
Last Updated: July 31, 2026
Introduction to Adipose Tissue
Adipose tissue is not merely an organ designed to passively store excess carbon in the form of fatty acids esterified to glycerol (triglycerides). Mature adipocytes synthesize and secrete numerous enzymes, growth factors, cytokines and hormones that are involved in overall energy homeostasis. Many of the factors that influence adipogenesis are also involved in diverse processes in the body including lipid homeostasis and modulation of inflammatory responses. In addition, a number of proteins secreted by adipocytes play important roles in these same processes. In fact recent evidence has demonstrated that many factors secreted from adipocytes are pro-inflammatory mediators and these proteins have been termed adipocytokines or adipokines. There are currently over 50 different adipokines recognized as being secreted from adipose tissue. These adipokines are implicated in the modulation of a range of physiological responses that globally includes appetite control and energy balance. Specific metabolic processes regulated by adipose tissue include lipid metabolism, glucose homeostasis, inflammation, angiogenesis, hemostasis (regulation of blood coagulation), and blood pressure.
The major form of adipose tissue in mammals (commonly referred to simply as “fat”) is white adipose tissue, WAT. Specialized adipose tissue that is primarily tasked with thermogenesis, especially in the neonate, is brown adipose tissue, BAT. BAT is so-called because it is darkly pigmented due to the high density of mitochondria rich in cytochromes. BAT specializes in the production of heat (adaptive thermogenesis), as well as the regulation of energy expenditure, via the process of fatty acid β-oxidation.
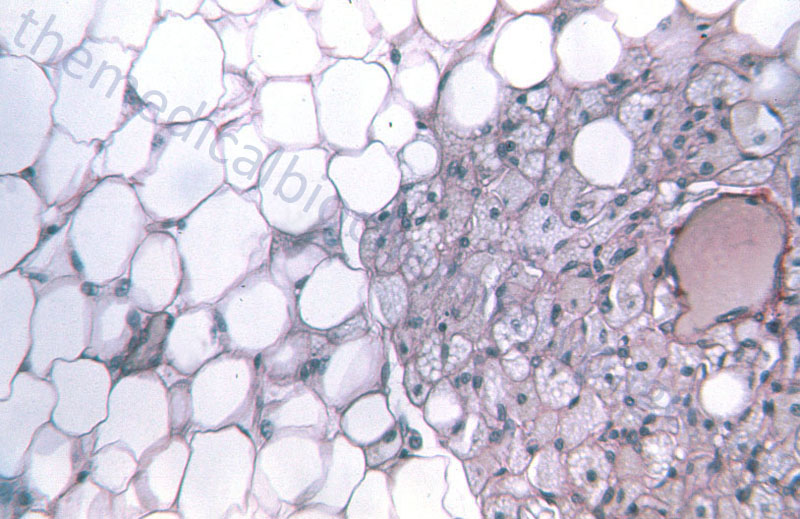
WAT is composed of adipocytes held together by a loose connective tissue that is highly vascularized and innervated. White adipocytes are rounded cells that contain a single large fat droplet that occupies over 90% of the cell volume. The mitochondria within white adipocytes are small and few in number. The mitochondria and nucleus of the white adipocyte is squeezed into the remaining cell volume. Molecular characteristics of white adipocytes include expression of leptin but no expression of uncoupling protein 1, UCP1 (designated UCP1–, leptin+).
BAT is composed of brown adipocytes that reside within an intricate network of blood vessels and sympathetic post-ganglionic nerves. The sympathetic innervation of BAT emanates from the ventromedial (VMH) and paraventricular nuclei (PVN) of the hypothalamus.
The high level of vascularization supplies the substrates and oxygen required for BAT to carry out its thermogenic functions. The sympathetic post-ganglionic neurotransmitter, norepinephrine, is the primary signal triggering lipolysis in brown adipocytes.
Brown adipocytes are smaller in overall size compared to white adipocytes. Brown adipocytes are polygonal in shape and contain numerous large mitochondria packed with cristae. Whereas, white adipocytes contain a single large fat droplet, brown adipocytes contain several small lipid droplets. Brown adipocytes are molecularly UCP1+ and leptin–. BAT is primarily visceral with highest concentrations around the aorta. BAT is highly vascularized and contains a very high density of noradrenergic nerve fibers.
In addition to adipocytes, WAT contains macrophages, leukocytes, fibroblasts, adipocyte progenitor cells, and endothelial cells. The presence of the fibroblasts, macrophages, and other leukocytes along with adipocytes, accounts for the vast array of proteins that are secreted from WAT under varying conditions. The highest accumulations of WAT are found in the subcutaneous regions of the body and surrounding the viscera (internal organs of the chest and abdomen).
Although WAT can be found associated with numerous organs its functions are more than just insulation of the organ and a ready reservoir of fat for energy production. Depending on its location WAT serves specialized functions. The WAT associated with abdominal and thoracic organs (excluding the heart), the so-called visceral fat, secretes several inflammatory cytokines and is thus involved in local and systemic inflammatory processes. WAT associated with skeletal muscle secretes free fatty acids, interleukin-6 (IL-6) and tumor necrosis factor-α (TNFα) and as a consequence plays a significant role in the development of insulin resistance. Cardiac tissue associated WAT secretes numerous cytokines resulting in local inflammatory events and chemotaxis that can result in the development of atherosclerosis and systolic hypertension. Kidney associated WAT plays a role in sodium reabsorption and therefore can affect intravascular volume and hypertension.
The major focus of this discussion will be on the biological activities associated with WAT, however, discussion of BAT is included at the end of this page. WAT serves many functions including insulating the viscera, storing excess carbon energy in the form of triglycerides and mediating glucose homeostasis. WAT also plays important roles as an endocrine/immune organ by secreting adipokines that includes inflammatory cytokines, complement-like factors, chemokines, and acute phase proteins. The endocrine functions of WAT regulate appetite, energy metabolism, glucose and lipid metabolism, inflammatory processes, angiogenesis, and reproductive functions.
Regulation of Adipogenesis
The process of adipocyte differentiation from a precursor preadipocyte to a fully mature adipocyte follows a precisely ordered and temporally regulated series of events. Adipocyte precursor cells emerge from mesenchymal stem cells (MSCs) that are themselves derived from the mesodermal layer of the embryo. The pluripotent MSCs receive extracellular cues that lead to the commitment to the preadipocyte lineage. Pre-adipocytes cannot be morphologically distinguished from their precursor MSCs but they have lost the ability to differentiate into other cell types. This initial step in adipocyte differentiation is referred to as determination and leads to proliferating pre-adipocytes undergoing a growth arrest. This initial growth arrest occurs coincident with the expression of two key transcription factors, CCAAT/enhancer binding protein alpha (C/EBPα) and peroxisome proliferator-activated receptor gamma (PPARγ). Following the induction of these two critical transcription factors there is a permanent period of growth arrest followed by expression of the fully differentiated adipocyte phenotype. This latter phase of adipogenesis is referred to as terminal differentiation.
Although PPARγ and C/EBPα are the most important factors regulating adipogenesis additional transcription factors are known to influence this process. These additional factors include sterol-regulated element binding protein 1c (SREBP1c, also known as ADD1 for adipocyte differentiation-1), signal transducers and activators of transcription 5 (STAT5), AP-1 and members of the Krüppel-like factor (KLF4, KLF5, and KLF15) family as well as C/EBP beta (β) and C/EBP delta (δ). More information about the roles of PPARγ and SREBP in metabolic homeostasis can be found in the PPAR page as well as the Cholesterol: Synthesis, Metabolism, and Regulation page. Although these numerous transcription factors have been shown to influence overall adipogenesis, either positively or negatively, PPARγ is the only one that is necessary for adipogenesis to take place. In fact, in the absence of PPARγ, adipocyte differentiation fails to occur and as yet no factor has been identified that can rescue adipogenesis in the absence of PPARγ. In spite of this fact, expression of PPARγ does not commence during the initial activation of adipocyte differentiation but only after the responses elicited by STAT5, KLF4, KLF5, AP-1, SREBP1c, and C/EBPβ and C/EBPδ are exerted.
PPARγ was originally identified as being expressed in differentiating adipocytes and as indicated above it is now recognized as a master regulator of adipogenesis. PPARγ was identified as the target of the thiazolidinedione (TZD) class of insulin-sensitizing drugs. The mechanism of action of the TZDs is a function of the activation of PPARγ and the consequent induction of genes necessary for differentiation of adipocytes. The human PPARγ gene (symbol PPARG) is located on chromosome 3p25 spanning over 100kb and composed of 9 exons encoding two biologically active isoforms as a consequence of alternative mRNAs and translational start codon usage.
The principal protein products of the PPARG gene are identified as PPARγ1 and PPARγ2. PPARγ1 is encoded for by exons A1 and A2 then common exons 1 through 6. PPARγ2 is encoded by exon B and common exons 1 through 6. PPARγ2 is almost exclusively expressed in adipocytes. Like all nuclear receptors the PPARγ proteins contain a DNA-binding domain (DBD) and a ligand-binding domain (LBD). In addition, like PPARα, the PPARγ proteins contain a ligand-dependent activation function domain (identified as AF-2) and a ligand-independent activation function domain (identified as AF-1). The AF-2 domain resides in the LBD and the AF-1 domain is in the N-terminal region of the PPARγ proteins. PPARγ2 protein contains an additional 30 N-terminal amino acids relative to PPARγ1 and these additional amino acids confer a 5–6-fold increase in the transcription-stimulating activity of AF-1 when compared to the same domain in the PPARγ1 protein. Expression of PPARγ1 is nearly ubiquitous. PPARγ2 is expressed near exclusively in white adipose tissue (WAT) where it is involved in lipid storage and in BAT where it is involved in energy dissipation.
As indicated above, during adipocyte differentiation several upstream genes are required for the activation of the PPARG gene. These include C/EBPβ and C/EBPδ, SREBP-1c, KLF5, KLF15, zinc-finger protein 423 (Zfp423), and early B-cell factor 1 (EBF1). In the process of adipocyte differentiation PPARγ activates nearly all of the genes required for this process. These genes include aP2 which is required for transport of free fatty acids (FFAs) and perilipin which is a protein covering the surface of mature lipid droplets in adipocytes. Additional genes regulated by PPARγ that are involved in lipid metabolism or glucose homeostasis include lipoprotein lipase (LPL), several acyl-CoA synthases (FATP1 and FATP2), acetyl-CoA acetyltransferase 1 (ACAT1), several phospholipase A (PLA) genes, adiponectin, the gluconeogenic enzyme PEPCK, and glycerol-3-phosphate dehydrogenase (GPD1). PPARγ also functions in macrophage lipid metabolism by inducing the expression of the macrophage scavenger receptor, CD36. The CD36 receptor is also known as fatty acid translocase (FAT) and it is one of the receptors responsible for the cellular uptake of fatty acids.
The role of SREBP-1c in activation of adipocyte differentiation is thought to be the result of this transcription factor initiating the expression of genes that, as part of their activities, generate PPARγ ligands. This fact explains the necessity for SREBP expression to precede that of PPARγ. In spite of this fact it has been shown that mice lacking SREBP-1 do not display significant reductions in the amount of WAT. However, levels of SREBP-2 are increased in these animals indicating that this may be a compensatory mechanism. Although loss of SREBP-1 expression does not result in a significant deficit in adipose tissue development, ectopic overexpression of SREBP-1c does enhance the adipogenic activity of PPARγ.
The C/EBP family of transcription factors were among the first to be shown to play a role in overall adipocyte differentiation. The three members of the family (C/EBPα, C/EBPβ and C/EBPδ) are highly conserved basic-leucine zipper containing transcription factors. The importance of these factors in adipogenesis has been demonstrated in knockout mouse models. for example whole body disruption of C/EBPα expression results in death shortly after birth due to liver defects, hypoglycemia, and failure of WAT or BAT accumulation. Using knockout mice it has been determined that the roles of C/EBPβ and C/EBPδ are exerted early in the process of adipocyte differentiation whereas those of C/EBPα are required later. In fact, expression of C/EBPα is induced late in adipogenesis and is most abundant in mature adipocytes. The expression of both C/EBPα and PPARγ is, in part, regulated by the actions of C/EBPβ and C/EBPδ. One of the major effects of the expression of C/EBPα in adipocytes is enhanced insulin sensitivity of adipose tissue. This later fact is demonstrated by the fact that C/EBPα knockout does not abolish adipogenesis but the WAT is not sensitive to the actions of insulin.
The general model of transcription factor activation of adipogenesis indicates that AP-1, STAT5, KLF4, and KLF5 are activated early and result in the transactivation of C/EBPβ and C/EBPδ. These latter two factors in turn activate the expression of SREPB-1 and KLF15 which leads to the activation of PPARγ and C/EBPα. It is important to keep in perspective that it is not only transcription factor activation of adipocyte precursors that controls adipogenesis. There is also a balance exerted at the level of transcription factor-mediated inhibition of adipogenesis. Some of the factors that are anti-adipogeneic include members of the Krüppel-like factor family, KLF2 and KLF3. GATA2 and GATA3 also exert anti-adipogenic activity. GATA factors are so-called because they bind DNA elements that contain a core GATA sequence. Two of the interferon regulatory factor family of transcription factors, IRF3 and IRF4, oppose the process of adipogenesis as well.
The changes in the pattern of expression of transcription factors that control the overall process of adipogenesis is associated with changes in chromatin dynamics. These changes in chromatin dynamics involve both histone protein methylation and DNA methylation events. The chromatin in pluripotent cells displays a highly dynamic nature with a high level of decondensed DNA. Once differentiation is induced there is a change in the overall pattern of methylated genes. Lineage-specific genes are demethylated whereas pluripotency genes are methylated resulting in transcriptional activation and silencing, respectively. As the process of adipocyte differentiation proceeds the genes encoding PPARγ and C/EBPα are observed to be repositioned into the interior of the nucleus coincident with their increased rates of transcription. Since MSCs can be induced to differentiate into bone and muscle, as well as adipocyte, it is necessary that adipocyte differentiation genes such as PPARγ and C/EBPα be silenced if the induced pathway is to bone or muscle.
Associated with transcriptional silencing are protein complexes termed co-repressors and associated with transcriptional activation are complexes termed co-activators. When MSCs are induced down the bone lineage the histone 3 proteins in the PPARγ promoter region are methylated on lysine 9 (identified as H3K9) by a co-repressor complex that includes the histone methyltransferase SETDB1 and the associated proteins NLK (Nemo-like kinase) and CHD7 (chromodomain helicase DNA binding protein-7). In addition to silencing of the PPARγ promoter, the activity of the PPARγ protein on its target genes is also restricted by association with co-repressor complexes. In preadipocytes PPARγ activity is repressed by association with pRB and HDAC3 (histone deacetylase 3). The induction of differentiation results in the phosphorylation of pRB which leads to its release from the repressive complex. This in turn results in the recruitment of histone acetyltransferases (HAT) and the co-activator proteins CBP and p300 to the PPARγ complex resulting in activation of PPARγ target gene transcription. CBP is derived from CREB Binding Protein, where CREB is cAMP-response element-binding protein. p300 is closely related to CBP and these two proteins form one family of histone acetyltransferases.
Numerous experiments have begun to define the large array of histone modifications that regulate the expression of genes involved in overall adipogenesis particularly the expression of PPARγ. These histone modifying complexes include HAT, HDAC, histone methyltransferases (HMT), and histone demethylases (HDM). The general consequences of activation of HAT and HMT is the activation of PPARγ expression and/or enhancement of PPARγ activity at its target gene promoters. Conversely, as expected, HDAC activation results in inhibited PPARγ activity at its target gene promoters.
Discrete Populations of Adipocytes within Adipose Tissue
The advent of single cell, specifically single nuclei, RNA sequencing (snRNA-Seq) technologies has allowed for the identification of several discrete populations of adipocytes that can be defined by their molecular signatures of gene expression. These different adipocyte populations carry out distinct subsets of metabolic and signaling processes, in part due to the differences in patterns of expressed genes. Using snRNA-Seq on cells isolated from human visceral and subcutaneous fat depots has allowed for the identification of at least seven discrete subpopulations of mature adipocytes. The seven different types of mature adipocytes have been designated Ad1 through Ad7. However, additional studies have defined mature adipocyte populations, based upon primary gene expression patterns, as AdipoLEP, AdipoPLIN, and AdipoSSA.
The different adipocyte populations can be grouped by primary functions into those that specialize in lipid metabolism and insulin signaling, those involved with lipid handling, retinol handling, and inflammation, those involved in leptin signaling, extracellular matrix remodeling, and stress responses, and those involved in calcium signaling and the organization of cell-substrate junctions.
Ad1 cells are molecularly defined by expression of the AFF3 (ALF transcription elongation factor 3) and WDPCP (WD repeat containing planar cell polarity effector) genes. Their functional characteristics are that they are involved in the organization of cell-substrate junctions.
Ad2 cells are molecularly defined by expression of the FABP4 (fatty acid binding protein 4) and NAV2 (neuron navigator 2) genes. The FABP4 encoded protein is also known as adipocyte fatty acid binding protein (A-FABP). Their functional characteristics are that they are involved in the immune responses to infection.
Ad3 cells are molecularly defined by expression of the CAV2 (caveolin 2) and SAA1 (serum amyloid A1) genes. Their functional characteristics are that they are involved in triglyceride biosynthesis and the metabolism of retinol.
Ad4 cells are molecularly defined by expression of the ANKRD30A (ankyrin repeat domain 30A) and GRIA4 (glutamate ionotropic receptor AMPA type subunit 4) genes. Their functional characteristics are that they are involved in lipid handling and the responses to leptin signaling.
Ad5 cells are molecularly defined by expression of the ATP1B3 (ATPase Na+/K+ transporting subunit beta 3) and PGAP1 (post-GPI attachment to proteins inositol deacylase 1) genes. Their functional characteristics are that they are involved in the regulation of lipolysis, responses to insulin signaling, and to HIF1α signaling.
Ad6 cells are molecularly defined by expression of the EBF2 (EBF transcription factor 2) and ZNF804A (zinc finger protein 804A) genes. Their functional characteristics are that they are involved in lipolysis, axon guidance, and thermogenic signaling.
Ad7 cells are molecularly defined by expression of the AGMO (alkylglycerol monooxygenase) and THSD7B (thrombospondin type 1 domain containing 7B) genes. Their functional characteristics are that they are involved in calcium ion handling and hormone secretion.
AdipoLEP cells are molecularly defined by expression of the LEP (leptin), DDR2 (discoidin domain receptor tyrosine kinase 2), and TNS1 (tensin 1) genes. Their functional characteristics are that they are involved in leptin signaling and the remodeling of the extracellular matrix.
AdipoPLIN cells are molecularly defined by expression of the PLIN1 and PLIN4 (perilipin 1 and 4), DGAT1 and DGAT2 (diacylglycerol O-acyltransferase 1 and 2), and LIPE (lipase E; more commonly called hormone-sensitive lipase, HSL) genes. Their functional characteristics are that they are involved in lipid biosynthesis and lipolysis.
AdipoSAA cells are molecularly defined by expression of the SAA1 and SAA2 (serum amyloid A1 and A2), AGPAT2 (1-acylglycerol-3-phosphate O-acyltransferase 2), and MGLL (monoglyceride lipase) genes. Their functional characteristics are that they are involved in handling of fatty acids and in the metabolism of retinol.
Regulation of Lipid Metabolism in Adipocytes
The overall regulation of adipose tissue lipid homeostasis represents a balance between triglyceride synthesis and triglyceride breakdown. The major mechanisms that regulate adipose tissue lipid homeostasis involve hormone-induced signal transduction. The primary hormones regulating adipose tissue lipid homeostasis are insulin, which stimulates triglyceride synthesis, and epinephrine, cortisol, and to some extent glucagon, which stimulate triglyceride breakdown. The regulation of adipose tissue lipid homeostasis by cortisol is more complex and involves both
Regulation of Adipocyte Triglyceride Breakdown
Rapid release of stored fatty acids from adipose tissue is triggered by epinephrine binding to, and activation of, β-adrenergic receptors (βAR). Adipocytes express all three β-adrenergic receptors, β1AR, β2AR, and β3AR. All three βAR are members of the G-protein coupled receptor (GPCR) family. The β1AR and β2AR predominantly activate Gs-type G-proteins whereas the adipocyte β3AR can interchangeably couple to both Gs– and Gi-type G-proteins.
The triglycerides (TG or TAG) found in WAT represent the major energy reserves of the body. The pool of TG is in a constant state of flux that is regulated by food intake and fasting and the consequences of those dietary states on the levels of pancreatic and adrenal hormones. In addition, adipose tissue fat pools change as a result of other hormonal fluctuations, inflammatory processes, and pathophysiology. The overall biochemistry of TG metabolism is covered in the Synthesis of Triglycerides page and the Lipolysis and the Oxidation of Fatty Acids page. The aim of this section is to discuss in more detail the enzyme activities that regulate overall adipose tissue TG homeostasis as well as the physiological and hormonal regulation of these processes.
It was originally believed that the liberation of fatty acids from adipose tissue TG stores was triggered exclusively via hormonal activation of hormone-sensitive lipase (HSL). However, when HSL-null mice were generated it was discovered that the process involves additional adipocyte HSL-independent TG lipase activities. Subsequent research has led to the identification of at least five adipose tissue TG lipases in addition to HSL.
HSL has demonstrated activity with a wide variety of substrates that includes TG, diglycerides (DG, also DAG), and cholesterol esters (CEs). When assayed in vitro the activity of HSL is at least 10-fold higher against DG than TG. When acting on TG or DG the activity of HSL is greatest against fatty acids that are in the sn-1 or sn-3 position of the glycerol backbone. Until recent experiments in knock-out mice demonstrated otherwise, HSL was believed to be the principal enzyme involved in adipocyte TG and DG hydrolysis as well as the primary neutral cholesteryl ester hydrolase (NCEH1) activity.
Although HSL-null mice still exhibit TG hydrolase activity, results from studies in these mice indicate that HSL-mediated lipolysis is a significant contributor to overall fatty acid liberation from adipocytes. In mice lacking HSL there is a reduced level of circulating free fatty acids and TG as well as reduced hepatic storage of TG. These results indicate that in the absence of HSL there is insufficient adipose tissue lipolysis to support the normal cellular demands for energy from fatty acids nor for adequate VLDL synthesis in the liver.
Results of studies on the role of HSL in overall adipose tissue lipolysis demonstrate that it is not strictly required for the initiation of TG hydrolysis as originally thought. However, in HSL-null mice there is an accumulation of DGs indicating that the critical role for HSL is in the liberation of fatty acid from DG which in turn generates monoglycerides (MG, also MAG). The rate of fatty acid release from DG is on the order of 10- to 30-times that the rate of release from TG. To date the only DG lipase identified in adipose tissue is HSL.
Further understanding of the role of HSL in overall adipose tissue lipolysis came from the identification of an additional TG lipase that was originally termed desnutrin. The overall structure of desnutrin indicates that it contains typical domains found in many other lipases. Subsequent to the identification of desnutrin another lipase was characterized and called adipose triglyceride lipase (ATGL). Desnutrin and ATGL are the same protein so it is often designated desnutrin/ATGL. The desnutrin/ATGL gene is expressed predominantly in adipose tissue but also at much lower levels in cardiac and skeletal muscle and the testes. The intracellular location of desnutrin/ATGL is the cytosol as well as in tight association with lipid droplets. The activity of desnutrin/ATGL is specific for TG as evidenced in cell culture experiments where over-expression of the gene results in increased free fatty acid release with no effect on phospholipid stores. In addition, desnutrin/ATGL has limited activity against DG since in these and similar in vitro experiments there is a significant accumulation of DG compared to the same types of experiments carried out with HSL.
Expression of desnutrin/ATGL is under the influence of dietary status. In fasting animals the level of desnutrin/ATGL increases and then declines following re-feeding. This dietary regulation of desnutrin/ATGL suggests that it may play a contributory role in the development of obesity, a hypothesis supported by the fact that in genetically obese mice (ob/ob and db/db) the level of desnutrin/ATGL expression is reduced. When experiments are performed that artificially reduce the level of desnutrin/ATGL RNA or protein there is a significant drop in the level of free fatt acid release. Demonstrating a synergy between HSL and desnutrin/ATGL activity, in cells where both enzymes are reduced there is an additive level of reduction in free fatty acid release. A critical role for desnutrin/ATGL in TG hydrolysis in tissues other than adipose tissue was shown by results of desnutrin/ATGL knock-out in mice. These animals died at around 12-weeks of age due to increased ectopic fat stores particularly in the heart. In addition, total lipase activities in several tissues in addition to WAT and BAT were altered in the desnutrin/ATGL-null mice. These data point to a critical role for desnutrin/ATGL in TG hydrolysis and fatty acid release not only from adipose tissue but also from tissues such as the heart, skeletal muscle, and testes.
In addition to HSL and desnutrin/ATGL, adipose tissue expresses a number of other TG hydrolases. Adipose tissue microsomes contain a non-HSL TG lipase that is identified as triacylglycerol hydrolase (TGH, also called carboxylesterase 3). TGH contains typical lipase motifs and displays catalytic activity against long-, medium-, and short-chain TG as well as neutral cholesteryl esters. However, TGH does not hydrolyze phospholipids . Expression of TGH is seen predominantly in the liver where its primary functions are to mobilize intracellular TG stores and participate in the synthesis of TG-rich VLDLs. TGH expression is also seen in adipocytes and the level of its expression increases dramatically when preadipocytes differentiate into mature adipocytes. The adipose tissue regulation of TGH expression is effected, in part, via the action of C/EBPα. A related protein, identified as TGH-2, has also been found predominantly expressed in the liver but is also present in adipose tissue and kidney.
There is another interesting protein expressed predominantly in adipose tissue with a significant degree of homology to desnutrin/ATGL. This protein is called adiponutrin. Whereas, adiponutrin shows TG lipase activity when assayed in vitro, when it is over-expressed in cells it has no effect on TG hydrolysis. In addition, whereas desnutrin/ATGL (as well as most other lipases) expression is increased in the fasting state and decreased following re-feeding, adiponutrin expression exhibits the opposite pattern. In fasted animals adiponutrin mRNA is essentially undetectable and its levels increase dramatically in the re-fed state. It appears that although this enzyme is a member of the lipase family of enzymes it plays an anabolic rather than a catabolic role in adipocyte lipid metabolism.
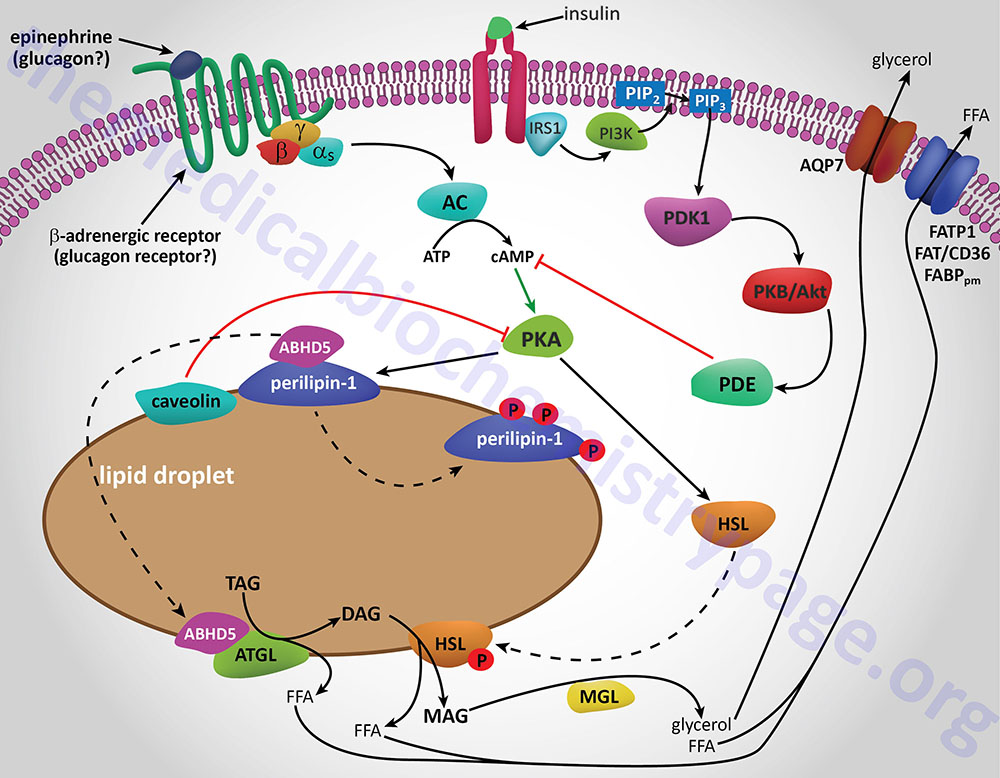
The final step in the complete hydrolysis of TG occurs when glycerol and the last fatty acid are released from MG by MAG lipase. This enzyme possesses no catalytic activity towards TG or DG nor cholesteryl esters. Numerous other proteins in addition to the lipases are involved in overall TG homeostasis in adipose tissue. Several of these other proteins are associated with the lipid droplets inside the cell such as the perilipins, adipose fatty acid-binding protein (aP2, also called FABP4 and aFABP), caveolin-1, and aquaporin 7 (a water and glycerol transport protein).
The perilipins play a role in restricting access of TG lipases to substrates in order to prevent unrestrained hydrolysis in the un-stimulated state. The role of aP2 is to carry free fatty acids from the fat droplet to the plasma membrane where they can be released to the plasma. The glycerol that is released from TG is exported via the action of aquaporin 7 as shown by experiments in mice lacking expression of the AQP7 gene. These mice release free fatty acids upon stimulation of adipose tissue with catecholamines but no glycerol is released.
Whether adipose tissue stores fatty acids as TG or releases them for energy production by other tissues is dependent upon the dietary, hormonal and physiological status of the organism. The primary mechanism for the stimulation of adipose tissue TG hydrolysis is discussed in the Lipolysis and the Oxidation of Fatty Acids page. In brief, catecholamines such as epinephrine and norepinephrine, as well as the pancreatic hormone glucagon, bind to their cognate receptors on adipocytes triggering activation of adenylate cyclase resulting in increased levels of cAMP. In turn the cAMP activates PKA which then phosphorylates and activates HSL. Of significance is the fact that activation of PKA in HSL-null mice also results in enhanced TG hydrolysis albeit at a level much lower than in the presence of active HSL. This indicates that there are PKA-mediated events in adipose tissue TG lipolysis that are distinct from the classic HSL-mediated process.
The primary change in adipose tissue TG lipolysis following feeding is exerted via the actions of insulin. The cAMP-dependent changes that occur in response to insulin binding are effected by activation of phosphodiesterase 3B which hydrolyzes cAMP rendering PKA much less active. The activation of phosphodiesterase 3B occurs via PKB/AKT-mediated phosphorylation which itself is activated following insulin binding its receptor. The principal cAMP-independent mechanism for insulin-mediated reduction in TG lipolysis is due to the stimulation of protein phosphatase-1 which removes the phosphate from HSL rendering it much less active.
The activity of HSL is also affected via phosphorylation by AMPK. In this case the phosphorylation inhibits the enzyme. Inhibition of HSL by AMPK may seem paradoxical since the release of fatty acids stored in TG would seem necessary to promote the production of ATP via fatty acid oxidation and the major function of AMPK is to shift cells to ATP production from ATP consumption. This paradigm can be explained if one considers that if the fatty acids that are released from TGs are not consumed they will be recycled back into TGs at the expense of ATP consumption. Thus, it has been proposed that inhibition of HSL by AMPK mediated-phosphorylation is a mechanism to ensure that the rate of fatty acid release does not exceed the rate at which they are utilized either by export or oxidation.
Role of Lactate in the Regulation of Adipocyte Lipolysis
Lactate is the byproduct of anaerobic glucose metabolism occurring in red blood cells, skeletal muscle, brain, and intestines. The vast majority of lactate (60%-70%) is disposed of by the liver via the pathway of gluconeogenesis. Although once considered a simple waste product of metabolism, lactate exerts numerous effects in many different tissues such as adipose tissue.
The ability of lactate to alter metabolic processes in adipocytes is the result of binding to specific cell surface receptors. The major lactate receptor was originally identified as the G-protein coupled receptor, GPR81. GPR81 is now known as hydroxycarboxylic acid receptor 1 (HCA1) which is encoded by the HCAR1 gene. HCA1 is coupled to a Gi-type G-protein that, upon lactate binding, results in a decrease in cAMP levels.
HCA1 is almost exclusively expressed in adipocytes and so lactate-induced activation of the receptor and the resultant decrease in cAMP leading to inhibition of lipolysis might at first seem counterintuitive. This is because an obvious cause of elevated blood lactate levels is intensive exercise and it would be expected that this type of exercise would require increased fatty acid release from adipocytes for skeletal muscle energy generation. However, adipocytes are another source of lactate and their reduction of glycolytic pyruvate to lactate increases as a result of insulin-stimulated glucose uptake into these cells. Therefore, the likely normal function of lactate-induced stimulation of HCA1 is to contribute to the insulin-induced inhibition of adipocyte lipolysis. Indeed, in HCA1 knock-out mice there is an associated decrease in insulin-mediated inhibition of lipolysis. This indicates that lactate functions in an autocrine and paracrine manner to mediate insulin-induced regulation of adipocyte lipolysis.
HCA1 expression is also found on cancer cells and dendritic cells of the immune system. These sites of expression contribute to the role of lactate in cancer metabolism and immune system evasion. Due to the altered metabolism of glucose in cancer cells they produce large amounts of lactate. The lactate is transported out of the cells via transporters of the monocarboxylate transporter (MCT) family (predominantly MCT4) which are encoded by genes of the SLC16 family. The lactate then binds to HCA1 receptors on these same cells resulting in the activation of a transcription program that contributes to immune evasion.
Adipose Tissue Hormones and Adipokines
Adipose tissue produces and releases a vast array of protein signals including growth factors, cytokines, chemokines, acute phase proteins, complement-like factors, and adhesion molecules. The Table below describes several of the adipocyte proteins in more detail with leptin, adiponectin, and resistin discussed in greater detail in the following sections. The proteins of the various signaling processes are listed below. Not all WAT secretes the same adipokines as is evident from studies of differences in adipose tissue function in various anatomical regions of the body as described above. This is most significant when considering the clinical risks associated with increased adipose tissue mass. For example, increases in visceral WAT, even when subcutaneous fat depots are not increased, carry a greater metabolic risk for insulin resistance and diabetes and cardiovascular disease.
Growth and angiogenic factors: fibroblast growth factors (FGFs), insulin-like growth factor-1 (IGF-1), hepatocyte growth factor (HGF), nerve growth factor (NGF), vascular endothelial cell growth factor (VEGF), transforming growth factor-β (TGFβ), angiopoietin-1, angiopoietin-2, tissue factor (TF, coagulation factor 3)
Cytokines: Interleukin-1beta (IL-1β), IL-4, IL-6, IL-8, IL-10, IL-18, tumor necrosis factor-alpha (TNFα), macrophage migration inhibitory factor (MIF)
Complement-like factors: adipsin, adiponectin, complement factor 3 (active form is C3adesArg and is commonly known as acylation stimulating protein, ASP)
Adhesion molecules and ECM components: α2-macroglobulin, vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), collagen types I, III, IV, and VI, fibronectin, matrix metalloproteinase 1 (MMP1), MMP7, MMP9, MMP10, MMP11, MMP14, MMP15, lysyl oxidase
Acute phase proteins: C-reactive protein (CRP), serum amyloid A3 (SAA3), plasminogen activator inhibitor-1 (PAI-1), haptoglobin
Chemokines: chemerin, monocyte chemotactic protein-1 (MCP-1), macrophage inhibitory protein-1 alpha (MIP-1α), regulated upon activation, normally T cell expressed and secreted (RANTES)
Metabolic processes: adipocyte fatty acid binding protein (FABP4, also known as aP2), apolipoprotein E (apoE), resistin, omentin, vaspin, apelin, retinol binding protein 4 (RBP4), visfatin, leptin
Other processes: COX pathway products PGE2 and PGI2 (prostacyclin), nitric oxide synthase pathway, renin-angiotensin system, tissue inhibitors of metalloproteinases (TIMPs)
In addition to these secreted factors adipose tissue produces several plasma membrane and nuclear receptors that can trigger changes in adipose tissue function. Plasma membrane receptors include those for insulin, glucagon, growth hormone, adiponectin, gastrin, and angiotensin-II. Nuclear receptors include peroxisome proliferator-activated receptor-γ (PPARγ), estrogens, androgens, vitamin D, thyroid hormone, progesterone, and glucocorticoids.
Table of Representative Adipose Tissue Hormones and Adipokines
| Factor | Principal Source | Major Action |
| adiponectin also called adipocyte complement factor 1q-related protein (ACRP30), and adipoQ | adipocytes | see below |
| adipsin (also called complement factor D) | adipocytes, liver, monocytes, macrophages | rate limiting enzyme in complement activation |
| apelin | white adipose tissue (WAT), skeletal muscle, vascular stromal cells, heart | apelin was identified as the ligand for the GPCR originally identified as APJ (Angiotensin-like receptor Protein J) and thus apelin is derived from APJ endogenous ligand; the APJ receptor is more commonly referred to as the apelin receptor (encoded by the APLNR gene); enhances skeletal muscle mass as well as density of mitochondria; levels increase with increased insulin; exerts positive hemodynamic effects; may regulate insulin resistance by facilitating expression of BAT uncoupling proteins (e.g. UCP1, thermogenin); also enhances expression of PGC-1α |
| asprosin | white adipose tissue (WAT) | released from white adipocytes in response to fasting; stimulates hepatic glucose production; also exerts effects in the hypothalamus to induce feeding behavior; released from the C-terminus (encompassing amino acids from exon 65 and 66) of the profibrillin protein by proteolytic processing; pathogenic variants that truncate the 3′-end of the FBN1 gene, that encodes profibrillin, are associated with neonatal progeroid syndrome (NPS) that is associated with, among other pathologies, reduced subcutaneous fat; binds to a receptor of the olfactory family of GPCR identified as olfactory receptor family 4 subfamily M member 1 (OR4M1; also identified as OLFR734); receptor is a Gs-type GPCR |
| chemerin | adipocytes, liver | also known as retinoic acid receptor responder protein 2 (RARRES2) and as such chemerin is encoded by the RARRES2 gene; modulates expression of adipocyte genes involved in glucose and lipid homeostasis such as GLUT4 and fatty acid synthase (FAS); potent anti-inflammatory effects on macrophages expressing the chemerin receptor (chemokine-like receptor-1, CMKLR1); the CMKLR1 protein is also known as ChemR23 |
| C-reactive protein (CRP) | hepatocytes, adipocytes | is a member of the pentraxin family of calcium-dependent ligand binding proteins; assists complement interaction with foreign and damaged cells; enhances phagocytosis by macrophages; levels of expression regulated by circulating IL-6; modulates endothelial cell functions by inducing expression of various cell adhesion molecules, e.g. ICAM-1, VCAM-1, and selectins; induces MCP-1 expression in endothelium; attenuates nitric oxide (NO) production by downregulating nitric oxide synthase (NOS) expression; increase expression and activity of PAI-1 |
| IL-6 | adipocytes, hepatocytes, activated Th2 cells, and antigen-presenting cells (APCs) | acute phase response, B cell proliferation, thrombopoiesis, synergistic with IL-1 and TNF on T cells |
| isthmin-1 (Ism1) | near exclusive to mature adipocytes | initially identified in Xenopus midbrain-hindbrain organizer called isthmus; increases adipocyte glucose uptake; similar to effects of insulin, Ism1 enhances GLUT4 mobilization to the plasma membrane in adipocytes; suppresses hepatic lipid synthesis; overexpression in mice prevents insulin resistance and hepatic steatosis |
| leptin | predominantly adipocytes, mammary gland, intestine, muscle, placenta | see below |
| monocyte chemotactic protein-1 (MCP-1) | leukocytes, adipocytes | is a chemokine defined as CCL2 (C-C motif, ligand 2); recruits monocytes, T cells, and dendritic cells to sites of infection and tissue injury |
| omentin (intelectin-1) | visceral stromal vascular cells of omental adipose tissue; intestinal enterocytes | the omentum is one of the peritoneal folds that connects the stomach to other abdominal tissues; the activity identified as omentin is also known as intestinal lactoferrin receptor; the protein is encoded by the intelectin-1 (ITLN1) gene; intelectins are members of the lectin family of carbohydrate-binding proteins; intelectin-1 exhibits galactofuranose, galactose, and N-acetylgalactosamine (GalNAc) binding; protein also enhances insulin-stimulated glucose transport; levels in the blood inversely correlated with obesity and insulin resistance |
| plasminogen-activator inhibitor-1 (PAI-1) | adipocytes, monocytes, placenta, platelets, endometrium | see the Hemostasis: Biochemistry of Blood Coagulation page for more details |
| resistin | adipocytes, spleen, monocytes, macrophages, lung, kidney, bone marrow, placenta | see below |
| TNFα | primarily activated macrophages, adipocytes | induces expression of other autocrine growth factors, increases cellular responsiveness to growth factors and induces signaling pathways that lead to proliferation |
| vaspin (serpinA12) | visceral and subcutaneous adipose tissue | is a serine protease inhibitor of the serpin peptidase inhibitor clade A family; levels decrease with worsening diabetes, increase with obesity and impaired insulin sensitivity; encoded by the SERPINA12 gene |
| visfatin; also called pre-B cell colony-enhancing factor (PBEF); these two independent activities are identical to the enzyme nicotinamide phosphoribosyltransferase (NAMPT) | ubiquitously expressed with highest levels of expression in visceral white adipose tissue | was originally reported to have insulin mimetic effects but that paper was subsequently retracted; the intracellular version of NAMPT (sometimes referred to as iNAMPT) has nicotinamide phosphoribosyltransferase activity; the extracellular version (eNAMPT) exhibits cytokine-like activity; conflicting results relative to insulin receptor binding but blocking insulin receptor signaling interferes with effects of eNAMPT; changes in NAMPT activity occur during fasting and positively regulate the activity of the NAD+-dependent deacetylase, SIRT1, leading to alterations in gene expression |
Leptin
Leptin is 16kDa peptide whose central function is the regulation of overall body weight by limiting food intake and increasing energy expenditure. However, leptin is also involved in the regulation of the neuroendocrine axis, inflammatory responses, blood pressure, and bone mass. The human leptin gene (symbol: LEP) is the homolog of the mouse “obese” gene (symbol: OB) that was originally identified in mice harboring a mutation resulting in a severely obese phenotype.
The LEP gene is located on chromosome 7q32.1 and is composed of 3 exons that encode a 167 amino acid precursor protein.
Leptin-deficient (ob/ob) and leptin receptor-deficient (db/db) mice exhibit numerous disruption in energy, hormonal, and immune system balance. These mice are obese, display hormonal imbalances, have defects in thermoregulation, have hematopoietic defects and are infertile. Levels of leptin increase in the serum in obese individuals and drop during weight loss. There is a direct correlation between the amount of body fat an individual carries and the circulating levels of leptin. Leptin activates the anorexigenic axis (appetite suppression) in the arcuate nucleus (ARC) of the hypothalamus by increasing the frequency of action potentials in the hypothalamic POMC neurons by depolarization through a nonspecific cation channel and by reduced inhibition by local orexigenic neuropeptide-Y (NPY) neurons.
Leptin functions by binding to its receptor which is a member of the cytokine receptor family. Leptin and the leptin receptors possess structural similarities to the IL-6 family of cytokines and the class I cytokine receptor family.
The leptin receptor is encoded by the LEPR gene. The LEPR gene is located on chromosome 1p31.3 and is composed of 24 exons that generate six alternatively spliced mRNAs. These six leptin receptor mRNAs encode six different protein isoforms. The leptin receptors are named Ob-R, OB-Rb, OB-Rc, Ob-Rd, Ob-Re, and Ob-Rf.
The OB-Rb mRNA encodes the long form of the leptin receptor (also called LEPR-B) and is expressed primarily in the hypothalamus but is also expressed in cells of the innate and adaptive immune systems as well as in macrophages. The other receptor subtypes are expressed in numerous tissues including muscle, liver, kidney, adrenal glands, leukocytes, and vascular endothelium.
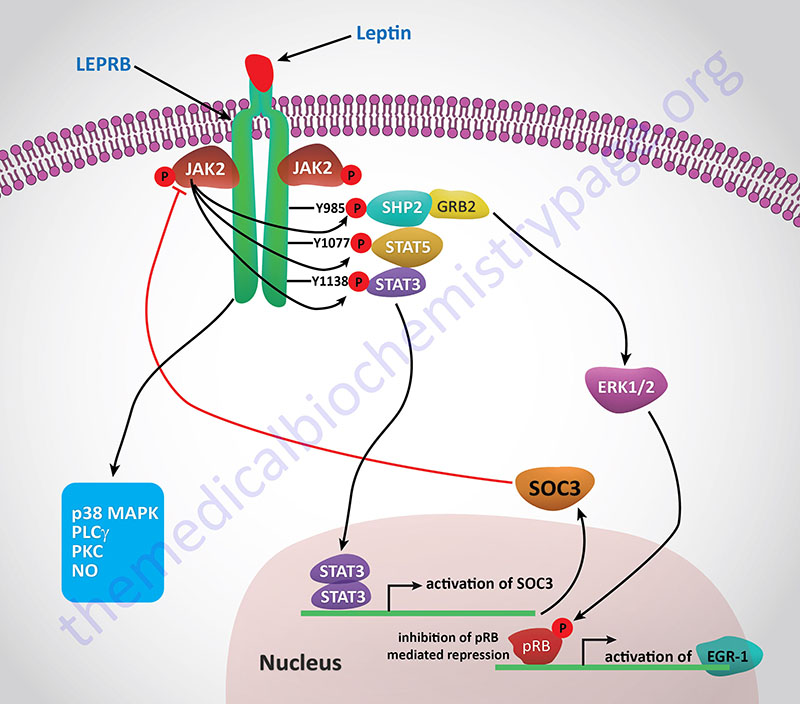
Activation of the receptor leads to increased phosphatidylinositol-3-kinase (PI3K) activity, while simultaneously inhibiting AMPK activity, via activation of the Jak/STAT signaling pathway. Within the hypothalamus, the inhibition of AMPK activity is a major contributor to the anorexigenic actions of leptin. Decreased hypothalamic AMPK activity results in reduced inhibition of the rate limiting enzyme of fatty acid synthesis, acetyl-CoA carboxylase, ACC. The resulting increased ACC activity leads to increased production of malonyl-CoA which is a potent modulator of hypothalamic inhibition of feeding behavior.
One effect of the activation of the Jak/STAT pathway is activation of suppressor of cytokine signaling 3 (SOCS3) which then inhibits leptin signaling in a negative feed-back loop. Leptin binding its receptor also results in the activation of mTOR (mechanistic target of rapamycin) both in the hypothalamus and in peripheral tissues. The role of mTOR in the regulation of protein synthesis is covered in both the Protein Synthesis (Translation): Processes and Regulation page as well as in the Insulin Function, Insulin Resistance, and Food Intake Control of Secretion page. The role of leptin in the activation of mTOR function is an important factor in the ability of leptin to activate macrophages. An interesting aspect of the role of leptin in mTOR function is that within mature adipocytes leptin synthesis itself is dependent on mTOR activation. Given that leptin levels rise in the serum of obese individuals and that leptin interaction with macrophages leads to increased macrophage inflammatory processes, it is not surprising that there is a direct correlation between leptin levels and the development of atherosclerosis.
Leptin expression is under complex control and a number of transcription factor binding sites have been identified in the promoter region of the leptin gene. Leptin levels are higher in age and weight-matched females compared with males. This is partially due to the inhibition of leptin expression by androgens and the stimulation of expression by estrogens. Leptin expression has been shown to be increased by sex steroids, glucocorticoids, cytokines, and toxins released during acute infection. The sympathetic nervous system triggers a reduction in circulating leptin levels via the release of catecholamines. This effect of catecholamines has been shown to be due to activation of β-adrenergic receptor signaling.
In addition to effects on appetite exerted via central nervous system functions, leptin is also known to exert effects on inflammatory processes. Leptin modulates peripheral T cell function leading to increased levels of T helper cell type 1 cytokines. In addition leptin reduces thymocyte apoptosis and increases thymic cellularity. These result correlate well with observations demonstrating a reduced capacity for immunologic defense when leptin levels are low. However, too much leptin is not beneficial as high concentrations can result in an abnormally strong immune response which predisposes an individual to autoimmune phenomena. Acute stimulation with pro-inflammatory cytokines results in increased serum levels of leptin, whereas, chronic stimulation by IL-1, IL-6, or TNFα leads to reduced levels of serum leptin.
Adiponectin
Adiponectin was independently isolated by four different laboratories leading to different names. However, adiponectin is considered the standard name for this adipose tissue-specific protein. Other names include adipocyte complement related protein of 30kDa (ACRP30) because of its homology to complement factor 1q (C1q), adipoQ, gelatin-binding protein 28kDa (BGP28), and adipocyte most abundant gene transcript 1 (apM1). The major biological actions of adiponectin are increases in insulin sensitivity and fatty acid oxidation.
Adiponectin is encoded by the ADIPOQ gene. The ADIPOQ gene is located on chromosome 3q27.3 spanning 16 kbp and composed of 4 exons that generate two alternatively spliced mRNAs encoding the same 244 amino acid precursor protein. The adiponectin gene does not contain a typical TATA-box promoter element upstream of the transcriptional start site. Expression of the ADIPOQ gene occurs exclusively in adipocytes of adipose tissue.
Adiponectin contains a C-terminal globular domain which harbors the homology to C1q and an N-terminal collagen-like domain. The globular domain allows for a homotrimeric association of the protein forming the functional structure of the protein. The association of the subunits is such that two trimeric globular domains interact with a single stalk of collagen domains formed from two trimers. This complex structure is similar to the TNF superfamily of proteins despite there being no amino acid sequence homology between adiponectin and TNF proteins. Within the circulation, adiponectin exists in both a full-length form as well as a globular form that is the result of proteolytic cleavage of the full-length protein. The hormone forms complex structures such that it can be found as a trimer, hexamer, and as the high molecular weight oligomer. In addition to the complex structure, adiponectin is glycosylated, a modification that is essential to its activity.
Adiponectin activity is inhibited by adrenergic stimulation and glucocorticoids. Expression and release of adiponectin is stimulated by insulin and inhibited by tumor necrosis factor-alpha (TNFα). Conversely, adiponectin exerts inflammatory modulation by reducing the production and activity of TNFα and IL-6. Unlike leptin, levels of adiponectin are reduced in obese individuals and increased in patients with anorexia nervosa. There are sex-related differences in adiponectin levels as well similar to that seen for leptin where age and weight matched males have lower levels than females. In patients with type 2 diabetes, levels of adiponectin are significantly reduced.
Adiponectin functions by interaction with specific cell-surface receptors and at least two receptors have been identified. AdipoR1 is expressed at highest levels in skeletal muscle and AdipoR2 in primarily expressed in the liver. Expression of adiponectin receptors is also seen in various regions of the brain. AdipoR1 is highly expressed in the medial prefrontal cortex, hippocampus, and the amygdala. Whereas, AdipoR2 expression in the brain is restricted to the hippocampus and specific hypothalamic nuclei. AdipoR1 is a high-affinity receptor for globular adiponectin and has low affinity for the full-length adiponectin. In contrast, AdipoR2 has intermediate affinity for globular and full-length adiponectin. Although these receptors contain seven transmembrane domains typical of the serpentine family of G-protein coupled receptors (GPCR), they are structurally distinct from the GPCR class. The AdipoR stimulate the phosphorylation and activation of AMPK. The adiponectin-mediated activation of AMPK results in increased glucose uptake, increased fatty acid oxidation, and increased phosphorylation and inhibition of acetyl-CoA carboxylase (ACC) in muscle. In the liver the result is reduced activity of gluconeogenic enzymes and glucose output.
Adiponectin also plays an important role in hemostasis by suppressing TNFα-mediated inflammatory changes in endothelial cell responses and inhibiting vascular smooth muscle cell proliferation. Activation of AMPK activity in endothelial cells results in increased fatty acid oxidation and activation of endothelial nitric oxide synthase (eNOS).
Resistin
Resistin is a 12 kDa protein that was originally identified in mice in a screen for genes suppressed by an agonist of the peroxisome proliferator-activated receptor-γ (PPARγ). The name resistin derives from the original observation that this protein induced insulin resistance in mice. Resistin belongs to a family of four proteins referred to as FIZZ proteins for “found in inflammatory zone”. Resistin is thus, also referred to as FIZZ3. Although resistin is expressed in adipocytes, in humans it appears that macrophages may be the most important source of the protein.
Resistin is encoded by the RETN gene. The RETN gene is located on chromosome 19p13.2 and is composed of 4 exons that generate five alternatively spliced mRNAs that collectively encode three distinct precursor protein isoforms.
In mice, resistin expression increases during adipocyte differentiation and levels of resistin increase in diet-induced obesity. Reduction in resistin levels is associated with increased AMPK activity in the liver which leads to decreased expression of gluconeogenic enzymes and consequent reduction in hepatic glucose production. Conversely, elevation in resistin levels is associated with increased hepatic glucose production and glucose intolerance. Whether these same responses to resistin are evident in human is still under investigation.
What is known is that overexpression of resistin in human hepatocytes impairs insulin-stimulated glucose uptake and glycogen synthesis. Part of the mechanism for impaired glycogen synthesis is that resistin decreases the expression of one of the insulin receptor substrates (IRS-2) which is involved in the activation of PI3K. The PI3K-activated signal pathway leads to the phosphorylation and inhibition of glycogen synthase kinase 3β (GSK3β). Un-phosphorylated GSK3β would normally phosphorylate and inhibit glycogen synthase activity. Loss of this pathway then leads to a higher rate of glycogen synthase inhibition by GSK3β-mediated phosphorylation.
Resistin also exerts effects on the immune system and the vasculature. Resistin modulates endothelial cell function by enhancing expression of the cell adhesion molecule VCAM-1 and the chemoattractant MCP-1. Resistin has also been shown to exert a pro-inflammatory effect on smooth muscle cells.
Inflammatory Functions of Adipose Tissue
The significance of inflammatory responses elicited via secretion of adipose tissue-derived (WAT) cytokines relates to the fact that their production and secretion is increased in obese individuals. There is a growing body of evidence that demonstrates a direct link between the changes in adipose tissue function in obesity and the development of type 2 diabetes and the metabolic syndrome. One key change in adipose tissue during obesity is an increase in the percentage of macrophages resident within the tissue. Macrophages are a primary source of pro-inflammatory cytokines secreted by adipose tissue. The primary adipokine responsible for this infiltration is monocyte chemotactic protein-1 (MCP-1).
As the level of macrophages increases in adipose tissue the level of pro-inflammatory cytokine secretion by the tissue increases. Circulating levels of both TNFα and IL-6 increase as adipose tissue expands in obesity and these changes are directly correlated with insulin resistance and the development of type 2 diabetes. As indicated above adiponectin plays an important anti-inflammatory role by suppressing the production of both TNFα and IL-6. However, as the level of macrophage infiltration increases in obesity the increased secretion of TNFα results in suppression of adiponectin production and secretion. Adipose tissue-derived IL-6 accounts for approximately 30% of the circulating level of this pro-inflammatory cytokine.
Visceral WAT has been shown to secrete a higher percentage of the circulating IL-6 than subcutaneous WAT and this fact correlates with the negative effects of a pro-inflammatory status (as is the case with obesity) on the organs. As WAT density increases there is an associated increase in IL-6 secretion which is correlated to an increase in the circulating levels of acute-phase proteins such as CRP. In addition to the effects of TNFα on adiponectin production the cytokine also directly affects insulin sensitivity by inhibiting insulin receptor signaling. This effect of TNFα is the result of increased insulin receptor serine phosphorylation.
TNFα (as well as IL-6) triggers the release of pro-inflammatory cytokines such as JUN N-terminal kinase (JNK) and nuclear factor kappa B (NF-κB). Activation of JNK leads to the insulin receptor serine phosphorylation as well as insulin receptor substrate (IRS) serine phosphorylation. Both of these TNFα-mediated serine phosphorylations lead to impaired insulin receptor signaling.
JNK and NF-κB activate pro-inflammatory genes which results in a self-perpetuating cycle of increased inflammatory cytokine release. TNFα also decreases endothelial nitric oxide synthase (eNOS) resulting in decreased levels of NO as well as decreased expression of mitochondrial oxidative phosphorylation genes. This leads increased oxidative stress, accumulation of reactive oxygen species (ROS), and increased endoplasmic reticulum stress. The responses of the cell to the increased oxidative stress is further increases in NF-κB releases and thus, increased inflammatory processes. The effects of adipocyte-derived TNFα and IL-6 demonstrates a clear correlation between obesity and a pro-inflammatory role for adipocytes.
Although lymphocytes (T-cells and B-cells) are not constituents of adipose tissue they are physically associated within the lymph nodes. Lymph tissue is surrounded by pericapsular adipose tissue which increases in density with increasing obesity. This close association allows for 2-way paracrine interactions between the lymph and adipose tissues. One important interaction between lymph tissue and WAT involves leptin. As indicated above, leptin plays a major role in the regulation of appetite and energy balance and the circulating levels of leptin increases as adipose tissue mass increases.
Leptin has also been shown to modulate T-cell function, thus demonstrating a pro-inflammatory role for leptin. Leptin protects T-cells from apoptosis and enhances the switching of T-cells to a Th1 response. Pro-inflammatory cytokine production and release from T-cells is increased as a result of leptin action.
Leptin induces a number of signal transduction pathways in immune and endothelial cells as outlined above. Leptin effects on the vascular endothelium are also pro-inflammatory. Expression of adhesion molecules is increased by leptin binding its receptor on endothelial cells. This results in an increased ability of neutrophils and other leukocytes to adhere to the endothelium leading to increased local inflammatory processes.
Metabolic Functions of Brown Adipose Tissue, BAT
Brown adipose It was originally thought that BAT was present in humans only during the neonatal period. However, evidence has demonstrated that adults retain some metabolically active BAT deposits that respond to cold and sympathetic nervous system activation. The most studied regulator of the metabolic and thermogenic properties of BAT is the catecholamine, norepinephrine.
Within BAT, norepinephrine interacts with all three types of adrenergic receptors (α1, α2, and β) each of which activates distinct signaling pathways in the brown adipocyte. With respect to the β-adrenoreceptors, the β3 subtype is the most significant, β1 receptors are expressed in brown preadipocytes but not mature adipocytes and β2 receptors are expressed in BAT but not brown adipocytes themselves.
Of significance to the role of BAT in thermogenesis and the major role of β3-adrenoreceptors is the fact that these receptors are not subject to desensitization as are the β1 and β2 receptors. However, this is not to say that continuous adrenergic stimulation has no negative effect in BAT. Indeed, the level of β3 receptor expression is downregulated during continuous adrenergic stimulation. However, this effect is transient and the mRNA rapidly re-accumulates after termination of the stimulatory signal.
Signal transduction events triggered by adrenergic stimulation of BAT are effected via the activation of Gs type G-proteins. Gs proteins couple to the activation of adenylate cyclase resulting in increased cAMP production and activation of PKA. For more information on the types of G-proteins go to the Signal Transduction Pathways: G-Proteins and GPCR page. Activation of HSL leads to release of free fatty acids which are taken up by the mitochondria similarly as they would be for purposes of oxidation, however, in BAT they interact with and activate the proton gradient uncoupling activity of uncoupling protein 1 (UCP1, also known as thermogenin). Uncoupling the proton gradient releases the energy of that gradient as heat. The thermogenic function of BAT is outlined in the Figure below. In addition to stimulating heat production in BAT, norepinephrine promotes the proliferation of brown preadipocytes, promotes the differentiation of mature brown adipocytes, inhibits apoptosis of brown adipocytes, and regulates the expression of the UCP1 gene.
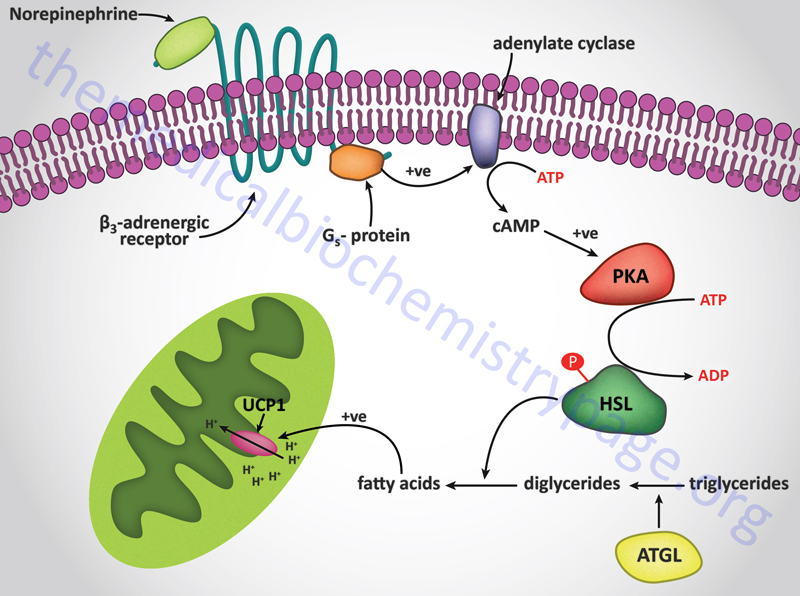
Interestingly, norepinephrine stimulation of α2-adrenergic receptors on BAT has the opposite effect to β3 receptor activation. This is due to the fact that the α2 receptors are coupled to Gi type G-proteins whose activation results in inhibition of adenylate cyclase. The precise physiological significance of this parallel response of BAT to norepinephrine is as yet not fully appreciated. It is unclear how, or when, BAT balances the thermogenic stimulatory actions of β3 receptors with the inhibitory actions of α2 receptors but it may allow the cells to modulate their responses to norepinephrine under varying physiological conditions.
In addition to norepinephrine acting through the adrenergic receptors, adenosine and secretin stimulate lipolysis and thermogenesis in BAT. Adenosine binds to the adenosine 2A (A2A) receptor (encoded by the ADORA2A gene) which is coupled to a Gs-type G-protein. Secretin binds to the secretin receptor (encoded by the SCTR gene) which is also coupled to a Gs-type G-protein. Thus, the effects of adenosine and secretin are similar to those of norepinephrine in BAT.
Another Gs-type GPCR that functions in BAT to regulate lipolysis and thermogenesis is GPR3. GPR3 is a constitutively active receptor meaning that it activates the production of cAMP in the absence of a ligand. In addition to being expressed in BAT, GPR3 is also expressed in the central nervous system and the ovaries.
Primary Factors Regulating the Metabolic Activity of BAT
Although the signaling molecules that includes norepinephrine, adenosine, and secretin, are the primary regulators of the lipolytic and thermogenic activities of BAT, numerous other factors contribute to the overall network of BAT regulators. These regulatory factors include, but are not limited to, the peroxisomal fatty acid metabolizing enzyme, acyl-CoA oxidase 2 (ACOX2), the acyl-CoA thioesterase encoded by the ACOT11 gene, the branched-chain amino acid import transporter encoded by the SLC25A44 gene, the DNAJC15 (DnaJ heat shock protein family (Hsp40) member C15) encoded enzyme, the SLIT3 (slit guidance ligand 3) encoded protein, and BMP8B.
Acyl-CoA oxidase 2 is also referred to as branched-chain acyl-CoA oxidase and this peroxisomal enzyme is primarily involved in the oxidation of 2-methyl (α-methyl) branched-chain fatty acids, also termed monomethyl branched-chain fatty acids (mmBCFA). In brown adipocytes ACOX2 functions in a futile cycle of fatty acid synthesis and oxidation that releases energy as heat in the absence of fatty acid interaction with UCP1.
The ACOT11 encoded acyl-CoA thioesterase functions in the process of fatty acid deactivation, releasing a free fatty acid and CoA-SH from fatty acyl-CoA molecules. The ACOT11 enzyme belongs to the thioesterase superfamily of enzymes and as such is also identified as THEM1. The ACOT11 encoded protein possesses multiple functional domains with one domain being the thioesterase domain and the other being a START (STeroidogenic Acute Regulatory protein-related lipid Transfer) domain. As such ACOT11 has also been identified as STARD14. Because the consequences of ACOT11 activity are reduced levels of activated fatty acids, the enzyme serves as a negative regulator of thermogenesis and leads to conservation of energy by suppressing fatty acid oxidation.
The role of the SLC25A44 encoded transporter in overall BAT function is discussed in the BAT Amino Acid Metabolism in the Regulation of Glucose Homeostasis section below.
The DNAJC15 encoded protein is a member of the large family of heat-shock 40 (Hsp40) proteins. DNAJC15 encoded protein is also identified as methylation-controlled J protein (MCJ). The DnaJ nomenclature is derived from the E. coli Hsp70 co-chaperone DnaJ. Hsp40 proteins are correctly referred to as obligate co-chaperones since they function in conjunction with proteins of the Hsp70 family whose ATPase activities require the interactions with Hsp40 proteins. The DNAJC15 encoded protein functions as a co-chaperone at the mitochondrial inner membrane to limit the activity of complex I of the electron transport chain by preventing the formation of supercomplexes. In this capacity the DNAJC15 protein functions as a negative regulator of BAT thermogenesis and fat oxidation.
The SLIT3 encoded protein is a member of the human SLIT family which is composed of three member, SLIT1, SLIT2, and SLIT3. The SLIT protein was originally identified in the fruit fly, Drosophila melanogaster, from a mutant associated with a midline defect in the nervous system resulting in a “slit” in the structure. The SLIT family of proteins are secreted proteins that are involved cell migration and axonal growth. Adipocyte precursor cells product and secrete SLIT3 that, when cleaved into N-terminal (SLIT3-N) and C-terminal (SLIT3-C) portions by the BMP1 (bone morphogenetic protein 1) protease, function independently to regulate angiogenesis and sympathetic innervation in BAT. The SLIT3-C fragment has been shown (2026) to bind to the plexin-1 (encoded by the PLXNA1 gene) protein. The role of SLIT3 in BAT-mediated thermogenesis was demonstrated by shRNA-mediated knock-down which is associated with increased fat storage, reduced expression of UCP1, and impaired cold-induced thermogenesis.
BMP8B (bone morphogenetic protein 8B) functions as a signaling molecule both centrally and peripherally to increase the thermogenic properties of BAT. Within the nervous system BMP8B induces an increase in sympathetic output to BAT. Sympathetic stimulation of BAT, via norepinephrine, results in increased production of BMP8B. Within BAT, BMP8B activates a signaling cascade that involves SMAD1, SMAD5, and SMAD8. In the absence of BMP8 the levels of these three SMAD proteins decreases significantly. The effect of BMP8B activity in BAT is an enhanced capacity for the tissue to respond to sympathetic stimulation and therefore induces enhanced thermogenesis and fatty acid oxidation.
BAT-Derived Adipokines
Similar to the endocrine role of WAT, BAT also synthesizes and secretes numerous factors. However, due to the relatively small overall size of brown fat compared to white fat, the role of factors secreted from BAT serve mainly autocrine and paracrine roles. This is not to say that BAT doesn’t have a potential endocrine role it is just not as well established as the endocrine role of WAT. Several proteins produced from BAT serve autocrine functions such as adipsin, fibroblast growth factor 2 (FGF2), insulin-like growth factor-1 (IGF-1), prostaglandins, and adenosine.
Adipsin (also known as complement factor D) cleaves the complement protein C3 into C3a and C3b. C3a is inactivated to C3desArg which is more commonly referred to as acylation-stimulating protein (ASP). ASP stimulates TG synthesis and glucose uptake in WAT. The level of C3 expression is much higher in WAT than in BAT and ASP has not been found in BAT so the precise function of adipsin in BAT is not yet well defined. BAT-derived adipsin could be functional when BAT is not participating in thermogenesis but is functioning in an anabolic state to store TG.
Acute and chronic cold exposure results in increased FGF2 expression in BAT as does norepinephrine. The FGF receptor 1 (FGFR1) gene is also expressed in BAT and the result of FGF2 activation of the receptor is increased brown adipocyte cell density. Similarly, during cold exposure the levels of IGF-1 mRNA increase and IGF-1 receptors are highly expressed on brown adipocytes. IGF-1 action can prevent TNFα-induced apoptosis in brown fat.
Paracrine factors synthesized by BAT include nerve growth factor (NGF), vascular endothelial cell growth factor (VEGF), angiotensinogen, and NO. The secretion of NGF occurs primarily from proliferating brown preadipocytes and in this capacity is believed to promote sympathetic innervation of the tissue which in turn permits increased norepinephrine stimulation of the cells in BAT.
Expression of VEGF is high in proliferating and mature brown adipocytes and the VEGF receptors, FLK-1 and FLK-4, are expressed in BAT. The expression of VEGF in BAT may promote and maintain the high level of vascularization in this tissue. Norepinephrine stimulation and cold stress both result in increased levels of VEGF expression in BAT.
Both inducible nitric oxide synthase (iNOS) and endothelial NOS (eNOS) are expressed in BAT. In addition to its expression in the endothelial cells of BAT, eNOS expression is seen in brown adipocytes. Norepinephrine stimulation of BAT, as well as brown adipocytes in culture, results in increased NO production. Within brown adipocytes the production of NO may lead to inhibition of mitochondrial oxidation. Within BAT the production of NO likely promotes rapid increases in blood flow to this tissure.
BAT Amino Acid Metabolism in the Regulation of Glucose Homeostasis
In humans the amount of BAT is vastly less than that of WAT. Nonetheless, evidence is clear that the metabolic processes exerted by BAT, outside the context of its role in thermogenesis, contribute to protection against the development of type 2 diabetes. One important metabolic function of BAT is the diversion of carbon and nitrogen from the branched-chain amino acids (BCAA) to the liver where they can be utilized for the production of the antioxidant peptide, glutathione. Indeed, metabolites from BCAA derived from BAT is higher than that from WAT.
Uptake of BCAA into the mitochondria of brown adipocytes requires the amino acid transporter encoded by the SLC25A44 gene, known as mitochondrial BCAA carrier (MBC). Within the mitochondria of brown adipocytes the BCAA are deaminated via the mitochondrial BCAA-specific transaminase encoded by the BCAT2 gene. The nitrogen is transferred to 2-oxoglutarate (α-ketoglutarate) generating glutamate. Indeed, the major product of BCAA metabolism in brown adipocytes is glutamate. Deamination of glutamate back to 2-oxoglutarate can take place through the action of mitochondrial alanine transaminase (ALT) which also yields alanine which is the second major byproduct of BCAA metabolism in brown adipocytes.
Metabolic byproducts of metabolism of BCAA that are enriched from BAT versus WAT include glutamate, N-acetylglutamate, N-acetylaspartate, glutathione, α-ketoisocaproic acid (KIC), and α-ketoisovaleric acid (KIV). In addition to glutamate and alanine, metabolism of BCAA in BAT yields the amino acids glycine and proline.
Several studies have demonstrated that the nitrogen from BCAA contributes to over 60% of the daily need of nitrogen for the synthesis of alanine. However, this represents whole body alanine synthesis, not just that derived from BCAA metabolism in BAT. With respect to the carbon atoms of the BCAA, only about 10% ends up in TCA cycle intermediates in BAT. Most of the BCAA-derived carbon atoms are released from brown adipocytes in the form of their respective α-keto acids (BCKA) and 3-hydroxyisobutyrate (3-HIB).
The delivery of BAT-derived metabolites of BCAA to the liver contributes to the synthesis of glutathione (GSH) in hepatocytes. Given the critical anti-oxidant function of GSH, the contribution of BAT to reductions in liver oxidative stress is of paramount importance and directly contributes to maintenance of hepatic insulin sensitivity. The ability of hepatocytes to robustly respond to insulin signaling is critical to the role of the liver in the regulation of circulating levels of glucose. A major response of the liver to insulin is increased uptake of glucose and storage in glycogen and the inhibition of endogenous glucose production via gluconeogenesis.
Since the liver does not express the branched-chain aminotransferases the role of BAT in delivering nitrogen carriers to the liver for the synthesis of glutathione is critical. With respect to the development of type 2 diabetes and its associated insulin resistance, it has been demonstrated that in obese individuals the BAT-mediated metabolism of BCAA is impaired. This impaired contribution of BAT metabolism to overall liver homeostasis and insulin sensitivity plays a significant role in the overall etiology of type 2 diabetes.