
Last Updated: November 15, 2022
Introduction to Sandhoff Disease
Sandhoff disease is an autosomal recessive disorder that is a member of a family of disorders identified as the GM2 gangliosidoses. The GM2 gangliosidotic diseases are severe psycho-motor developmental disorders caused by the inability to properly degrade membrane associated gangliosides of the GM2 family (see the Sphingolipids page). Because neural cell membranes are enriched in GM2 gangliosides, the inability to degrade this class of sphingolipid results in neural cell death. In addition to Sandhoff disease the family includes Tay-Sachs disease and the GM2 activator deficiencies.
Hexosaminidases
GM2 ganglioside degradation requires the enzyme β-hexosaminidase and the GM2 activator protein (encoded by the GM2A gene). Hexosaminidases are a dimer composed of two subunits, either the α subunit (encoded by the HEXA gene) and/or the β subunit (encoded by the HEXB gene). The various isoforms of β-hexosaminidase result from the combination of α and β subunits. The HexS protein is a homodimer of αα, HexA is a heterodimer of αβ and HexB is a homodimer of ββ. It is the α-subunit that carries out the catalysis and only the HexA form of β-hexosaminidase can catalyze the cleavage of GM2 gangliosides (the bond cleaved is shown by the arrow in the Figure below). The activator first binds to GM2 gangliosides followed by hexosaminidase and then digestion occurs. The degradation of GM2 gangliosides takes place in the lysosomes. Failure to degrade these sphingolipids results in the lysosomes becoming engorged filling the cell, eventually choking off normal cellular functions. Because of the deposition of abnormal sphingolipids in the lysosomes the GM2 gangliosidotic diseases are also referred to as lysosomal storage diseases.
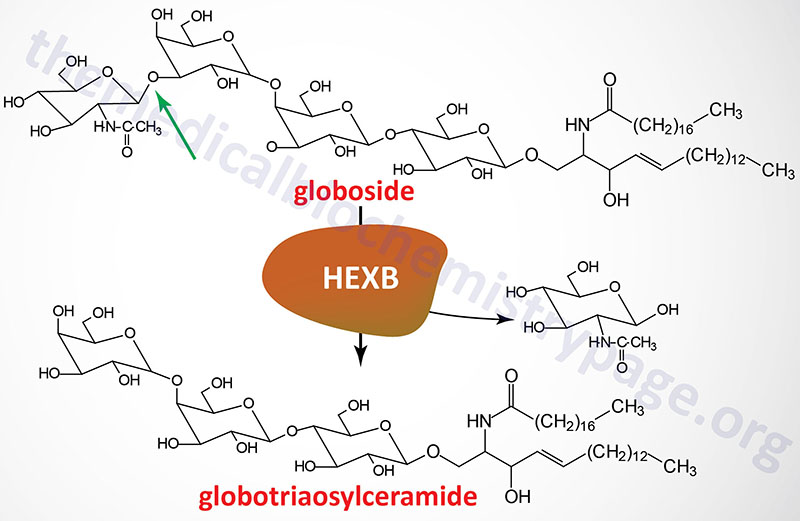
Molecular Biology of Sandhoff Disease
Sandhoff disease results from defects in the HEXB gene encoding the β-subunit of β-hexosaminidase. As such, Sandhoff disease, and variants, are defective in both the HexA and HexB forms of the enzyme. Deficiencies in the HEXA gene result in Tay-Sachs disease and deficiencies in the GM2A gene result in GM2 activator deficiency diseases (also referred to as the AB variant of Tay-Sachs disease).
The HEXB gene resides on chromosome 5q13.3 spanning 45 kb and composed of 15 exons that generate two alternatively spliced mRNAs. These mRNAs encode preproproteins of 556 amino acids (isoform 1) and 331 amino acids (isoform 2).
At least 26 mutations have been identified in the HEXB gene resulting in Sandhoff disease. Mutations in the HEXB gene will result in defective synthesis of the β-subunit of β-hexosaminidases and thus defective formation of both the HexA and HexB forms of the enzyme. The infantile forms of Sandhoff disease are very severe and infants will not normally survive beyond 2 years of age.
Clinical Features of Sandhoff Disease
The clinical phenotypes associated with the infantile forms of Sandhoff disease, Tay-Sachs disease, and GM2 activator deficiency are for the most part indistinguishable. The initial discrimination of Sandhoff disease from Tay-Sachs disease was accomplished by enzyme assay not by clinical differential.
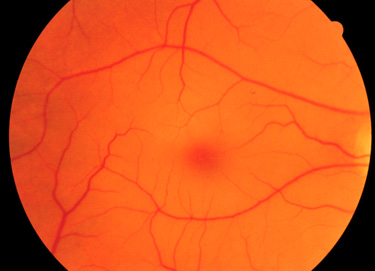
Infants with Sandhoff disease appear normal at birth. Symptoms usually begin with mild motor weakness by 3 to 5 months of age. Parents will begin to notice that their afflicted child has a dull response to outside stimuli. Another early symptom is an exaggerated startle response (sudden extension of arms and legs) to sharp sounds. By 6 to 10 months of age infants will begin to show regression of prior acquired motor and mental skills. It is the loss of these activities that will normally prompt parents to seek a medical opinion. A progressive loss in visual attentiveness may lead to an ophthalmological consultation which will reveal macular pallor and the presence of the characteristic “cherry-red spot” on the fundus of the eye (see Figure below).
At around 8 to 10 months of age the symptoms of Sandhoff disease begin to progress rapidly. Infants are progressively non-responsive to parental stimulation. The exaggerated startle response becomes quite pronounced. Most frightening to parents is the onset of seizures which initially can be controlled by anti-seizure medication. However, the seizures become progressively more severe and are very frequent by the end of the first year. Psycho-motor deterioration increases by the second year and invariably leads to decerebrate posturing (typical of patients in persistent vegetative states), difficulty in swallowing and increased seizure activity. Ultimately, the patient will progress to an unresponsive vegetative state with death resulting from bronchopneumonia resulting from aspiration in conjunction with a depressed cough.