4). The role of Ca2+ ions in the function of NOS is related to the calmodulin subunits of NOS enzymes (see below). All NOS enzymes also require Zn2+ ions for function. The role of zinc in NOS function is a structural one not a catalytic one.
All three NOS enzymes function as homodimers and the interactions of the subunits is required for the process of electron flow from NADPH (bound in the reductase domain of the enzyme), through the various co-factors to the heme moiety which is bound to the N-terminal oxygenase domain of the enzyme. It is the oxygenase domain that binds the required oxygen and arginine substrates. The oxygenase domain also binds the H4B co-factor. The actual process of NO production by NOS enzymes involves two steps. In the first step the enzyme hydroxylates arginine to Nω-hydroxy-L-arginine. During the second step the enzyme oxidizes the Nω-hydroxy-L-arginine to L-citrulline and NO.
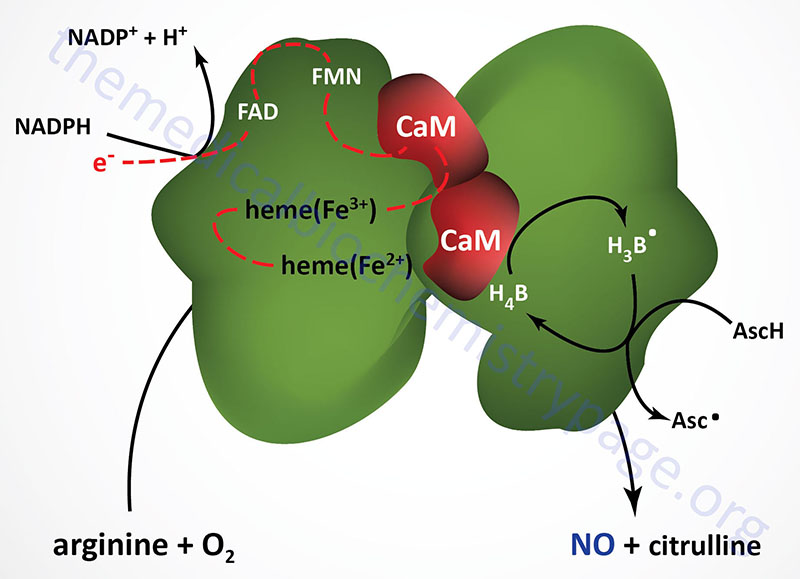
Both eNOS and nNOS are constitutively expressed and regulated by Ca2+. The calcium regulation is imparted by the associated calmodulin subunits, thus explaining how vasodilators such as acetylcholine and bradykinin effect smooth muscle relaxation as a consequence of increasing intracellular endothelial cell calcium levels. Although iNOS interacts with calmodulin, its activity is unaffected by changes in Ca2+ concentration. iNOS is transcriptionally activated in macrophages, neutrophils, and smooth muscle cells. When intracellular Ca2+ levels rise the calmodulin subunits have an increased affinity for eNOS and nNOS. The calmodulin subunits facilitate the flow of electrons from NADPH to the heme moiety in the oxygenase domain of the enzyme.
Endothelial NOS, eNOS
Endothelial NOS (eNOS) is most highly expressed within endothelial cells. However, expression of eNOS is also detected in platelets, cardiac myocytes, specific renal tubular epithelial cells, certain neurons within the brain, and in placental syncytiotrophoblast cells. As indicated, the major regulator of eNOS (and nNOS) activity is Ca2+ ion. However, other factors regulate the activity of eNOS that are not related to changes in calcium concentrations. Several proteins, such as caveolin-1 and hsp90, interact with and affect the overall activity of eNOS.
In addition, eNOS activity can be modulated by phosphorylation. The predominant trigger for eNOS phosphorylation is fluid shear stress in the vasculature. Additional triggers for eNOS phosphorylation include vascular endothelial growth factor (VEGF)-induced signal transduction, insulin-mediated activation of PKB/AKT, and bradykinin-induced activation of Ca2+/calmodulin-dependent protein kinase II (CaMKII). PKB (protein kinase B)/AKT (AK strain transforming) was originally identified as the tumor inducing gene in the AKT8 retrovirus found in the AKR strain of mice. Humans express three genes in the AKT family identified as AKT1 (PKBα), AKT2 (PKBβ), and AKT3 (PKBγ). Phosphorylation of eNOS enhances the flow of electrons through the reductase domain of the enzyme. The predominant effects of NO produced in response to eNOS activation are vasodilation and inhibition of platelet adhesion and aggregation.
Endothelial NOS-derived NO induces the dilation of all types of blood vessels by both S-nitrosylation of proteins and through the stimulation of soluble guanylate cyclase (sGC) and increasing cGMP production in smooth muscle cells. S-nitrosylation of proteins alters their activity directly. Increases in cGMP result in the inhibition of plasma membrane localized L-type Ca2+ channels resulting in decreasing overall intracellular Ca2+ which normally serves to activate myosin light chain kinase leading to vasoconstriction. The same L-type Ca2+ channels can be directly inhibited by NO via S-nitrosylation. The cGMP also activates PKGI isoforms resulting in the phosphorylation and activation of myosin light chain phosphatase resulting in phosphate removal from myosin light chains contributing to vasodilation. The cGMP also activates K+ channels inducing hyperpolarization of the smooth muscle resulting in relaxation.
The ability of eNOS produced NO to prevent platelet adhesion and aggregation reduces the propensity for the development of abnormal fibrin clots (thrombi) and, therefore, will contribute to a reduced potential for the development of atherosclerosis. Endothelial cell produced NO can also interfere with leukocyte adhesion to the endothelium thus, further contributing to a reduction in the potential for atherosclerosis.
Neuronal NOS, nNOS
Neuronal NOS (nNOS) is constitutively expressed in specific neurons within the brain and spinal cord. Neurons that express nNOS and thus produce NO, are called nitrergic nerves. Neuronal NOS is also constitutively expressed within the adrenal glands, pancreatic islet cells, epithelial cells of numerous tissues, vascular smooth muscle, skeletal muscle, and renal macula densa cells. Within the brain the principal functions of nNOS produced NO are in the mediation of long-term synaptic transmission. Production of NO within the brain, via nNOS, is also important for the central regulation of blood pressure.
Expression of nNOS in the autonomic nervous system innervating the heart as well as the heart tissue itself plays a central role in cardiac physiology. In addition to the nerves, nNOS is expressed in the aortic and pulmonary arteries, the coronary arteries and the atrial and ventricular myocardium. The expression of nNOS in cardiac tissue allows it, through the production of NO, to play essential roles in the modification of sympathetic and parasympathetic tone within the heart itself, in the regulation of heart rate, and in the delivery of essential nutrients through the coronary arteries as well as through the regulation of myocardial contractility. Cardiac nNOS is localized to the sarcoplasmic reticulum (SR) where it is involved in the Ca2+ handling processes of cardiac excitation-contraction coupling.
Neuronal NOS is also expressed within smooth muscle cells of the penile corpus cavernosum and thus, NO produced by this enzyme is involved in penile erection. Smooth muscle relaxation induced by NO in the corpus cavernosum is mediated by cyclic GMP (cGMP). The cGMP that is produced via NO activation of soluble guanylate cyclase (sGC) in this tissue is degraded by a specific cyclic nucleotide phosphodiesterase (PDE) identified as PDE5A. Residual corpus cavernosum nNOS activity is essential for the erectile promoting actions of selective PDE5 inhibitors such as sildenafil (Viagra), vardenafil (Levitra), and tadalafil (Cialis).
Inducible NOS, iNOS
The major functions of NO production through activation of iNOS are associated with the bactericidal and tumoricidal actions of macrophages. Overproduction of NO via iNOS is associated with cytokine-induced septic shock such as occurs post-operatively in patients with bacterial infections. Bacteria produce endotoxins such as lipopolysaccharide (LPS) that activate iNOS in macrophages.
Although predominately induced in macrophages, under appropriate stimulation conditions many other cell types will express the iNOS gene. Once expressed, the iNOS enzyme is catalytically active and does not respond with changes in enzyme activity to changes in intracellular calcium levels as is the case for nNOS and eNOS. The production of large amounts of NO by activation of iNOS expression in macrophages represents the major cytotoxic activity of these cells. The NO produced binds to the iron in numerous enzymes that contain the metal in their active sites. Examples of NO-inhibited enzymes include the Fe-S cluster-dependent activities of mitochondrial oxidative phosphorylation and ribonucleotide reductase.
Macrophage generated NO can also cause DNA strand breaks and fragmentation. These effects of macrophage produced NO explain, to a large degree, the cytotoxic and cytostatic effects of NO on invading parasites as well as on certain tumor cells.
Generation of Reactive Nitrogen Species
Like reactive oxygen species (ROS), reactive nitrogen species (RNS) can be toxic if allowed to accumulate in cells. The major source of various RNS is the synthesis of nitric oxide, NO (correctly identified as NO●). NO reacts with superoxide anion (O2●–) to form a stronger oxidant identified as peroxynitrite anion (ONOO–). The peroxynitrite anion will then react with other molecules to generate additional RNS that includes as nitrogen dioxide (●NO2) and dinitrogen trioxide (N2O3). The reduction of NO also results in the formation of another RNS identified as the nitroxyl anion (NO–). The oxidation of NO in the presence of ●NO2 can lead to the generation of the RNS identified as the nitrosomium cation (NO+).
Reactive nitrogen species (RNS) are neutralized by the same systems that are responsible for the neutralization of reactive oxygen species, ROS. These systems include the various low molecular weight antioxidants such as reduced glutathione (GSH), vitamin C and vitamin E, bilirubin, and urate. There are also several non-catalytic antioxidant proteins such as the glutaredoxins, the thioredoxins (Trx), and the metallothioneins (MT). The catalytic antioxidant proteins include the
superoxide dismutases (SOD), catalase, the peroxiredoxins (Prx), and the glutathione peroxidases (GPx).
Nitric Oxide Pharmacology
Pharmacologic vasodilators include nitrate compounds such as glycerin trinitrate (nitroglycerin, NTG), nitroprusside, isosorbide dinitrate, and isosorbide mononitrate. Nitroglycerin and the isosorbides are converted to nitric oxide through the action of mitochondrial aldehyde dehydrogenase (encoded by the ALDH2 gene). At the low doses of nitroglycerin used to treat angina pectoris (cardiac pain associated with ischemic heart disease) there is a greater effect of vasodilation in veins compared to arteries which in turn reduces the volume of blood returning to the heart (venous return is referred to as preload). With reduced preload the heart does not have to exert as much contractile force which reduces the oxygen requirements of the heart and also reduces ventricular transmural pressure resulting in reduced compression of the coronary arteries allowing for more efficient blood flow through the heart.
Nitroglycerin can exert vasodilation in the arteries at higher doses which results in a decrease in the pressure the heart must exert (termed afterload) to expel blood from the left ventricle, thereby, adding to reduced cardiac oxygen demand.
Sodium nitroprusside is disodium pentacyanonitrosylferrate, most often in the dihydrate form: Na2[Fe(CN)5NO]·2H2O. Nitroprusside interacts with hemoglobin leading to the release of nitric oxide and cyanide, and the formation of methemoglobin [ferric iron (Fe3+) containing hemoglobin]. Because of the release of cyanide (CN–) nitroprusside administration can result in toxicities.
Tetrahydrobiopterin Synthesis
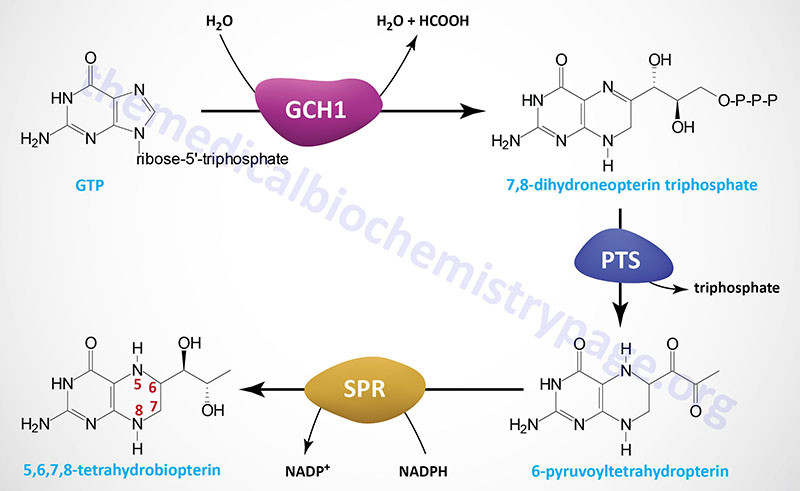
Tetrahydrobiopterin (6R-L-erythro-5,6,7,8-tetrahydrobiopterin) is most often abbreviated BH4 but also can be written as HB4. Depending upon cell type BH4 can be generated by several different (alternative) de novo biosynthesis pathways as well as by two different salvage pathways. The major de novo pathway is diagrammed in the Figure below. The synthesis of BH4 occurs de novo from the nucleotide, GTP. There are three principal enzymes involved in the major de novo pathway of BH4 synthesis. In reaction order these enzymes are GTP cyclohydrolase 1 (GTPCH), 6-pyruvoyltetrahydropterin synthase (PTPS), and sepiapterin reductase (SR).
GTP cyclohydrolase 1 is encoded by the GCH1 gene which is located on chromosome 14q22.2 and is composed of 10 exons that generate four alternatively spliced mRNAs that collectively encode three distinct protein isoforms.
The 6-pyruvoyltetrahydropterin synthase enzyme is encoded by the PTS gene which is located on chromosome 11q23.1 and is composed of 6 exons that encode a 145 amino acid protein.
Sepiapterin reductase is encoded by the SPR gene which is located on chromosome 2p13.2 and is composed of 3 exons that encode a 261 amino acid protein.
The reaction catalyzed by GTP cyclohydrolase 1 (GTPCH) is considered the rate-limiting and committed reaction of BH4 synthesis. Functional GTP cyclohydrolase 1 exists as a homodecamer that consists of two tightly associated dimers that each comprise a pentameric complex of the GTP cyclohydrolase 1 monomers. Through the actions of GTP cyclohydrolase 1 GTP is converted to 7,8-dihydroneopterin triphosphate.
The second step in BH4 synthesis, catalyzed by 6-pyruvoyltetrahydropterin synthase (PTPS), involves the Zn2+– and Mg2+-dependent rearrangement of 7,8-dihydroneopterin triphosphate, with concommitant loss of the triphosphate, to 6-pyruvoyltetrahydropterin. Functional 6-pyruvoyltetrahydropterin synthase is a homoheaxmeric complex that is formed via the head-to-head interaction of a pair of trimers. The final step of BH4 synthesis actually involves three distinct steps all of which are catalyzed by the NADPH-dependent enzyme, sepiapterin reductase (SR). Functional sepiapterin reductase is a homodimeric enzyme which first converts 6-pyruvoyltetrahydropterin to 1′-hydroxy-2′-oxopropyltetrahydropterin, which is then reduced to 1′-oxo-2′-hydroxypropyltetrahydropterin, and then finally to 5,6,7,8-tetrahydrobiopterin, BH4.
Creatine Biosynthesis
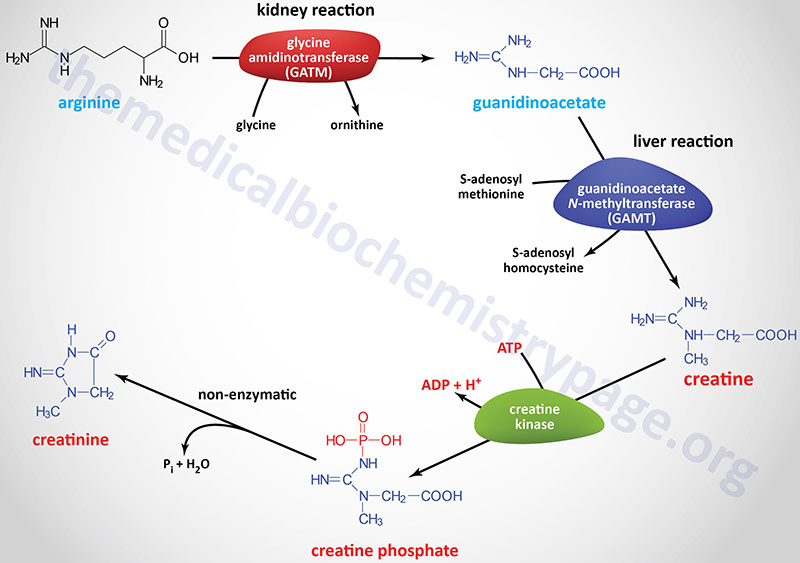
Creatine synthesis occurs predominantly in the kidneys and the liver. The synthesis of creatine begins in the kidneys using the amino acids arginine and glycine. The formation of guanidinoacetate (GAA) from these two amino acids is catalyzed by the enzyme glycine amidinotransferase, also called L-arginine:glycine amidinotransferase (AGAT).
Glycine amidinotransferase is encoded by the GATM gene. The GATM gene is located on chromosome 15q21.1 and is composed of 13 exons that generate two alternatively spliced mRNAs that encode proteins of 423 amino acids (isoform 1) and 294 amino acids (isoform 2). The GAMT encoded proteins localize to the mitochondria.
Guanidinoacetate it transported to the blood and picked up by hepatocytes where it is methylated forming creatine. The methyl donor for this reaction is S-adenosylmethionine (SAM or AdoMet) and the reaction is catalyzed by the enzyme guanidinoacetate N-methyltransferase.
Guanidinoacetate N-methyltransferase is encoded by the GAMT gene. The GAMT gene is located on chromosome 19p13.3 and is composed of 6 exons that generate two alternatively spliced mRNAs that encode proteins of 236 amino acids (isoform a) and 269 amino acids (isoform b).
Following synthesis creatine is released to the blood where is is picked up, predominantly, by the brain and skeletal muscle cells through the action of the SLC family member transporter, SLC6A8.
The SLC6A8 gene is located on the X chromosome (Xq28) and is composed of 14 exons that generate three alternatively spliced mRNAs, each of which encode a distinct protein isoform.
Within cells, creatine is phosphorylated by creatine kinases (CK; also called creatine phosphokinase, CPK) that generate the high-energy storage compound, creatine phosphate. There are two creatine kinase genes in humans identified as the muscle creatine kinase gene (symbol: CKM) and the brain creatine kinase gene (symbol: CKB).
The CKM gene is located on chromosome 19q13.32 and is composed of 8 exons that encode a protein of 381 amino acids.
The CKB gene is located on chromosome 14q32.33 and is composed of 7 exons that generate two alternatively spliced mRNAs encoding proteins of 381 amino acids (isoform 1) and 405 amino acids (isoform 2)..
Different combinations of the proteins encoded by these two genes generate tissue-specific isoforms of the functional enzymes. For details on the different forms of CK go to the Enzyme Kinetics and Diagnostic Uses of Enzymes page.
Although creatine, synthesized by the kidneys and liver, can be transported from the blood into the brain, during both embryonic development and during postnatal life the brain can directly synthesize its own creatine. The GATM and GAMT genes are expressed in neural tissues. However, the level of creatine synthesis in the embryonic nervous system is not sufficient for needs and so the developing brain requires maternally and peripherally synthesized creatine.
In the adult brain, glial cells and neurons continue to express the GATM and GAMT genes, however, populations of cells express only the GATM gene while others express only the GAMT gene. Therefore, similar to the transport of GAA from the kidneys to the liver for creatine synthesis, the GAA produced in certain neural cells must be transported into GAMT expressing cells in order for brain creatine synthesis. This latter transport is likely catalyzed by the SLC6A8 encoded transporter. This explains why, in patients with mutations in the SLC6A8 gene, there is a lack of sufficient brain creatine. Like in skeletal muscle, brain creatine phosphate plays an important role in the regulation of a high energy balance in the brain. Creatine also functions as a neurotransmitter in the brain by acting as a partial agonist at postsynaptic GABAA receptors.
Creatine is used as a storage form of high energy phosphate. The phosphate of ATP is transferred to creatine, generating creatine phosphate, through the action of creatine phosphokinase (CPK; also called creatine kinase, CK). The reaction is reversible such that when energy demand is high (e.g. during muscle exertion) creatine phosphate donates its phosphate to ADP to yield ATP. Both creatine and creatine phosphate are found in muscle, brain, and blood.
Creatinine is formed in muscle from creatine phosphate by a non-enzymatic dehydration and loss of phosphate. The amount of creatinine produced is related to muscle mass and remains remarkably constant from day to day. Creatinine is excreted by the kidneys and the level of excretion (creatinine clearance rate: CCR) is a measure of renal function.
Creatine Deficiency Syndromes
Creatine deficiency syndromes (CDS) represent a group of disorders that result from defects in the synthesis and transport of creatine. These disorders are also referred to as primary metabolism disorders of creatine. As discussed in the previous section, there are two enzymes of creatine synthesis, glycine amidinotransferase (encoded by the GATM gene ) and guanidinoacetate N-methyltransferase (encoded by the GAMT gene), and one major creatine transporter encoded by the SLC6A8 gene.
The first disorder in creatine metabolism to be characterized involved mutations in the GAMT gene. The initial GAMT deficient patient experienced severe psychomotor impairment, refractory epilepsy, and movement disorder. The patient exhibited no cerebral creatine along with high guanidinoacetate (GAA) concentrations in the brain and body fluids. Treatment of this patient was effected by oral administration of creatine monohydrate.
Following this initial patient characterization, mutations in the GATM gene and the SLC6A8 gene were found in patients exhibiting similar pathology to that seen with mutations in the GAMT gene. The GATM and GAMT mutation-associated disorders are inherited as autosomal recessive diseases. The SLC6A8 gene is located on the X chromosome and, therefore, creatine transporter deficiencies are inherited in an X-linked recessive manner. Few GATM deficient patients have been identified, 40 GAMT deficient patients have been identified, and around 150 SLC6A8 deficient patients have been identified to date.
Although skeletal muscle creatine synthesis will be impaired in individuals with GATM and GAMT mutations, impaired muscle function is not the major symptom in creatine deficiency syndrome patients. All of these disorders exert dramatic effects on central nervous system function and, therefore, neurological symptoms are the main clinical complications in these patients. The clinical features in creatine deficiency syndromes include intellectual disability, speech and language delay, epilepsy and autism spectrum disorder.
Due to the different levels of enzyme defect with various mutations and the different genes there is wide variability in the severity of the clinical symptoms. Patients with GATM deficiencies have milder clinical manifestations which are generally limited to moderate intellectual disability and language disorder. Patients with GAMT deficiencies have high concentrations of GAA in body fluids and manifest with refractory epilepsy and severe movement disorders. Patients with SLC6A8 deficiencies exhibit intellectual impairment, epilepsy, autism spectrum disorder, and severe language delay.
Treatment of both GATM and GAMT deficient patients can be carried out with oral creatine monohydrate. However, this therapy does not lower the levels of the intermediate metabolite, GAA, seen with GAMT deficiency. In these latter patients the addition of ornithine supplementation and dietary restriction in arginine help to reduce the production of GAA. The reductions in GAA provide better control of the refractory epilepsy that is common in GAMT deficiency. Currently there is no effective treatment for patients with SLC6A8 deficiency.
Glutathione Synthesis and Functions
Glutathione (abbreviated GSH) is a tripeptide composed of glutamate, cysteine and glycine that has numerous important functions within cells. Glutathione serves as a potent antioxidant eliminating reactive oxygen species (ROS) such as hydroxy radicals, peroxynitrites, and hydroperoxides. Glutathione is conjugated to drugs to make them more water soluble. Glutathione is involved in amino acid transport across cell membranes via a process known as the glutathione cycle (historically but incorrectly referred to as the γ-glutamyl cycle). Glutathione is a substrate for the synthesis of the peptidoleukotrienes. Glutathione serves as a cofactor for several enzymatic reactions. Glutathione also serves as an aid in the rearrangement of protein disulfide bonds both in the cytosol and in the endoplasmic reticulum, ER.
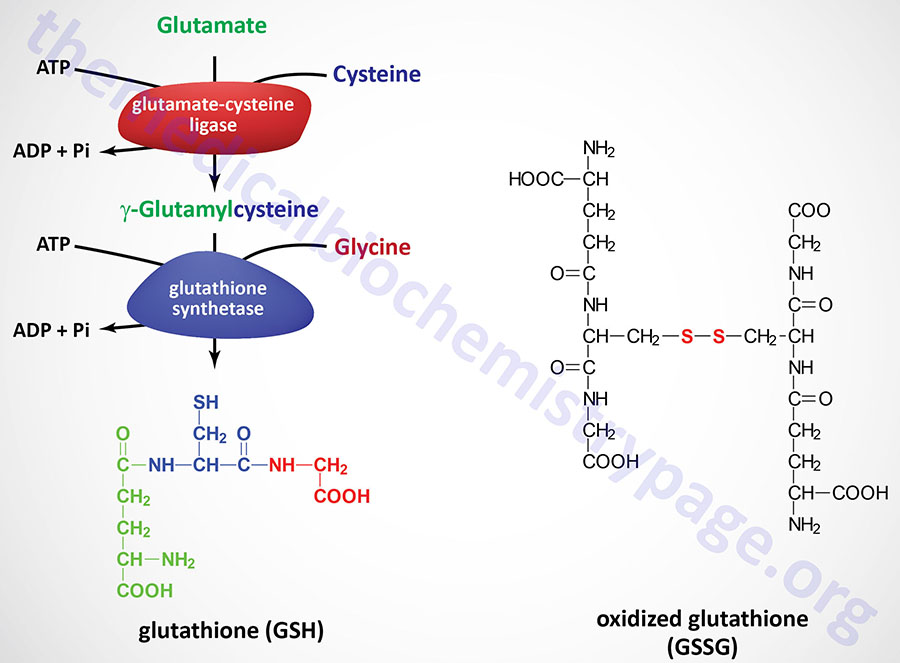
GSH is synthesized in the cytosol of all mammalian cells via the two-step reaction shown in the Figure above. The rate of GSH synthesis is dependent upon the availability of cysteine and the activity of the rate-limiting enzyme, γ-glutamylcysteine synthetase (also called glutamate-cysteine ligase, GCL). The second reaction of GSH synthesis involves the enzyme, glutathione synthetase, which condenses γ-glutamylcysteine with glycine. Both reactions of GSH synthesis require ATP.
Functional GCL is a heterodimer composed of the catalytic subunit and a regulatory subunit called the modifier subunit. The GCL catalytic subunit is encoded by the GCLC gene and the modifier subunit is encoded by the GCLM gene.
The GCLC gene is located on chromosome 6p12.1 and is composed of 16 exons that generate two alternatively spliced mRNA that encode GCL isoform a (637 amino acids) and GCL isoform b (599 amino acids).
The GCLM gene is located on chromosome 1p22.1 and is composed of 9 exons that generate two alternatively spliced mRNAs that encode proteins of 274 amino acids (isoform 1) and 252 amino acids (isoform 2).
The glutathione synthetase gene (symbol: GSS) is located on chromosome 20q11.22 and is composed of 15 exons that generate three alternatively spliced mRNAs, each of which encode the same 474 amino acid protein.
In the liver, major factors that determine the availability of cysteine are diet, membrane transport activities of the three sulfur amino acids cysteine, cystine, and methionine, and the conversion of methionine to cysteine (see the Amino Acid Biosynthesis page). Numerous conditions can alter the level of GSH synthesis via changes in glutamate-cysteine ligase activity and expression of the GCLC gene such as oxidative stress, antioxidant levels, hormones, cell proliferation, and diabetes mellitus.
The role of GSH as a antioxidant is extremely important particularly in the highly oxidizing environment of the erythrocyte. The sulfhydryl of GSH can be used to reduce peroxides formed during oxygen transport. Endogenously produced hydrogen peroxide (H2O2) is reduced by GSH in the presence of selenium-dependent GSH peroxidase. Hydrogen peroxide can also be reduced by catalase, which is present only in the peroxisomes. In the mitochondria, GSH is particularly important because mitochondria lack catalase.
The resulting oxidized form of GSH consists of two molecules disulfide bonded together (abbreviated GSSG). The enzyme glutathione reductase utilizes NADPH as a cofactor to reduce GSSG back to two moles of GSH. Hence, the pentose phosphate pathway is an extremely important pathway of erythrocytes for the continuing production of the NADPH needed by glutathione reductase. In fact as much as 10% of glucose consumption, by erythrocytes, may be mediated by the pentose phosphate pathway.
Glutathione also functions within the endoplasmic reticulum (ER) where it performs the critical function of reducing oxidized chaperones of the protein disulfide isomerase (PDI) family. During the process of correct protein folding in the ER, the PDI enzymes catalyze disulfide bond isomerization and reduction which aids in correct protein folding. During this process the PDI enzymes are themselves oxidized, requiring ER-localized GSH to carry out their reduction back to a function state. Since the ER lacks glutathione reductase the oxidized glutathione (GSSG) must be transported to the cytosol for reduction. This transport is carried out by the SLC33A1 encoded transporter. Clinical significance of the SLC33A1 encoded transporter is evident from the fact that mutation in the gene are associated with a lethal disorder identified as Huppke-Brendel syndrome (HPBDS). HPBDS is associated with severe psychomotor impairment, congenital hearing loss, bilateral cataracts, and low serum ceruloplasmin and copper.
Detoxification of xenobiotics or their metabolites is another major function of GSH. These compounds form conjugates with GSH either spontaneously or enzymatically in reactions catalyzed by members of the glutathione S-transferase (GST) family. The conjugates formed are usually excreted from the cell and, in the case of the liver they are excreted in the bile.
There are several GST enzymes that are divided into three major families that comprise cytosolic, mitochondrial, and microsomal family members. The cytosolic GST enzymes comprise the largest family and based upon amino acid sequences and substrate specificities this family has been divided into seven subclasses identified as alpha (α, A), mu (μ, M), omega (ω, O), pi (π, P), sigma (σ, S), theta (θ, T), and zeta (ζ, Z).
The human genome contains five GST alpha genes, five mu genes, two omega genes, one pi gene, one sigma gene, two theta genes, and one zeta gene. The cytosolic enzymes are functional as dimers and since the alpha and mu proteins can form functional heterodimers, the complexity of functional enzymes is greater than the number of individual subunit genes. Another soluble GST enzyme that is not found in the cytosol is the human GST kappa (κ, K) enzyme (encoded by the GSTK1 gene) that is localized to the peroxisomes. The GST enzymes function not only in detoxification and antioxidant reactions but also in the biosynthesis and metabolism of eicosanoids and steroids as well as in the modulation of signal transduction processes.
Table of Human Cytosolic Glutathione S-Transferases
| Class | Enzyme Subunits | Encoding Genes | Comments |
| Alpha | 1, 2, 3, 4, 5 | GSTA1, GSTA2, GSTA3, GSTA4, GSTA5 | the GSTA genes are clustered on chromosome 6; most abundantly expressed class of GSTs in the liver; also expressed in kidney and testis |
| Mu | 1, 2, 3, 4, 5 | GSTM1, GSTM2, GSTM3, GSTM4, GSTM5 | the GSTM genes are clustered on chromosome 1; the mu genes are highly polymorphic accounting for a high degree of variability in a given individuals ability to detoxify certain toxins and carcinogens; GSTM2 expression highest in muscle; GSTM3 expression highest in brain; polymorphisms in GSTM1 gene associated with hypertension and increased risk for schizophrenia |
| Omega | 1, 2 | GSTO1, GSTO2 | both genes found on chromosome 10 |
| Pi | 1 | GSTP1 | expression is highest in the brain, lung, and heart; polymorphisms in GSTP1 gene associated with increased risk for schizophrenia |
| Sigma | 1 | HPGDS | the sigma GST enzyme is a prostaglandin-D synthase encoded by the hematopoietic prostaglandin D synthase gene; catalyzes the conversion of PGH2 to PGD2 |
| Theta | 1, 2 | GSTT1, GSTT2, GSTT2B | the GSTT genes are clustered on chromosome 22; the GSTT1 gene is absent in 38% of the population; in some populations the GSTT2B gene is a pseudogene; polymorphisms in GSTT1 associated with hypertension and increased risk for schizophrenia |
| Zeta | 1 | GSTZ1 | this GST catalyzes the conversion of maleylacetoacetate to fumarylacetoacetate in the catabolism of phenylalanine and tyrosine; originally identified as 4-maleylacetoacetate isomerase or maleylacetoacetate cis–trans-isomerase |
Protein S-Glutathionylation
In response to increased production of reactive oxygen species (ROS) glutathione forms disulfide bonds with cysteines in proteins in a process termed protein S-glutathionylation. The process of protein S-glutathionylation has been found to be involved the regulation of metabolism, signal transduction, transcription, inflammation, physiological processes, and cytoskeletal organization.
Glutathione not only functions as the substrate for protein S-glutathionylation but can also reverse the process by reducing the cysteine in proteins to which glutathione is disulfide bonded forming oxidized glutathione, GSSG. However, the primary pathway for glutahionylation and its reversal involves enzymes of the glutathione S-transferase family and glutaredoxin. Humans express two glutaredoxin encodeing genes, GLRX and GLRX2. The GLRX encoded enzyme is localized to the cytosol while the GLRX2 encoded enzyme is predominantly localized to the mitochondria.
Glutathione S-transferase pi (encoded by the GSTP1 gene) catalyzes protein S-glutathionylation while glutathione S-transferase omega (encoded by the GSTO1 gene) and glutaredoxin catalyze the deglutathionylation of cysteines in proteins.
The Glutathione Cycle
A mechanism originally proposed in 1970 for the role of glutathione in amino acid transport across membranes was termed the γ-glutamyl cycle and was proposed to involve the enzyme, γ-glutamyl transpeptidase (γ-glutamyltransferase, GGT). However, it has now been demonstrated that the GGT enzyme does not play a role in glutathione-mediated amino acid transport.
The glutathione cycle begins with the synthesis of glutathione. The glutathione can then be degraded by the cytoplasmic ChaC family of γ-glutamylamine cyclotransferases [ChaC1 (ChaC glutathione specific gamma-glutamylcyclotransferase 1) and ChaC2 (ChaC glutathione specific gamma-glutamylcyclotransferase 2)] generating 5-oxoproline and the dipeptide Cys-Gly. The 5-oxoproline is then cleaved by a 5-oxoprolinase to yield glutamate, while Cys-Gly is cleaved by specific cys-gly peptidases to yield cysteine and glycine. Glutathione equilibrates and determines the redox environment. When oxidized glutathione (as well as glutathione conjugates) can be transported out of the cell by transporters that are specific for the oxidized glutathione or glutathione conjugates. The extracellular glutathione, as well as the glutathione conjugates, can be salvaged by membrane bound γ-glutamyltransferase (GGT). The action of the GGT enzyme yields glutamate and Cys-Gly or bis-Cys-Gly. The glutamate can be transported back into the cell as can the Cys-Gly dipeptide. Alternately, Cys-Gly or bis-Cys-Gly can catabolized by membrane bound peptidases to yield cysteine (or cystine) and glycine. The cysteine, cystine, and glycine can then be transported back into the cell by specific transporters.
Polyamines
The polyamines are highly cationic molecules that tend to bind nucleic acids with high affinity. Because of this interaction activity, it is believed that the polyamines are important participants in DNA synthesis, or in the regulation of that process. In addition to their function in DNA replication, polyamines and polyamine derivatives are involved in the processes of protein synthesis, ion channel function (particularly the inwardly rectifying potassium channels), regulation of gene expression, cell proliferation, and apoptosis (programmed cell death).
Although de novo polyamine synthesis from ornithine represents the major source of intracellular polyamines, transport of extracellular polyamines into cells is accomplished via the SLC45A4 encoded transporter which is a member of the glycoside-pentoside-hexuronide cation symporter family of transporters. Extracellular polyamines are also taken up into cells via caveolin-coated pit-mediated endocytosis process. These polyamines are released into the cell via the action of the ATP13A2 transporter present in the membranes of late endosomes.
Following their synthesis, polyamines can be exported from cells following incorporation into secretory vesicles. Uptake of polyamines into secretory vesicles is accomplished via the activity of the SLC18B1 encoded vesicular amine transporter.
Polyamine Biosynthesis
One of the earliest signals that cells have entered their replication cycle is the appearance of elevated levels of mRNA for ornithine decarboxylase 1 (ODC1), and then increased levels of the enzyme. Ornithine decarboxylase is the first enzyme in the pathway to synthesis of the polyamines. The human genome contains two ODC sequences with only the ODC1 gene being functional. The other locus, identified as ODCP, is a pseudogene.
The ODC1 gene is located on chromosome 2p25.1 and is composed of 13 exons that generate four alternatively spliced mRNAs. Three of the ODC1 splice variants encode the same enzyme identified as isoform 1 (461 amino acids). ODC isoform 2 is composed of 332 amino acids.
Functional ODC is a homodimer and requires pyridoxal phosphate (PLP), derived from vitamin B6, as a cofactor. The catalytic activity of ODC produces the 4-carbon saturated diamine, putrescine.
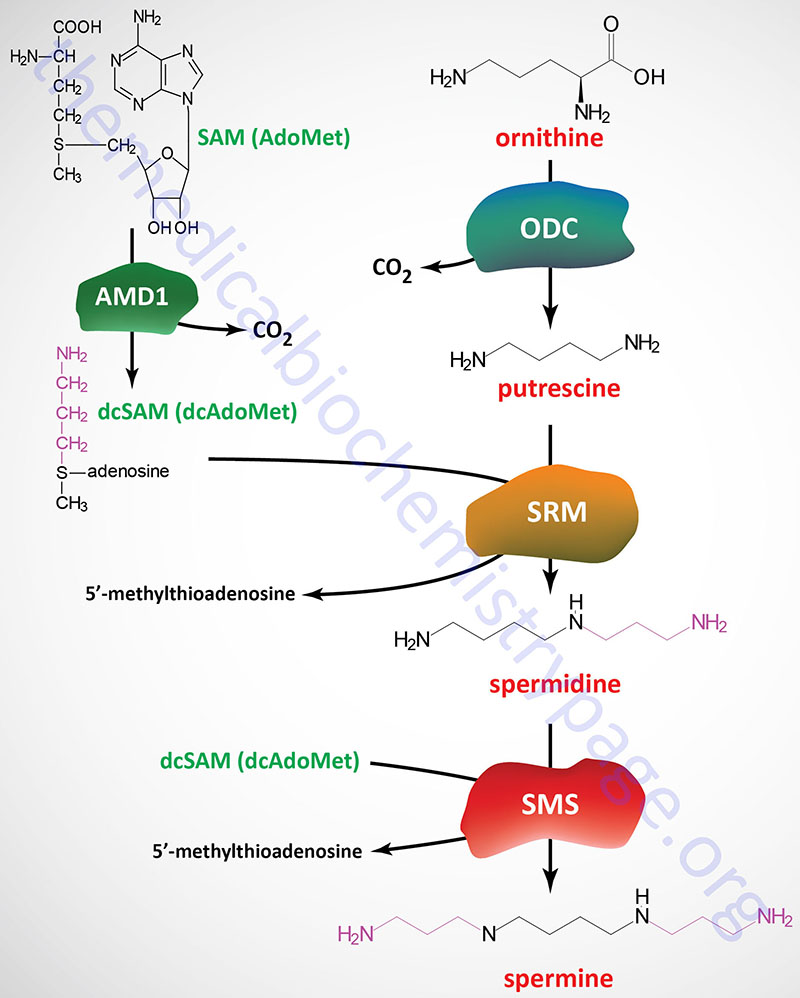
An important intermediate required for the synthesis of spermidine and spermine from putrescine is derived from S-adenosylmethionine (SAM). The enzyme, adenosylmethionine decarboxylase 1 (also known as S-adenosylmethionine decarboxylase or SAM-decarboxylase) cleaves the SAM carboxyl residue, producing decarboxylated SAM (S-adenosylmethylthiopropylamine; also known as decarboxylated S-adenosylmethionine, dcAdoMet or dcSAM), which retains the methyl group usually involved in SAM methyltransferase activity. Adenosylmethionine decarboxylase 1 activity is regulated by product inhibition and allosterically stimulated by putrescine.
Adenosylmethionine decarboxylase 1 is encoded by the AMD1 gene. The AMD1 gene is located on chromosome 6p21 and is composed of 11 exons that generate seven alternatively spliced mRNAs that encode five distinct isoforms of the enzyme.
Spermidine synthase catalyzes a condensation reaction between dcAdoMet and putrescine, producing spermidine and 5′-methylthioadenosine. Spermidine synthase is encoded by the SRM gene (also known as SPDSY) which is located on chromosome 1p36.22 and is composed of 8 exons that encode a protein of 302 amino acids.
A second propylamine residue from dcAdoMet is added to spermidine producing spermine by the enzyme spermine synthase encoded by the SMS gene (also known as SPMSY). The SMS gene is located on the X chromosome (Xp22.11) and is composed of 13 exons that generate two alternatively spliced mRNAs encoding SMS isoform 1 (366 amino acids) and SMS isoform 2 (313 amino acids).
Transcriptional Regulation of ODC
The signals for regulating the expression of ODC are complex. The promoter of the ODC1 gene contains elements that are regulated by growth factors, hormones, and tumor promoters. Production of ODC1 protein is also regulated at the level of translation by the product of the reaction, putrescine. The ODC1 mRNA contains a long 5′-untranslated region (5′-UTR) that folds into a complex secondary structure. Within the 5′-UTR there are two domains that reduce translational efficiency. One domain is a GC-rich sequence, the other is a small functional upstream open reading frame (uORF).
Translational Regulation of ODC
Another significant mechanism for regulating ODC1 mRNA translation is particularly important during mitosis. Cap-dependent translation is frequently repressed during mitosis and the ODC1 mRNA contains an internal ribosome entry site (IRES) that allows the mRNA to be translated under these conditions.
Antizyme Regulation of ODC Activity
The stability of the ODC1 protein also contributes to the level of enzyme activity in cells. The half-life of ODC1 protein is short (< 1hr) and this half-life is controlled by proteasomal degradation. However, entry into the proteasome, in the case of ODC1, does not require prior ubiquitylation. The ability of the proteasome to recognize, and degrade, non-ubiquitylated ODC1 involves a family of proteins called antizymes. The antizyme proteins interact with ODC1 monomers preventing formation of functional homodimers while simultaneously altering the conformation of the monomers so that they are recognized by the proteasome.
Humans express three distinct ODC antizymes identified as antizyme 1, 2, and 3.
Antizyme 1 is encoded by the OAZ1 (ornithine decarboxylase antizyme 1) gene. The OAZ1 gene is located on chromosome 19p13.3 and is composed of 5 exons that generate two alternatively spliced mRNAs. These two mRNAs encode antizyme 1 isoform 1 (228 amino acids) and antizyme 1 isoform 2 (226 amino acids). The translation of the functional antizyme 1 proteins is regulated by a polyamine-regulated ribosome frameshifting mechanism referred to as programmed ribosomal frameshifting (PRF).
The antizyme 1 mRNAs contain two overlapping open reading frames (ORFs). These ORFs are identified as ORF1 which is the shorter of the two, and ORF2. The reading frame of ORF2 is in the +1 frame relative to reading frame of ORF1. The ORF2 reading frame does not contain an AUG translation initiation codon and as such translation through ORF2 requires the failure of the ribosome to terminate at the end of ORF1 and instead shift to the +1 reading frame. This ribosomal frameshifting event is stimulated by polyamines. In the presence of high concentrations of polyamines the ribosome frameshifting occurs resulting in the synthesis of functional antizyme 1 which comprises amino acids from both ORF1 and ORF2. In addition to the two different isoforms, the antizyme 1 mRNA contains two in-frame AUG codons that have been shown, in rodents, to be alternatively utilized resulting in differentially localized antizyme 1 isoforms.
Antizyme 2 is encoded by the OAZ2 gene. The OAZ2 gene is located on chromosome 15q22.31 and is composed of 5 exons that generate two alternatively spliced mRNAs that encode two distinct protein isoforms. Although widely expressed like the OAZ1 gene, the level of OAZ2 expression is much lower.
Antizyme 3 is encoded by the OAZ3 gene. The OAZ3 gene is located on chromosome 1q21.3 and is composed of 7 exons that generate three alternatively spliced mRNAs encoding three distinct protein isoforms. Antizyme 3 isoform 1 is the longest protein encoded by the OAZ3 gene and the translation of this form of the enzyme begins from a CUG codon and not an AUG codon. Expression of the OAZ3 gene is restricted to haploid germ cells in the testis.
Regulation of Antizyme Activity
The ability of the antizyme proteins to bind to and inhibit the activity ODC is regulated by a family of antizyme inhibitors. The antizyme inhibitors are homologs of ODC but that have lost the ability to decarboxylate ornithine. The antizyme inhibitors bind to the antizyme proteins thereby blocking their ability to bind ODC and target its degradation by the proteasome. Humans express two antizyme inhibitor genes identified as AZIN1 and AZIN2.
The AZIN1 gene is located on chromosome 8q22.3 and is composed of 13 exons that generate 10 alternatively spliced mRNAs that collectively encode four distinct protein isoforms. The AZIN1 gene is ubiquitously expressed. The AZIN1 protein localizes to the cytosol and the nucleus. Editing of AZIN1 mRNAs occurs via the activity of the ADAR1 enzyme that edits A residues to I residues. This editing alters a Ser codon to a Gly codon resulting in increased affinity of the AZIN1 protein for the antizymes, particularly the OAZ1 encoded antizyme. Increased levels of AZIN1 mRNA editing has been shown to occur in certain types of cancer.
The AZIN2 gene is located on chromosome 1p35.1 and is composed of 16 exons that generate 18 alternatively spliced mRNAs that collectively encode six distinct protein isoforms. Expression of the AZIN2 gene is highest in the brain, ovaries, testes, pancreas, and adrenal glands. The AZIN2 protein localizes to the endoplasmic reticulum (ER) and the Golgi.
Role of Spermidine in Aging Processes
Numerous studies have found that spermidine has the capacity to extend life in invertebrates and vertebrates such as fruit flies and mice. With respect to the polyamines, spermidine is the only molecule in this family that has been shown to exhibit these effects. Supplementation with spermidine exerts these effects, in part, through the prevention of obesity and the metabolic syndrome in the context of a high-fat diet. Spermidine has also been shown to be associated with a delay is age-related cognitive decline.
One of the mechanisms by which spermidine exerts anti-aging effects is through activation of autophagy. Aging is known to be accompanied by a decline in autophagy. Spermidine-mediated induction of autophagy requires that the pro-autophagic transcription factor identified as TFEB (transcription factor EB, where the EB refers to enhancer box binding site) be activated. TFEB is considered a master regulator of the biogenesis of lysosomes through its ability to regulate the expression of genes encoding lysosomal hydrolases and lysosomal membrane proteins. The activation of TFEB requires modification of a specific lysine residue in the translation initiation factor 5A (eIF-5A) generating what is called hypusine (derived from hydroxyputrescine and lysine) which involves spermidine as outlined in the Polyamines in Protein Modification below.
Polyamine Catabolism
Synthesis of the polyamines is regulated by several independent mechanisms. In addition to these mechanism of control, intracellular polyamine levels are also regulated via their catabolism. Putrescine is catabolized by amine oxidase, copper containing 1 encoded by the AOC1 gene. The AOC1 encoded enzyme is most commonly identified as diamine oxidase, DAO.
The AOC1 gene is located on chromosome 7q36.1 and is composed of 7 exons that generate two alternatively spliced mRNAs. The AOC1 isoform 1 encoding mRNA encodes a precursor protein of 770 amino acids. The AOC1 isoform 2 encoding mRNA encodes a precursor protein of 751 amino acids. In addition to putrescine, the AOC1 encoded enzyme is responsible for the catabolism of histamine and other related biogenic amines. Because the AOC1 enzymes are inhibited by amiloride (a potassium-sparing diuretic), they are also identified as amiloride-sensitive amine oxidase.
Spermidine and spermine are catabolized by spermidine/spermine N1-acetyltransferase 1 (also known as diamine acetyltransferase 1) encoded by the SAT1 gene (also identified as SSAT). SAT1 catabolism of spermine yields N1-acetylspermine and its catabolism of spermidine yields N1-acetylspermidine. N1-acetylspermine and N1-acetylspermidine are converted to spermidine and putrescine, respectively, by polyamine oxidase (encoded by the PAOX gene) along with the production of H2O2 and 3-acetylaminopropanal (3Ac-AP or 3-AAP). Both H2O2 and 3Ac-AP contribute to oxidative stress as a result of polyamine catabolism.
The acetylated polyamines can be exported from cells via heteromeric transporters containing the SLC3A2 encoded heavy chain subunit.
The SAT1 gene is located on the X chromosome (Xp22.11) and is composed of 7 exons that generate two alternatively spliced mRNAs. Only one of the mRNAs encodes a functional SAT1 enzyme as the other mRNA is subjected to nonsense-mediated mRNA decay. The functional SAT1 enzyme is a 171 amino acid protein.
The PAOX gene is located on chromosome 10q26.3 and is composed of 8 exons that generate three alternatively spliced mRNAs, each of which encode a distinct protein isoform. Although ubiquitously expressed, the highest levels of expression of the PAOX gene occur in the testis.
Spermine catabolism can also be accomplished by its conversion back to spermidine via the activity of the SMOX (spermine oxidase) encoded FAD-dependent enzyme. The conversion of spermine to spermidine generates not only spermidine but also H2O2 and 3-aminopropanal (3-AP). 3-AP will spontaneously convert to the toxic compound acrolein. The production of H2O2 and acrolein as a result SMOX-mediated catabolism of spermine contributes to increased cellular oxidative stress.
Polyamines in Protein Modification
The butylamino group of spermidine is used in a posttranslational modification reaction important to the process of translation. A specific lysine residue in the translational initiation factor eIF-5A (eIF-5α) is modified. Following the modification the residue is hydroxylated yielding a residue in the protein termed hypusine. The term hypusine is derived from hydroxyputrescine and lysine. The hypusination of eIF-5A is necessary for the promotion of translational elongation.
The hypusination of eIF-5A involves a two-step reaction process. The first reaction involves the enzyme deoxyhypusine synthase which transfers the butylamine moiety from spermidine to the specific eIF-5A lysine residue. The second reaction involves the enzyme deoxyhypusine hydroxylase which hydroxylates the deoxyhypusine to hypusine.
Deoxyhypusine synthase is encoded by the DHPS gene. The DHPS gene is located on chromosome 19p13.13 and is composed of 12 exons that generate six alternatively spliced mRNAs, each of which encode a distinct isoform of the enzyme.
Deoxyhypusine hydroxylase is encoded by the DOHH (deoxyhypusine hydroxylase/monooxygenase) gene. The DOHH gene is located on chromosome 19p13.3 and is composed of 6 exons that generate two alternatively spliced mRNAs that both encode the same 302 amino acid protein.