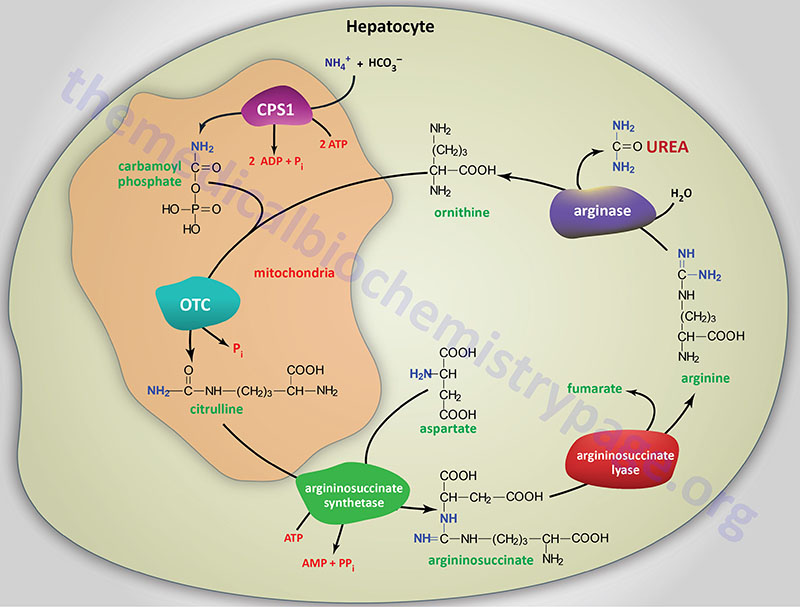
Last Updated: May 9, 2023 Introduction to Arginase Deficiency Arginase deficiency (AD) represents one of the disorders that result from defects in the processes of the urea cycle. Arginase deficiency is a rare autosomal recessive disorder. Arginase deficiency is the...