

Last Updated: October 28, 2025
Introduction to Phenylketonuria: PKU
Phenylketonuria (PKU) is an autosomal recessive disorder associated with hyperphenylalaninemia that results from defects in the metabolism of phenylalanine. PKU represents the most severe form of the hyperphenylalaninemias (HPA). There are several hyperphenylalaninemias that are not PKU and are called non-PKU hyperphenylalaninemias (non-PKU HPA).
Normal serum levels of phenylalanine are 50 – 110 μM (0.8 – 1.8 mg/dL). A serum phenylalanine concentration of 120 – 600 μmol/L is classified as mild hyperphenylalaninemia. Serum concentrations of 600–1200 μmol/L are classified as mild PKU. Moderate PKU is classified as having a serum phenylalanine concentration of 900–1200 μmol/L. Serum phenylalanine concentrations above 1,200 μmol/L (20 mg/dL) denote classic PKU.
PKU, often termed classic PKU, is caused by mutations in the gene (PAH) encoding phenylalanine hydroxylase.
The HPA are disorders of phenylalanine hydroxylation. Because the reaction catalyzed by PAH involves tetrahydrobiopterin (BH4) as a co-factor, the HPAs can result from defects in any of the several genes required for synthesis and recycling of BH4. Removal of excess phenylalanine normally proceeds via the tyrosine biosynthesis reaction and then via tyrosine catabolism. The first reaction in this process is the PAH catalyzed hydroxylation of phenylalanine.
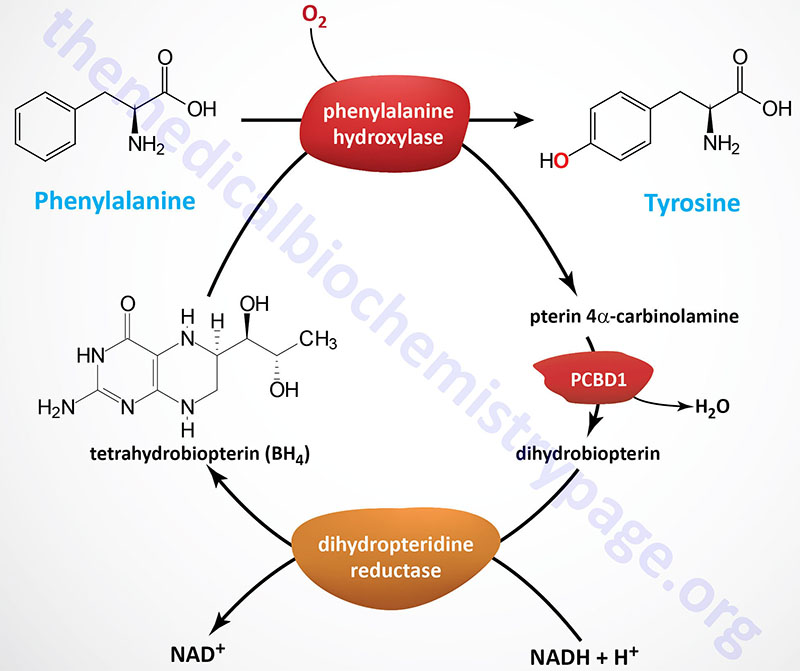
Molecular Biology of PKU
The PAH gene spans 170 kb located on chromosome 12q23.2 and is composed of 15 exons that generate two alternatively spliced mRNAs, both of which encode the same 452 amino acid protein.
The 5′-untranslated region of the PAH gene contains numerous cis-acting regulatory elements. There are many restriction fragment length polymorphisms (RFLP) and single nucleotide polymorphisms (SNP) associated with the PAH gene (for an explanation of RFLP and SNP see the Molecular Biology in Medicine page). Several hundred disease-causing mutations have been identified in the PAH gene.
As of 2016 more than 950 mutations have been identified mutations in the PAH gene that were associated with the development of PKU. The majority (50%) of these mutations are missense mutations and around 10% are splice site mutations. The originally identified PAH mutation in a PKU patient was a mutation of a single nucleotide in the splice site of the 5′ donor site of exon 12.
Molecular Biology of non-PKU Hyperphenylalaninemias
The non-PKU HPA can result from any defect in phenylalanine hydroxylation. Many are the result of defects in the metabolism of the tetrahydrobiopterin (BH4) co-factor of PAH. Approximately 1-2% of all cases of hyperphenylalaninemia are the result of defects in the pathway of BH4 synthesis or regeneration.
During the PAH catalyzed reaction, BH4 is converted to 4α-hydroxytetrahydrobiopterin. The regeneration of BH4 is catalyzed by reactions that involve 4α-carbinolamine dehydratase and dihydropteridine reductase (DHPR). In addition to these latter two enzymes, BH4 can be synthesized in a pathway that requires guanosine triphosphate cyclohydrolase, 6-pyruvoyltetrahydrobiopterin synthase, and sepiapterin reductase. A deficiency in any of the five enzymes will lead to defects in phenylalanine hydroxylation and consequently hyperphenylalaninemia.
Because BH4 is involved in additional critical hydroxylation reactions, notably tryptophan hydroxylation by tryptophan hydroxylase and tyrosine hydroxylation by tyrosine hydroxylase it is important to correctly diagnose any HPA as being the result of abnormal BH4 homeostasis or defects in PAH. These two hydroxylation reactions are critical processes in the brain in the formation of serotonin and the catecholamines, respectively. Both tryptophan hydroxylase and tyrosine hydroxylase catalyze the rate-limiting reactions in the synthesis of serotonin and dopamine, respectively.
Clinical Features of PKU
The major clinical manifestation associated with PKU, and many HPA, is impaired cognitive development and function. The precise mechanism for the intellectual impairment associated with PKU has not been worked out but is thought to be the result of the accumulation of phenylalanine in the brain, which becomes a major donor of amino groups in aminotransferase activity and depletes neural tissue of 2-oxoglutarate (α-ketoglutarate). The absence of 2-oxoglutarate in the brain shuts down the TCA cycle and the associated production of aerobic energy, which is essential to normal brain development.
Another hypothesis proposed to explain the severe intellectual impairment that can result in untreated PKU patients is that the accumulating phenylalanine impairs the transport of tyrosine into the brain. The reduction in brain tyrosine levels would then negatively impact the synthesis of the neurotransmitters, dopamine and norepinephrine. Transport of phenylalanine, tyrosine, and tryptophan into the brain is the function of the heterodimeric transporter composed of a heavy chain encoded by the SLC3A2 (4F2hc) gene and a light chain encoded by the SLC7A5 (LAT1) gene.
The product of phenylalanine transamination, phenylpyruvic acid, is reduced to phenylacetate and phenyllactate, and all three compounds appear in the urine. The presence of phenylacetate in the urine and sweat imparts a “mousy” odor.
Currently in this country (and many others as well), newborns are routinely screened for PKU by measurement of serum phenylalanine levels. All HPA, including PKU, occur with a frequency of 5 to 350 cases/million live births. In order to classify the phenotype of PKU into severe and less severe forms, as well as to exclude BH4-deficient HPA, it is necessary to measure the plasma, urine, and cerebrospinal fluid levels of phenylalanine, pterins, and the derivatives of the neurotransmitters derived from tryptophan and tyrosine.
Treatment of PKU
The mainstay in treatment of PKU is the low-phenylalanine diet. Optimal treatment requires both early onset (hence the utility of the post-natal assessments in this country) and continuous treatment throughout adolescence and possibly for life. Because of the necessity for phenylalanine in protein synthesis and neurotransmitter synthesis (via conversion to tyrosine) it is important to carefully control the intake of the amino acid. Too little and developmental impairment will occur, too much and severe neurological dysfunction will result.
An adjunct to the low phenylalanine diet is glycomacropeptide, a protein derived from cheese whey. Glycomacropeptide is rich in essential amino acids but contains no phenylalanine, tyrosine, or tryptophan. Studies of patients with PKU suggest that foods containing glycomacropeptide are palatable and the majority of patients prefer a diet supplemented with glycomacropeptide to their usual amino acid formula.
Some PAH mutations are associated with a BH4-sensitive phenotype of PKU. In individuals with these mutations are administered pharmacological doses of exogenous BH4 there is an increase in the activity of PAH that is sufficient to reduce circulating phenylalanine by a therapeutically relevant extent. The mechanisms of this BH4-responsiveness are multifactorial as determined by the fact that the mutations in PAH that respond to BH4 administration are found throughout the protein. However, 70% of these BH4-responsive mutations are found within the catalytic domain of PAH. The main mechanism of BH4-responsiveness appears to be the result of the stabilization of the PAH tetramer by preventing misfolding, subunit aggregation, proteolytic degradation, and thermal inactivation.
The current US FDA approved BH4 related drug is KUVAN® which is sapropterin dihydrochloride. KUVAN is an orally administered drug. Sapropterin dihydrochloride functions similarly to BH4 and thus, stimulates any residual PAH activity in patients with BH4-responsive forms of PKU.
Another approved drug for the treatment of PKU is a form of enzyme replacement therapy (ERT) and is identified as Palynziq® (pegvaliase). Palynziq is the enzyme phenylalanine ammonia lyase attached to polyethylene glycol (PEG). Palynziq is administered by injection. The consequences of injection of Palynziq is reduced circulating levels of phenylalanine. Phenylalanine ammonia lyase is a plant enzyme that converts phenylalanine to ammonia and trans-cinnamic acid in the process of the synthesis of phenyl propanoids. Phenylalanine ammonia lyase is also found in some yeasts, bacteria, and fungi but is not a mammalian enzyme.