
Last Updated: November 9, 2025
Introduction to Sialidosis
Sialidosis is an autosomal recessive disease that belongs to the family of disorders identified as lysosomal storage disorders. This disorder is characterized by the lysosomal accumulation of sialyloligosaccharides derived from glycoproteins and glycolipids as a consequence of defects in the lysosomal hydrolase, neuraminidase (also referred to as sialidase). There are two distinct subtypes of sialidosis identified ad type 1 (adult) and type 2 (infantile). Type 2 sialidosis is also referred to as mucolipidosis I, ML-I.
Humans express four genes encoding neuraminidase, NEU1, NEU2, NEU3, and NEU4. The NEU1 encoded enzyme is lysosomal, the NEU2 encoded enzyme is cytosolic, and the NEU3 encoded enzyme is membrane localized. Mutations in the NEU1 gene are the causes of sialidosis.
Deficiency in neuraminidase 1 is also associated with galactosialidosis (also referred to as Goldberg syndrome).
Molecular Biology of Sialidosis
The neuraminidase 1 gene (NEU1) is located on chromosome 6p21.33 and is composed of 6 exons that encode a 415 amino acid precursor glycoprotein.
As of 2025 there have been a total of 52 pathogenic mutations identified in the NEU1 gene associated with sialidosis.
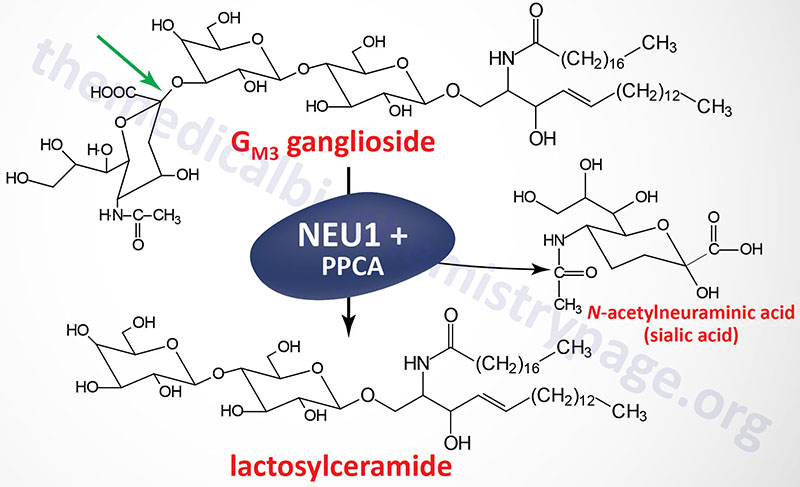
Neuraminidases have dual functions, they are involved in lysosomal degradation of sialylated glycoconjugates as well as in cellular immune responses. Functional neuraminidase is found in a multisubunit complex with β-galactosidase (GLB1) and protective protein/cathepsin A (PPCA). Deficiency in β-galactosidase results in GM1 gangliosidosis.
Neuraminidase functions in the regulation of immune responses primarily through it’s action on specialized antigen-presenting cells called dendritic cells (DCs). The action of neuraminidase on DCs leads to their activation and maturation, thus enhancing immune responses to foreign antigen.
Clinical Features of Sialidosis
Clinically, sialidosis patients can be divided into two types. Type 1 sialidosis is the more mild form of the disease. Type 2 sialidosis is further divided into congenital, infantile and juvenile forms.
Type 1 Sialidosis
Patients with type 1 sialidosis appear normal for the first 20 to 30 years of life. Symptoms that bring attention to the this form of the disease is the development of walking difficulties and/or the loss of visual acuity. As a consequence of ophthalmic examination to address the vision problems, the presence of ocular cherry-red spots (as described for Tay-Sachs disease) becomes apparent. In fact, so common is this clinical finding in type 1 sialidosis that the disease is often referred to as the cherry-red spot-myoclonus syndrome.
Type 2 Sialidosis
Type 2 sialidosis symptoms are more severe and present much earlier than in type 1 disease. All three forms of type 2 sialidosis are associated with coarse facial features and intellectual impairment. The congenital form is evident in utero, the infantile form manifests between birth and 12 months of age and the juvenile form manifests after 2 years of age. Because deficiencies in neuramindase are associated with sialidosis and galactosialidosis it may be that those patients defined as type 2 juvenile may in fact be afflicted with galactosialidosis. The congenital form of the disorder is associated with hydrops fetalis and afflicted fetuses are stillborn.
All patients with type 2 sialidosis will develop a progressive severe phenotype including intellectual impairment and dystosis multiplex. Dystosis multiplex represents a constellation of skeletal abnormalities characterized by an enlarged skull, thickened calvarium, premature closure of lamboid and sagittal sutures, shallow orbits, enlarged J-shaped sella turcica (a saddle-shaped skull structure into which sits the bottom of the pituitary gland), and abnormal spacing of the teeth with dentigerous cysts. There is anterior hypoplasia of the lumbar vertebrae, the long bone diaphyses are enlarged and an irregular appearance of the metaphyses. The epiphyseal centers not well developed, the pelvis is poorly formed with small femoral heads and coxa valga. The clavicles are short, thick and irregular and the ribs are oar shaped. Phalanges are shortened and trapezoidal in shape.