
Last Updated: October 30, 2025
Introduction to Acute Intermittent Porphyria
Acute intermittent porphyria (AIP) is a disorder that is a member of a family of disorders referred to as the porphyrias. Each disease in this family results from deficiencies in a specific enzyme involved in the biosynthesis of heme (also called the porphyrin pathway). The term porphyria is derived from the Greek term porphura which means “purple pigment” in reference to the coloration of body fluids in patients suffering from the originally described porphyria which is now known as porphyria cutanea tarda (PCT).
The porphyrias are classified on the basis of the tissue that is the predominant site of accumulation of metabolic intermediates. These classifications are hepatic or erythroid. Each disease is also further characterized as being acute or cutaneous dependent upon the major clinical features of the disease.
Acute intermittent porphyria is inherited as an autosomal dominant disorder with late onset of symptoms. AIP is classified as an acute hepatic porphyria and results from defects in gene (HMBS) encoding the enzyme commonly called porphobilinogen deaminase (PBGD). PBGD is also known as hydroxymethylbilane synthase (HMBS). Mutations in the HMBS gene that result in 50% or less of normal enzyme activity cause the symptoms of AIP.
Molecular Biology of AIP
PBG deaminase is encoded by the HMBS (hydroxymethylbilane synthase) gene. The HMBS gene is located on chromosome 11q23.3 spanning 11 kb and encompassing 15 exons that generate four mRNAs through alternative promoter usage and alternative splicing. PBG deaminase isoform 1 is composed of 361 amino acids. PBG deaminase isoform 2 is composed of 344 amino acids. PBG deaminase isoform 3 is composed of 321 amino acids. PBG deaminase isoform 4 is composed of 304 amino acids. One of the HMBS mRNAs is erythroid cell-specific and is composed of exons 2 through 15 and encodes isoform 2 (344 amino acid) of the enzyme.
The classic form of AIP results from mutations in the HMBS gene that alter the function of both the ubiquitous (housekeeping) non-erythroid cell form and the erythroid-specific form of the enzyme. A small percentage (5%) of AIP patients have functional erythroid-specific PBG deaminase but defective ubiquitous PBG deaminase. Mutations in the HMBS gene that result in AIP encompass splice site mutations, missense, frameshift, and nonsense mutations. Approximately 60% of all mutations are found in exons 10, 12, and 14.
Once produced, hydroxymethylbilane has two main fates, one is due to enzymatic action, the other is non-enzymatic. Non-enzymatic alteration in hydroxymethylbilane is a cyclization. More than 150 mutations have been identified in the HMBS gene resulting in AIP. These mutations include missense, nonsense, and splicing mutations and insertions and deletions. Most of the identified mutations are unique to a given AIP patient. AIP is prevalent in Swedish populations, particularly northern Swedes where the frequency approaches 100 cases per 100,000 population. Because of this prevalence, AIP is also known as Swedish porphyria.
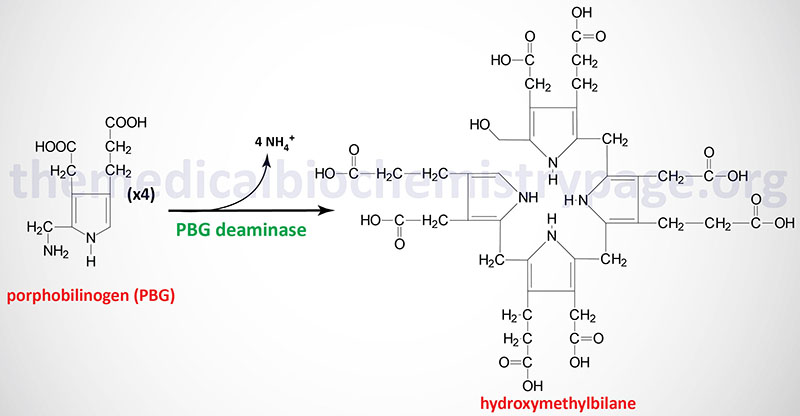
Using immunological assays for the presence of PBGD activity, mutations in the HMBS gene have been classified into three distinct categories. Type I mutations result in about 50% detectable PBGD protein and enzyme activity. These mutations are referred to as CRIM-negative (or CRM-negative) mutations where CRIM means “cross-reactive immunologic material”. Type I mutations represent the largest percentage of mutations in AIP patients, consisting of approximately 85% of mutations. Type II mutations are also CRIM-negative and result in absence of the house-keeping form of the enzyme but normal amounts of the erythroid-specific isozyme. These mutations are found in less than 5% of AIP patients. Type III mutations are CRIM-positive and result in decreased PBGD activity but do not alter the stability of the mutant enzyme.
Clinical Features of AIP
The clinical manifestations of AIP are related to the visceral, autonomic, peripheral and central nervous system involvement of the disease. In fact almost all of the symptoms of AIP result from neurologic dysfunction. The symptoms of AIP result from a dramatic increase in the production and excretion of porphyrin precursors. Although accumulation of δ-aminolevulinc acid is a significant contributor to the pathology of AIP, the exact mechanism leading to neural cell damage is not clearly understood. The pain associated with the neurologic dysfunction can be so severe as to require treatment with opiates.
A reduced ability to synthesize heme in a state of increased demand results in loss of heme-mediated control of the rate-limiting enzyme of heme synthesis, synthase, ALAS1. In this circumstance, unregulated ALAS1 results in large increases in synthesis of δ-aminolevulinic acid (ALA). The large increases in ALA drive the ALA dehydratase (also called porphobilinogen synthase) reaction to produce large amounts of porphobilinogen (PBG). With the block in heme synthesis at the PBG deaminase reaction the resulting increases in ALA and PBG synthesis lead to marked increases in urinary excretion of both compounds. Urinary ALA and PBG contributes to the deep red urine, a hallmark of AIP.
Clinical manifestation in AIP becomes apparent in the event of an increased demand for hepatic heme production. This most often occurs due to drug exposure or some other precipitating factor such as an infection. In fact, an infection is the leading cause of attacks of acute porphyria. If a drug is the cause of an attack its use should be discontinued immediately.
The neurovisceral signs and symptoms of AIP are nonspecific and highly variable resulting in patients being diagnosed with an unrelated illness. Because certain factors such as hormonal or nutritional influences or drug interactions can affect the clinical manifestation of AIP, as well as other hepatic porphyrias, these diseases are referred to as ecogenetic or pharmacogenetic disorders. Numerous drugs are known to lead to acute attacks of porphyria, in particular barbiturates. The use of barbiturates is strongly contraindicated for the treatment of pain in AIP patients, and for that matter, any individual with a porphyria. This is due to the inductive effect of barbiturates on the hepatic cytochrome P450 (CYP) drug metabolism pathway. Induction of CYP synthesis leads to increased utilization of heme which then results in reduced feed-back inhibition of ALAS1. The de-repression of ALAS1 results dramatic increases in the heme biosynthetic intermediates that were the cause of the porphyric symptoms. Other drugs that are know to exacerbate the symptoms of AIP include those used to treat hypertension such as ACE inhibitors (e.g. enalapril: Vasotec®) and calcium channel blockers (e.g. nifedipine: Procardia®), drugs used to treat hyperglycemia associated with type 2 diabetes such as the sulfonylureas (e.g. glipizide: Glucotrol®), and the sulfonamide class of antibiotics.
The most common symptom (occurring in 85%–95% of cases) during an acute attack is diffuse, poorly localized abdominal pain which can be quite severe. The abdominal pain is usually accompanied by constipation, nausea and vomiting. The most common physical sign that is observed in up to 80% of AIP cases is tachycardia which may be due to sympathetic nervous system over activity. Additional symptoms of AIP are pain in the limbs, head, neck or chest, muscle weakness, and sensory loss. Often, without any precipitating influences, AIP remains latent and there may be no family history of the disease. The symptoms of AIP are almost never observed in pre-pubescent children and are seen more frequently in women than in men.
Treatment of AIP
Treating the symptoms of AIP involves a high carbohydrate diet and during a severe attack an infusion of 10% glucose is highly recommended. The metabolic and molecular logic behind the use of glucose infusion in acute porphyric attacks stems from the fact that hypoglycemia induces the expression of the transcriptional co-activator PGC-1α (peroxisome proliferator activated receptor-γ co-activator 1α).
PCG-1α is a major transcriptional regulator of hepatic genes involved in gluconeogenesis and is also an activator of the ALAS1 gene. Thus, hypoglycemia can precipitate acute attacks in AIP patients (as well as other porphyrias) due to increased synthesis of ALAS1.
Hemin (Panhematin®, a form of alkaline heme) is used to treat attacks of porphyria as it acts like heme to inhibit ALAS thereby reducing the production of ALA and subsequently PBG. Many physicians with experience in treating porphyrias recommend early use of hemin without waiting to see if glucose administration alone eases symptoms.
Newer methods of treatment for AIP patients that are being developed involve RNA interference. The therapy identified as givosiran (trade name Givlaari) is an RNA interference therapy that targets the ALAS1 encoded mRNA. This technique has been found to reduce the rate of acute attacks and improves other disease manifestations in patients with AIP. The US FDA approved the use of Givlaari in patients with acute hepatic porphyria in 2019.