
Last Updated: July 13, 2026
Introduction to ALA Dehydratase Deficient Porphyria, ADP
ALA dehydratase deficient porphyria (ADP) is an autosomal recessive disorder that is a member of a family of disorders referred to as the porphyrias. Each disease in this family results from deficiencies in a specific enzyme involved in the biosynthesis of heme (also called the porphyrin pathway). The term porphyria is derived from the Greek term porphura which means “purple pigment” in reference to the coloration of body fluids in patients suffering from the originally described porphyria which is now known as porphyria cutanea tarda (PCT).
The porphyrias are classified on the basis of the tissue that is the predominant site of accumulation of metabolic intermediates. These classifications are hepatic or erythroid. Each disease is also further characterized as being acute or cutaneous dependent upon the major clinical features of the disease. ADP is an autosomal recessive disorder that results from defects in gene encoding the enzyme, δ-aminolevulinic acid dehydratase (also called porphobilinogen synthase: PBG synthase). ADP is classified as an acute hepatic porphyria.
Molecular Biology of ADP
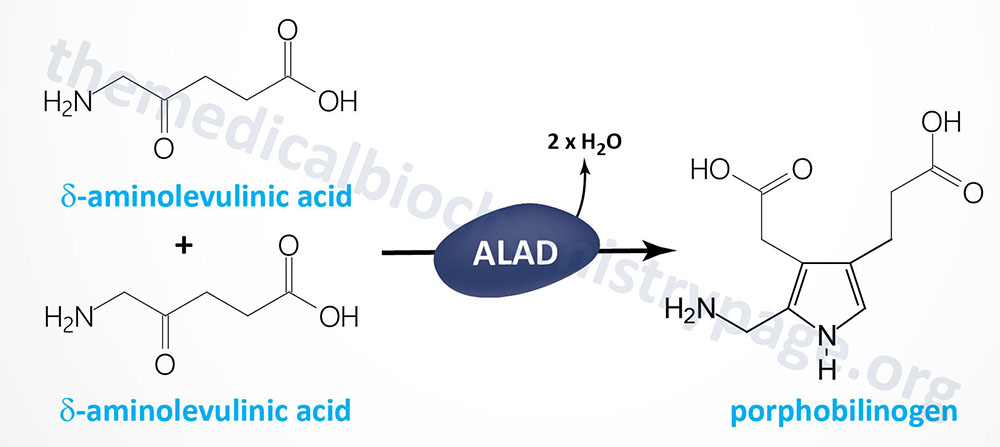
ALA dehydratase is encoded by the ALAD gene. The ALAD gene is located on chromosome 9q32 spanning 16 kb and is composed of 15 exons that generate three alternatively spliced mRNAs, each of which encode a distinct protein isoform. One of the alternatively spliced ALAD mRNAs is erythroid cell-specific and contains exon 1B and then exons 2 through 12. Functional ALA dehydratase is a complex composed of eight identical subunits derived from the ALAD encoded proteins. Very few cases of ADP have been reported and in those few individuals missense and frameshift pathogenic variants were identified in the ALAD gene.
Clinical Features of ADP
The clinical manifestations of ADP are heterogeneous and somewhat similar to those seen in patients with acute intermittent porphyria, AIP. Common symptoms include abdominal pain and neuropathy. The symptoms of ADP are due to the accumulation and urinary excretion of large amounts of δ-aminolevulinic acid, ALA. Acute attacks of ADP are believed to occur in association with overexpression of δ-aminolevulinic acid synthase 1 (ALAS1) in the liver. ALAS is the rate-limiting enzyme of heme biosynthesis. ADP heterozygotes are at increased risk of acute attacks of porphyria if exposed to certain compounds such as iron, trichloroethylene, and styrene that have adverse effects on the activity of ALAD.